International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288
- Current Issue
- Volume 21; 2025
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Archive
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
1. Pig genetic resources
2. Pig genome mapping and...
3. Approaches to understanding...
4. Future expectations of...
Acknowledgements
References
Tables and Figures
Introduction
1. Pig genetic resources
2. Pig genome mapping and...
3. Approaches to understanding...
4. Future expectations of...
Acknowledgements
References
Tables and Figures

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(3):153-165. doi:10.7150/ijbs.3.153 This issue Cite
Review
Genetic Resources, Genome Mapping and Evolutionary Genomics of the Pig (Sus scrofa)
Kefei Chen1, Tara Baxter1, William M. Muir2, Martien A. Groenen3, Lawrence B. Schook1 4 ![]()
1. Department of Animal Sciences, University of Illinois at Urbana-Champaign, 1201 W. Gregory Dr., Urbana, IL 61801, USA
2. Department of Animal Science, Purdue University, West Lafayette, Indiana 47907-1151, USA
3. Animal Breeding and Genetics Group, Wageningen University, PO Box 9101, Wageningen, 6701 BH, The Netherlands
4. Institute for Genomic Biology, University of Illinois at Urbana-Champaign, 1206 W. Gregory Dr., Urbana, IL 61801, USA
Received 2006-10-16; Accepted 2007-1-9; Published 2007-2-10
Citation:
Chen K, Baxter T, Muir WM, Groenen MA, Schook LB. Genetic Resources, Genome Mapping and Evolutionary Genomics of the Pig (Sus scrofa). Int J Biol Sci 2007; 3(3):153-165. doi:10.7150/ijbs.3.153. https://www.ijbs.com/v03p0153.htm
Other stylesAbstract
The pig, a representative of the artiodactyla clade, is one of the first animals domesticated, and has become an important agriculture animal as one of the major human nutritional sources of animal based protein. The pig is also a valuable biomedical model organism for human health. The pig's importance to human health and nutrition is reflected in the decision to sequence its genome (3X). As an animal species with its wild ancestors present in the world, the pig provides a unique opportunity for tracing mammalian evolutionary history and defining signatures of selection resulting from both domestication and natural selection. Completion of the pig genome sequencing project will have significant impacts on both agriculture and human health. Following the pig whole genome sequence drafts, along with large-scale polymorphism data, it will be possible to conduct genome sweeps using association mapping, and identify signatures of selection. Here, we provide a description of the pig genome sequencing project and perspectives on utilizing genomic technologies to exploit pig genome evolution and the molecular basis for phenotypic traits for improving pig production and health.
Keywords: Alternative splicing, Association mapping, Domestication, Genetic diversity, Genome sequencing, QTL, Selection, Selective sweeps, SNPs
Introduction
The recent completion of the human genome sequence provides a starting point for understanding genetic complexity and elucidating genetic variations contributing to diverse traits and diseases. Pigs are even-toed ungulates belonging to the order artiodactyla, an order phylogenetically closer to primates than rodentia [1]. A separate suborder, the suina includes hippopotamuses, peccaries and pigs. All pigs are members of the suidae family. The pig is of particular interest in evolutionary studies not only because existing pig breeds show great phenotypic varieties for morphological, physiological and behavior traits but also because the wild ancestors of domesticated pigs and a convenient number of outgroup species are still present in the world. The pig (S. scrofa domesticus) was domesticated from S. scrofa, a wild boar, approximately 9,000 years ago in multiple regions of the world [2-4]. These domestication events were separated not only by 1000s of kilometers but also by 1000s of years. During the past decade, there has been an increasing interest in detecting genes and genomic regions in human and other organisms. Domestic animal species have experienced strong selective pressures directed at genes or genomic regions controlling traits of biological, agricultural, or medical importance following their domestication and subsequent episodes of selective breeding. Consequently, these genes or genomic regions are expected to exhibit signatures of selective breeding. Pigs offer a unique opportunity to identify genes or genomic regions encoding quantitative trait loci (QTLs) since they have been through recent and strong selective sweeps targeted at phenotypes to improve agricultural performance and disease resistance.
The pig whole genome sequencing project has been launched in the early of 2006 initiated by the Swine Genome Sequencing Consortium (SGSC) (http://www.piggenome.org/). In addition to providing important evolutionary information, the availability of the pig whole genome sequence will contribute toward revealing the molecular mechanisms controlling phenotypes and play an increasingly significant role in pork production, by integrating 'omics' techniques and bioinformatics tools to reduce the incidence of disease and respond more rapidly to the changing demands of consumers.
1. Pig genetic resources
S. scrofa is one of the most globally widespread mammalian species. It has long been assumed that the force driving evolution was domestication and natural selection. Domestic pigs are found in a globally wide range of environments. Several features, including teeth and skull morphology, external proportions, hair and colour patterns, biochemical and molecular polymorphisms, ecology and behaviour, reproductive isolation and natural areas, are used for discriminating the many species in the genus Sus. S. scrofa is classed into a large number of subspecies, but the number is uncertain and depends on the definition of the subspecies. It has been possible to discriminate more than 16 distinct subspecies, each occupying distinct geographical areas [5-8].
1.1. Pig domestication
Domestication is the process of genetically adapting a wild biological organism to better suit the needs of human beings, as a result of living and breeding conditions under careful human control for multiple generations [9]. Pig domestication has been an integral part of the rise of agriculture and the adoption of the agricultural practices throughout much of the world. Insights into the evolution and spread of the pig are likely to deepen our understanding of the origins and spread of livestock agriculture and the rise of early human civilization. The earliest remains of domesticated pigs have been excavated at Çayönü in southeast Anatolia dated to 7,000 BC [10]. According to most traditional but arguable views based on extensive zooarcheological record [6], the domestic pig originated in the near east and spread west to Europe and east to China. However, recent preliminary research using mitochondrial DNA (mtDNA) sequences from samples of Eurasian wild boars and various breeds of domestic pigs has provided evidence to support a “multiple and independent domestication” hypothesis [2, 3]. Additional recent mtDNA data from the analysis of 685 individuals including wild boars, feral and domestic pigs across Eurasia also support the hypothesis that the pig domestication occurred independently in the world at diverse geographic locations across Eurasia: three from Far-East (two in China, additional ones in Thailand/Burma and northern India), one from Island South-East Asia (Wallacea), and two from Europe [4]. These results also suggest that the S. scrofa as a species originated from islands in South-East Asia (Phillippines, Indonesia), where they dispersed across Eurasia, and with little or no importation of Near East domestic pigs into Europe by early farmers.
Domestication also provides rapid phenotypic evolution through artificial selections. Pig domestication has resulted in highly modified morphological architectures and has caused several major changes in physical types, e.g. one of the earliest results of domestication was a decrease in skeletal size [6]. However, it could be argued that size differences in various areas of the world may have arisen from environmental diversity such as feed resources. Improvement after domestication has also resulted in striking changes in yield, biochemical composition, and other traits. Most domesticated animals have experienced a “domestication bottleneck” with reduced genetic diversity relative to their wild ancestor(s). This bottleneck affects all genes in the genome and modifies the distribution of the genetic variation among loci. The magnitude and variance of the reduction in genetic diversity across loci provide insights into the demographic history of domestication.
The pig represents a domesticated animal that has both a convenient number of outgroup species nicely spaced in evolutionary distance, as well as surviving wild conspecifics (see Figure 1). This renders the pig as perhaps one of the most suitable animal species for inferring ancestral mutations as well as determining the fate of derived states and selective processes. Ancestral mutations are important because: (i) the probability that an allele is ancestral is equal to its frequency and (ii) strong positive selection results in regions with reduced heterozygosity and an excess of derived alleles. Since in the case of the pig, it is still unclear as to what constitutes the nearest living relative (likely S. barbatus) and the age of the species S. scrofa relative to some of it's nearest relatives, it is critical to compare S. scrofa with several related species (e.g. S. barbatus, S. celebensis, S. verrucosus, African warthog) that fall within a range of 1 to 6 million years ago (MYA) of inferred evolution [11-14] (Figure 1).
1.2. Natural and artificial selections
Darwin (1859) clearly believed both nature and artificial selection shaped breeds, “The key (to domestic breeding) is man's power to accumulative selection: nature gives successive variations; man adds them up in certain directions useful to him” [15]. Human and novel environmental pressures during pig domestication have been principally responsible for the generation of inter-breed genetically variation and for the formation of many unique breeds. Domestic pig diversity has evolved over millions of years through the processes of natural and artificial selections forming and stabilizing each of the species used in food and agriculture. Over the more recent millennia, interactions between environmental and human selection have led to the development of genetically distinct breeds. Artificial selection in a targeted gene is similar to a more severe bottleneck that removes most of the genetic variation from a targeted locus.
Over the centuries, global pig farming in different environmental conditions has resulted in breeds with traits such as heat/cold tolerance and disease resistance, which favor their survival under environmental stresses. Farmers have also been breeding for a variety of attributes with a major focus on productivity traits such as meat yields and fertility. To date, there are likely over 730 pig breeds or lines worldwide of which two thirds reside in China and Europe and over 270 are considered as endangered or critical (Table 1 and Figure 2) [8]. Currently, 58 pig breeds are recorded as “transboundary” (occuring in more than one country) including 25 regional transboundary breeds and 33 international transboundary breeds. The worldwide distribution of pigs is dominated by five international transboundary pig breeds from the United States (US) or Europe i.e. Large white (117 countries), Duroc (93 countries), Landrace (91 countries), Hampshire (54 countries) and Pietrain (35 countries) [16]. Pig breeds vary greatly in size, color, body shape, ear carriage, behavior, prolificacy, and other traits. In order to meet future challenges in the agricultural and food industries, special efforts are required to conserve genetic resources. Therefore, phylogenetic studies aimed to evaluate the genetic uniqueness and pig breed diversity will assist in developing a rational plan for breed conservation programs. A set of criteria in an attempt to choose specifically breeds for conservation has been suggested including two essential criteria. These include the degree of endangerment and the genetic uniqueness of the breed [17]. In addition, the origin and history of domestic pigs can also be explained by phylogenetic analysis. Independent domestication has occurred from wild boar subspecies in Eurasia, and through the introgression of Asian germplasm into European domestic breeds that occurred during the 18th and early 19th centuries [9, 18].
1.3. Selective sweep detection
When selective pressure is applied to individuals, it ultimately leads to the changes in the underlying genetic content of the population [19]. Individuals that carry a more favorable genotype would outcompete their peers, resulting in the fixation of beneficial alleles in the population with concomitant removal of inferior alleles. Two primary approaches have been utilized to identify and study genes or gene pathways. First is a conventional candidate gene approach which represents a gene selection based on comparative mapping and gene function. The second approach is whole genome scans to identify genomic regions under selection through association mapping, i.e. associating phenotypes with genotypes. A third approach involves identification of genomic patterns due to selective sweeps whereby large-scale high density single nucleotide polymorphism (SNP) haplomap on a specific region from diverse populations along with wild ancestral outgroup species or a panel of genes that might be associated with traits. The identification of the causative mutation for the insulin-like growth factor 2 (IGF2) QTL in pigs is an excellent application using these combined approaches [20]. Furthermore, by using comparative genomic data sets from different breeds containing wild ancestral species, several interesting genotype-phenotype relationships in domestic animals have been recently illustrated [21-28].
A selective sweep results in the elimination of surrounding variation in regions linked to a recently fixed beneficial mutation. For instance, the muscle-favoring mutation in the porcine IGF2 gene (intron3-3072G/A) has swept through commercial pig populations, but is not present in the tested Asian or European wild boars [20]. More recently, a naturally occurring G to A transition in the 3' untranslated region of the myostatin gene creates a target site for mir1 and mir206 microRNAs (miRNAs) affecting muscularity in sheep, and a selective sweep has been detected in the hypermusculed Texel sheep [28]. The identification of selective sweeps is interesting, not only because it elucidates important evolutionary questions, but also because of the increasing evidence linking selection and disease genes [29, 30]. The beneficial substitution of an allele shapes patterns of genetic variation at linked sites, and may provide important insights into (i) the mechanisms of evolutionary change; (ii) guide selection of loci for population genetic studies; (iii) facilitate significant genomic regions; and (iv) help elucidate genotype-phenotype correlations in complex traits [31].
Genome scans for detecting signatures of selective sweeps in natural populations have been proposed as a phenotype independent approach to identifying adaptive trait loci even when gene function or phenotype of interest are unknown [32]. There are many different methods available for detecting selective sweeps from DNA sequence data [29, 33-36]. Hitchhiking mapping provides a universal approach for the identification of important mutations and selective sweeps. Hitchhiking is a phenomenon known as neutral variants linked to the beneficial mutation are also affected by a selective sweep [37]. This approach has been very successful for identification of selective sweeps at several genes [38, 39]. More information about genes causing the sweep can be obtained if divergent populations are compared, particularly if the populations have been exposed to well-known selection regimes. Similar comparisons could be performed for hitherto uncharacterized, commercially important traits, such as fat content in pigs. The most ambitious goal of hitchhiking mapping is the identification of quantitative trait nucleotides (QTNs) that confers the selective advantage [32].
1.4. Integrated global pig biodiversity
Comparative genomic analysis of different domestic breeds can prove an efficient way of exploiting the genetic basis of phenotypic variation [40]. Phylogenetic studies can reconstruct the correct genealogical ties between species and estimate the time of divergence between two organisms since they last shared a common ancestor.
To help understand the animal evolutionary history and genetic diversity, a variety of genetic markers can be utilized. Genetic markers can generally be grouped into two types based on their association with functionality: type I markers are DNA segments encoding for expressed DNA sequences which possess a relatively low degree of polymorphism but high evolutionary conservation, whereas type II markers usually have no identifiable biological function but they are highly polymorphic and not well conserved between species. The comparison of the characteristics of main classes of genetic markers is shown in Table 2 [41-43]. As one of the most widely used marker types, microsatellites (also called simple sequence repeats, SSRs), are characterized as having a short motif, generally from 1 to 6 bp, are commonly regarded as “junk DNA”; however, SSRs have served as one of the most important markers for genome mapping as well as phylogenic studies. SSRs have been more recently proposed to modify genes with which they are associated. The influence of SSRs on gene regulation, transcription and protein function typically depends on the number of repeats, while mutations that add or subtract repeat units are both frequent and reversible. Over the past decade, it have been demonstrated that SSR variation has been tapped by natural and artificial selection to affect almost every aspect of gene function [44]. In addition, mtDNA is a widely used molecular tool in domestication studies, but it suffers from the limitations of poor information for the whole genome and the loss of male-mediated gene flows by its maternal inheritance patterns.
To date, a number of molecular markers have been used for genetic diversity and phylogenetic analysis in pigs including SSRs [45-49], AFLPs [50, 51], SNPs [52, 53] and mtDNA genotyping [2-4, 54-61]. SSR markers have been largely used in phylogenetic studies and to measure differences within breeds, however due to their neutral properties, they are poorly correlated with phenotypic changes due to selection. Very recently the use of gene markers has attracted more researchers as variation in these allele frequencies may provide information related to functional differences between breeds. Phylogenetic studies using gene markers or SNPs associated with traits of interest are relevant for breed conservation and potential breeds efficiently for the future production markets. Moreover, mtDNA maternally inherited is useful for tracing the maternal lineages in populations. Alternatively, variable sequences on the Y chromosome are useful to measure breed history and phylogenetic origins, although it is much less variable within species than most other genomic sequences [62]. The largest ongoing project on biodiversity studies of pig breeds is the European Union (EU) pig biodiversity project II (PigBioDiv II), which will evaluate and compare genetic diversity among at least 100 pig breeds originated from China and Europe [49-51, 53, 60, 61]. The project not only determines the relationships between breeds by estimating genetic distances, based on SSR markers and haplotypic relationships from mtDNA and Y chromosome polymorphisms, but also determines functional differences among breeds by characterizing trait gene loci and QTL regions.
2. Pig genome mapping and sequencing
Over the past years, our understanding of the pig genome has rapidly evolved from the localization of genes on specific chromosomes to high density marker maps, and now the pig whole genome is being completely sequenced which represents a key milestone to exploit the pig genome evolution and decipher the molecular basis of various phenotypic traits.
2.1. Genome positioning system (GPS)
The availability of large-insert libraries [63-68] allows for a more targeted approach to physical and comparative mapping. Over 620K BAC end-sequences (BES) with an average read length of 635 bp have provided a previously untapped source of both coding and noncoding porcine sequence information [69]. The first high-resolution, physically anchored, contiguous whole genome radiation hybrid (RH) comparative maps of the porcine autosomes were constructed by using physically anchored sequences derived from BACs [70]. Furthermore, a physical map of the pig genome by integrating 265K restriction fingerprints and BES generated from 4 BAC libraries with RH markers, and contig alignments to the human genome was recently constructed with coverage across the 18 pig autosomes and the X chromosome in 176 contigs with an average length of 15 Mb as well as localised representation of the gene rich regions on Y. The map represents an entry point for rapid electronic positional cloning of genes and fine mapping of QTLs, and also provides a platform for the selection of an efficient minimum tiling path (MTP) through the genome to support clone-based sequencing and targeted functional genomics studies (http://www.sanger.ac.uk/Projects/S_scrofa/WebFPC/porcine/large.shtml). Exploitation of this resource as well as the complete human sequence and bioinformatics tools permit the establishment of an ordered list of unique sequences from which to select evenly spaced markers prior to mapping [69].
With the development of molecular markers, porcine genomic maps have been largely enriched in the last few years. The pig genome database has entries for over 4,000 loci including more than 1,588 genes and 2,493 markers (http://www.animalgenome.org/pig/). However, while the average distance between markers is about 2 - 3 cM, some large gaps still exist in the pig genetic linkage map (http://www.marc.usda.gov/genome). The physical map for pigs as for other farm animals lagged behind initially. With the use of a somatic cell hybrid panel [71] and a 7,000 rad (IMpRH) or recently of a 12,000 rad (IMNpRH2) RH panel [72-74], the physical map has been growing rapidly and contains now over 10,000 genes and markers [75]. The publicly available information related to pig genomics and proteomics is shown in Table 3.
2.2. The pig genome project
The pig whole genome is currently being sequenced by The Wellcome Trust Sanger Institute through funding provided by Cooperative State Research, Education and Extension Service at the United States Department of Agriculture (CSREES-USDA) (target of 3X genome coverage sequencing by January 2008) [76]. This project uses a clone-by-clone sequencing strategy, based on the MTP of BAC clones. The planned order of contig selection for sequencing is: (i) SSC7, SSC14 and SSC4 are highest priority since additional EU funding targeting these chromosomes started earlier; (ii) SSCX, since it will be more challenging to complete and require increased depth sequencing; and (iii) SSC1, SSC11, SSC17, SSC5, SSC6, SSC2, SSC3, SSC8, SSC9, SSC10, SSC12, SSC13, SSC15, SSC16, and SSC18. To date, a total of 7,321 CHORI-242 clones have been selected and used to generate initial shotgun sequencing data (> 52% of the swine genome) (Table 4). Since the CHORI-242 represents a female Duroc pig, 495 additional BACs with at least one BES anchored on chromosome X or Y from the French National Institute for Agricultural Research (INRA) BAC library was selected for sequencing the chromosome Y. A total of 1,660 accessioned clones have generated > 287 Mb of sequence. A pre-finishing strategy is being employed for gap closure and ambiguity resolution. Automated annotation will be used after the entire chromosome has been sequenced (http://www.piggenome.org/).
To take advantage of the emerging genome sequence and the characterization of new QTLs, there is an increasing need for improving the process of SNP discovery to define haploblocks in unique germplasms. Thus, a discovery platform that exploits ancestral chromosomes for unique SNP discovery would expedite SNP discovery for exploitation in breeding. Also there is a need for a united, global initiative that captures and utilizes the broadest porcine germplasms. Porcine SNP discovery is ongoing and several large projects have been completed (Sino-Danish) or are currently being initiated by INRA-Genescope in conjunction with SGSC pig genome sequencing project [76]. Within the Sino-Danish initiative [77], 3.84 million sequences have been generated using 5 different breeds (Duroc, Erhuanlian, Hampshire, Landrace and Yorkshire) and within the Genescope initiative, 1 million sequences are being generated from 7 different breeds (Iberian, Landrace, Meishan, Minipig, Pietrain, Wild boar and Yorkshire) [77, 78]. However, the discovery of SNPs using a limited pool of independent germplasm limits the potential to identify QTLs using genome-wide SNP sweeps and the ability to identify traits highly difficult to phenotype (reproduction, disease resistance) or marker-associated introgression of traits from wild-type alleles into commercial breeding populations. This supports the need for an alternative strategy to generate informative SNPs for use in commercial populations. In addition, the EU PigBioDiv II has provided significant insights into the multiple origins of the pig and phenotypic variation associated with geography, breeding and husbandry practices. Using 1,536 SNPs, distributed across the genome for genotyping 672 DNA samples, it has been demonstrated that the utility of SNPs is being able to define haploblock structure and extending linkage disequilibrium (LD) into genomic regions where genes controlling agricultural traits have been selected [53].
3. Approaches to understanding genome evolution
The relationship between genome size and organismal complexity remains unanswered. The C-value (genome size) paradox is that genome size does not correlate closely with organismal complexity [79]. However, the genomes of more complex organisms are, on average, larger than the genomes of less complex. The C-value of the domestic pig varies from 2.81-3.51 measured using various cell types and by different methods [80-82]. The pig genome comprises 18 autosomes and X/Y sex chromosomes with a size of 2.7 gigabases (Gb) estimated by integration of BES and fingerprints [69, 76]. Comparative genomic analysis indicates that organismal complexity arises from progressively more elaborate regulation of gene expression, and physiological/ behavioral complexity correlates with the likely number of gene expression patterns exhibited during an animal's life cycle [83]. The unexpectedly high frequency of alternative splicing (AS) events has been proposed to be an attractive mechanism for increasing gene expression patterns and consequently for the organismal complexity in eukaryotes [84, 85]. As one of the most exciting recent discoveries in the field of genomics, the ultraconserved regions that are not functionally transcribed in mammalian genomes, has been suggested to play important role as transcriptional regulatory elements, and account for the complexity of gene regulation [86-89]. This is particularly evident for some genes involved in embryonic development. Another mechanism for increasing organismal complexity was suggested to be DNA arrangement where genes themselves are rearranged during cellular differentiation [90].
3.1. Comparative cytogenetics and genomics
Genome organization has traditionally been inferred using two approaches: cytogenetics mapping and genetic-linkage or physical mapping [91]. Comparisons of G-banded chromosome patterns were first used to infer homologies of whole chromosomes or subregions between species and even across mammalian orders. Gene mapping utilizing somatic cell hybrids subsequently confirmed the large tracts of mammalian genomes were remarkably conserved, suggesting that transferring information from species such as human and mouse, which have gene-rich maps, to the gene-poor developing maps of domestic animals is feasible [92]. Chromosome painting [or Zoo-fluorescence in situ hybridization (Zoo-FISH)] permits rapidly detecting entire chromosomal homologies across mammalian orders. Genetic linkage map are best suited to ordering polymorphic SSR markers, but less efficient for developing comparative maps since the limited degree of coding locus (type I markers) polymorphism observed within most interspecies crosses. Radiation hybrid (RH) mapping has proven to be an effective approach for the rapid ordering of evolutionarily conserved type I coding gene markers over the whole genome of various species [70, 74, 92, 93]. Genome sequence based comparative mapping is becoming a powerful approach to reveal the molecular basis for phenotypic variation as well as the evolutionary forces that have contributed to speciation, including underlying mutational processes and selective constraints [94-96]. In addition to comparative genome mapping, with the integration of genomics and phylogenetics, phylogenomic studies are progressing to resolve long-standing evolutionary/phylogenetic controversies, to refine dogma on how chromosomes evolve, and to guide annotation of human and other mammalian genomes [97].
3.2. Exploiting varieties of genomic architectures
Genome rearrangements: In eukaryotes, genome rearrangements, such as inversion, translocations and duplications, are common and range from gene segments to hundreds of genes. In most eukaryotes, there is a strong association between rearrangement breakpoints and repeat sequences. Rearrangement polymorphisms in eukaryotes are correlated with phenotypic differences, and proposed to confer varying fitness in different environments. There is little evidence that chromosomal rearrangements causes speciation, but probably intensify reproductive isolation between species that have formed by other routes [98]. A relatively large number of chromosomal abnormalities including inversion, translocation, duplication, fission and fusion have been identified in pig [93, 99, 100]. The chromosomal abnormalities are often responsible for a considerable decrease in prolificacy of the carrier animals. Recently, a bioinformatics tool was created to permit multi-species comparisons between the genomes of humans, horses, cats, dogs, pigs, cattle, rats, and mice (http://evolutionhighway.ncsa.uiuc.edu/). This provides a useful resource for evaluating pig evolution. A large set of reuse breakpoints were discovered and more than 20% of the discovered breakpoints have been reused during mammalian evolution. The eight species comparison showed that the historical rate of chromosome evolution in mammals was different than previously thought. The study demonstrated that evolutionary changes has been moving faster during the last 65 million years than for the prior 35 or so million years [92].
Transposable elements: Evolutionary biologists hypothesized that the earliest life originated via a system based on a self-replicating RNA genome and RNA catalysts [101]. The advent of polymerases that make DNA copies of RNA templates allowed the conversion of information from unstable ribose-based polymers to more stable deoxyribose-based polymers through the process of reverse transcription. It is now known that only approximately 1-2% of the human genome is comprised of exonic sequences. The remainder, so-called “junk DNA”, is composed largely of introns, simple repeat sequences and transposable elements or their remnants. In mammals, transposable elements account for nearly 50% of the genome [102, 103]. Transposable elements were historically dismissed as junk or selfish sequences parasitizing the genome of living organisms [104, 105]. This view has been challenged through a wave of new information demonstrating their emergence as contributors to the evolution and function of genes and genomes, and have a tremendous impact on an organism's phenotype [106-108]. These effects include drug response, disease susceptibility and evolution novelties between species. The most common genomic effect of transposable elements is the induction of mutation. Through their mobility and ability to recombine, transposable elements can generate various types of rearrangements and lead to insertions, deletions, duplications and inversions. In mammals, retrotransposon have been proposed to act as general modulators of gene expression and to play a role in X-chromosome inactivation [109, 110]. Transposable elements, first recognized as potential causal agents of human disease in 1988 [111], have evolved over millions of years and have achieved a balance between detrimental effects on the individual and long-term beneficial effects on a species through genome modification. It has been suggested that transposable elements play an important role through diverse ways in the event of shaping the genome to speciation [107].
Single nucleotide mutations: SNPs are abundant and widespread throughout the pig genome (coding and non-coding regions), and are rapidly becoming the marker of choice for many applications in population genomics, evolutionary analysis, conservation genetics, because of the potential for higher genotyping efficiency, data quality, genome coverage and cost-effective high throughput genotyping techniques. In most species, SNPs occur typically on average every 200-500 bp [43, 112-114]. About 90% of genetic variation has been ascribed to SNP allelic variants that occur at a frequency of > 1%. Within coding regions (~1-2%), nonsynonymous SNPs can be considered candidates for functional changes. The phenotypic effect of any particular SNP is rarely known and often can only be inferred based on the evolutionary dynamics of the variant or on its effect on protein function. The nonsynonymous (dN) : synonymous (dS) SNPs ratio (dN/dS also known as Ka/Ks) can then be taken as a measure of the strength of purifying selection on a gene or the entire genome. Even synonymous SNPs in protein-encoding genes can have functional implications. Although multiple codons can encode the same amino acid, some occur more frequently in the genome than is predicted by random (i.e. codon usage bias). Therefore, a SNP that causes a change from a more common or preferred codon to a rare or unpreferred codon can affect the efficiency of protein synthesis and expression. Most SNPs occurs in the non-coding portion of the genome, but can nevertheless be evaluated with regard to function. For example, the IGF2-intron3-G3072A substitution causes a major QTL effect on muscle growth in the pig [20], and explains a major imprinted QTL effect on backfat thickness in a Meishan × European white pig intercross [115, 116].
A substantial fraction of the non-coding genome is conserved between species, suggesting that purifying selection acts on a large portion of the genome. Thus, SNPs can be evaluated based on their location in conserved versus non-conserved non-coding regions. Moreover, the regulatory regions of genes (e.g. promoters, enhances, silencers, insulators, miRNA binding sites) have been annotated using comparative and predictive algorithms, and thereby enabling the assessment of non-coding regulatory SNPs. For instance, SNPs that occur in the transcription factor binding sites of a promoter are more likely to affect function than SNPs that occur outside the regulatory region of a gene [28, 117]. Although ascertainment bias can be a problem with some applications, SNPs can generate equivalent statistical power whilst providing broader genome coverage and higher quality data than can either SSRs or mtDNA, suggesting that SNPs could become an efficient and cost-effective genetic tool.
3.3. Alternative splicing (AS) events and evolutionary impacts
Alternative splicing (AS), one of the most important and nearly ubiquitous mechanisms regulating gene expression in many organisms, occurs in the coding sequence, coordinates physiologically meaningful changes in protein structure and function and is a key mechanism to generate the complex proteome of multicellular organisms. AS results in two ways: (i) through skipping exons that encode a certain protein feature; and (ii) by introducing a frameshift that changes the downstream protein sequences. Recently, novel types of AS events have been proposed that either join two non-consecutive exons (creating a protein feature) or insert an exon into the protein body (destroying a feature) [118]. The effects of AS range from a complete loss of function or acquisition of a new function to very subtle modulations, which are observed in the majority of cases reported such as binding properties, enzymatic activity, intracellular localization, protein stability, phosphorylation and glycosylation patterns [119].
It has been estimated that 30-70% of mammalian genes are alternatively spliced [120-122], and that mammalian AS events frequently arise from the evolutionarily rapid loss or gain of exons from genomes [121, 123-125]. Variant splice patterns are often specific to different stages of development, particular tissues or a disease state [126]. Utilizing a highly predictive computational method over 11% of human and mouse alternative exons were estimated to represent species-specific AS events [127]. By comparing gene structure of orthologous genes in human and mouse genomes, it has been revealed that the majority (98%) of human constitutive and major forms of alternative exons are conserved in the genomic sequences of their mouse and rat orthologues [121]. By contrast, nearly 75% of the minor forms of alternative exons are not conserved, suggesting that AS is associated with a significant increase in the rate of exon creation and deletion in mammals, and plays a role on speciation events.
Splicing mutations have long been proposed to be the basis for a number of human diseases [128]. More recently, based on the disease-gene propensity of human genes in terms of their coding region length and intron number, it was estimated that ~60% of human disease mutations represent splicing mutations, the most frequent cause of hereditary diseases [129]. Although the importance of AS in various biological processes such as sex determination [130] and apoptosis has been known for a long time, genomics and in particular the shotgun sequencing expressed sequence tags (ESTs), have revealed its nearly ubiquitous role in gene regulation [85]. Genome sequencing has made it possible to study the evolutionary impact and constraints of AS [131].
3.4. Exploring functional portion of the genome
Recently, it was estimated that according to sequence conservation patterns, the actual functional portion of the mammalian genome is at least 5% [103]. In mammals, using comparative evolutionary approaches it appears that functional elements are clustered mostly within ~2 kb surrounding protein-coding sequence [132, 133]. These observations help to paint a general picture of noncoding conservation and structure in the genome and are likely to be extremely helpful in focusing future detailed investigation. Given that the protein-coding fraction is approximately 1.5%, there is significant opportunity for identification of additional functional elements. Sequence conservation does not reveal the total fraction of the functional genome, but simply the fraction of the genome that has remained functional within the group of species compared. An additional fraction that is not conserved across larger evolutionary distances such as across all vertebrate lineages represent species-specific or lineage-specific genes. The best known functional fraction is the class of protein-coding genes. Regulatory elements and noncoding RNAs such as small interfering RNAs, (siRNAs) and miRNAs are considered two other significant functional classes of the mammalian genomes. Analysis of the human and mouse genomes has identified an abundance of conserved non-genic sequences (CNGs). The significance and evolutionary depth of their conservation remain unknown. A striking extremely high number of such elements is found in vertebrate gene deserts, defined as long regions (> 500 kb) containing no protein-coding sequences and without obvious biological functions [87-89]. It has been suggested that a global role of CNGs in genome function and regulation, through long-distance cis or trans chromosomal interactions [134].
4. Future expectations of facilitating pig genome navigation
Exploring the complete functional information encoded in a genome is a major challenge in biological research. Comparative genome analysis between the pig and related mammals could provide a powerful and general approach to identifying functional elements without previous knowledge of function and detect phylogenetic footprinting of pig genome evolution. A principal goal of genetic research is to identify specific genotypes that are associated with phenotypes and to conduct genome-wide genotyping on a massive scale. The advent of the complete genome sequencing along with gene prediction has resulted in the development of technologies that allow the assignment of genes to particular biological modules. Integration of 'omic' technologies including genomics, transcriptomics, proteomics and metabolomics will link genomics and system biology and accelerate the acquisition of fundamental knowledge about biology systems. The outputs of 'omics' research will change our approach to solving biological problems and result in novel uses of biotechnology to develop and improve products for agriculture. Advances in genome-phenome research will contribute to agriculture and food, bioengineering, biomedicine and health, conservation and the environment. Genome to phenome research for the pig is still at a very early stage, and requires enormous amount of work to understand the genetics and development of shape, specialization and organization at levels from cells to the whole individual.
Since the whole genome sequence of the pig will soon be available, comparative studies with the completed human genome, and other mammalian genomes having moderate to deep genome coverage (i.e. cow, horse, dog, mouse, rat and chimpanzee) will yield new information about the pig genome evolution. In the next decade, by utilizing approaches of comparative genomics, it will be possible to effectively select animals for agricultural purposes, create appropriate biodiversity conservation programs and create pig models for medical research. The utility of the pig in biomedical research affords many advantages compared with other animals such as mouse and rat i.e. (i) its similar size to humans (ii) sharing high similarities with human both anatomically and physiologically; and (iii) the ability to target gene manipulation and clone using nuclear transfer.
Acknowledgements
We would like to acknowledge the funding from USDA/NRI-CSREES AG2006-35216-16668, AG2005-4480-15939, AG2004-35205-14187, AG2002-3448-11828, AG2002-35205-12712, AG2001-3520-11698; USDA-ARS AG58-5438-2-313.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Jorgensen FG, Hobolth A, Hornshoj H, Bendixen C, Fredholm M, Schierup MH. Comparative analysis of protein coding sequences from human, mouse and the domesticated pig. BMC Biol. 2005;3:2
2. Giuffra E, Kijas JM, Amarger V, Carlborg O, Jeon JT, Andersson L. The origin of the domestic pig: Independent domestication and subsequent introgression. Genetics. 2000;154(4):1785-1791
3. Kijas JM, Andersson L. A phylogenetic study of the origin of the domestic pig estimated from the near-complete mtDNA genome. J Mol Evol. 2001;52(3):302-308
4. Larson G, Dobney K, Albarella U. et al. Worldwide phylogeography of wild boar reveals multiple centers of pig domestication. Science. 2005;307(5715):1618-1621
5. Groves CP. Ancestors for the Pigs. Canberra, Australia: Australian National University Press. 1981
6. Epstein J, Bichard M. Pig. In: (ed.) Mason IL. Evolution of Domesticated Animals. New York: Longman. 1986:145-162
7. Ruvinsky A, Rothschild MF. Systematics and evolution of the pig. In: (ed.) Ruvinsky A, Rothschild MF. The Genetics of the Pig. Oxon, UK: CAB International. 1998:1-16
8. FAO SoW-AnGR. The State of the World's Animal Genetic Resources for Food and Agriculture. 1st ed. Rome: FAO. 2006
9. Darwin C. The Variation of Animals and Plants Under Domestication. 1st ed. Lodon, UK: John Murray. 1868
10. Reed CA. The pattern of animal domestication in the prehistoric Near East. In: (ed.) Ucko PJ, Dimbleby GW. The Domestication and Exploitation of Plants and Animals. London, UK: Duckworth. 1969:361-380
11. Randi E, Lucchini V, Diong CH. Evolutionary genetics of the suiformes as reconstructed using mtDNA sequencing. J Mamm Evol. 1996;3:163-194
12. Groves CP, Schaller GG, Amato G, Khounboline K. Rediscovery of the wild pig sus bucculentus. Nature. 1997;386:335
13. Fokkinga A. Het Varkensboek. The Netherlands: Uitgeverij Thoth. 2004
14. Robins JH, Ross HA, Allen MS, Matisoo-Smith E. Taxonomy: Sus bucculentus revisited. Nature. 2006;440(7086):E7
15. Darwin C. On the Origins of the Species by Means of Natural Selection, Or the Preservation of Favoured Races in the Struggle for Life. UK: John Murray. 1859
16. Domestic Animal Diversity Information System. 2006 DAD-IS. http://www.fao.org/dad-is/
17. Ruane J. A critical review of the value of genetic distance studies in conservation of animal genetic resources. J Ani Breed Genet. 1999;116:317-323
18. Jones GF. Genetic aspects of domestication, common breeds and their origin. In: (ed.) Rothschild MF, Ruvinsky A. The Genetic of the Pig. Oxon, UK: CAB International. 1998:17-50
19. Nielsen R. Molecular signatures of natural selection. Annu Rev Genet. 2005;39:197-218
20. Van Laere AS, Nguyen M, Braunschweig M. et al. A regulatory mutation in IGF2 causes a major QTL effect on muscle growth in the pig. Nature. 2003;425(6960):832-836
21. Grobet L, Martin LJ, Poncelet D. et al. A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle. Nat Genet. 1997;17(1):71-74
22. Milan D, Jeon JT, Looft C. et al. A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science. 2000;288(5469):1248-1251
23. Galloway SM, McNatty KP, Cambridge LM. et al. Mutations in an oocyte-derived growth factor gene (BMP15) cause increased ovulation rate and infertility in a dosage-sensitive manner. Nat Genet. 2000;25(3):279-283
24. Mulsant P, Lecerf F, Fabre S. et al. Mutation in bone morphogenetic protein receptor-IB is associated with increased ovulation rate in booroola merino ewes. Proc Natl Acad Sci U S A. 2001;98(9):5104-5109
25. Pailhoux E, Vigier B, Chaffaux S. et al. A 11.7-kb deletion triggers intersexuality and polledness in goats. Nat Genet. 2001;29(4):453-458
26. Freking BA, Murphy SK, Wylie AA. et al. Identification of the single base change causing the callipyge muscle hypertrophy phenotype, the only known example of polar overdominance in mammals. Genome Res. 2002;12(10):1496-1506
27. Grisart B, Coppieters W, Farnir F. et al. Positional candidate cloning of a QTL in dairy cattle: Identification of a missense mutation in the bovine DGAT1 gene with major effect on milk yield and composition. Genome Res. 2002;12(2):222-231
28. Clop A, Marcq F, Takeda H. et al. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet. 2006;38(7):813-818
29. Sabeti PC, Reich DE, Higgins JM. et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419(6909):832-837
30. Clark AG, Glanowski S, Nielsen R. et al. Inferring nonneutral evolution from human-chimp-mouse orthologous gene trios. Science. 2003;302(5652):1960-1963
31. Teshima KM, Coop G, Przeworski M. How reliable are empirical genomic scans for selective sweeps? Genome Res. 2006;16(6):702-712
32. Schlotterer C. Hitchhiking mapping--functional genomics from the population genetics perspective. Trends Genet. 2003;19(1):32-38
33. Tajima F. The effect of change in population size on DNA polymorphism. Genetics. 1989;123(3):597-601
34. Fay JC, Wu CI. Hitchhiking under positive darwinian selection. Genetics. 2000;155(3):1405-1413
35. Akey JM, Zhang G, Zhang K, Jin L, Shriver MD. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002;12(12):1805-1814
36. Przeworski M. Estimating the time since the fixation of a beneficial allele. Genetics. 2003;164(4):1667-1676
37. Smith JM, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res. 1974;23(1):23-35
38. Kohn MH, Pelz HJ, Wayne RK. Natural selection mapping of the warfarin-resistance gene. Proc Natl Acad Sci U S A. 2000;97(14):7911-7915
39. Wootton JC, Feng X, Ferdig MT. et al. Genetic diversity and chloroquine selective sweeps in plasmodium falciparum. Nature. 2002;418(6895):320-323
40. Andersson L, Georges M. Domestic-animal genomics: Deciphering the genetics of complex traits. Nat Rev Genet. 2004;5(3):202-212
41. O'Brien SJ. Mammalian genome mapping: Lessons and prospects. Curr Opin Genet Dev. 1991;1(1):105-111
42. Dodgson JB, Cheng HH, Okimoto R. DNA marker technology: A revolution in animal genetics. Poult Sci. 1997;76(8):1108-1114
43. Morin PA, Luikart G, Wayne RK, the SNP workshop group. SNPs in ecology, evolution and conservation. Trends Ecol Evol. 2004;19(4):208-216
44. Kashi Y, King DG. Simple sequence repeats as advantageous mutators in evolution. Trends Genet. 2006;22(5):253-259
45. Paszek AA, Flickinger GH, Fontanesi L. et al. Evaluating evolutionary divergence with microsatellites. J Mol Evol. 1998;46(1):121-126
46. Laval G, Iannuccelli N, Legault C. et al. Genetic diversity of eleven european pig breeds. Genet Sel Evol. 2000;32(2):187-203
47. Fan B, Wang ZG, Li YJ. et al. Genetic variation analysis within and among chinese indigenous swine populations using microsatellite markers. Anim Genet. 2002;33(6):422-427
48. Fang M, Hu X, Jiang T. et al. The phylogeny of chinese indigenous pig breeds inferred from microsatellite markers. Anim Genet. 2005;36(1):7-13
49. SanCristobal M, Chevalet C, Haley CS. et al. Genetic diversity within and between european pig breeds using microsatellite markers. Anim Genet. 2006;37(3):189-198
50. Foulley JL, van Schriek MG, Alderson L. et al. Genetic diversity analysis using lowly polymorphic dominant markers: The example of AFLP in pigs. J Hered. 2006;97(3):244-252
51. SanCristobal M, Chevalet C, Peleman J. et al. Genetic diversity in european pigs utilizing amplified fragment length polymorphism markers. Anim Genet. 2006;37(3):232-238
52. Ciobanu DC, Day AE, Nagy A, Wales R, Rothschild MF, Plastow GS. Genetic variation in two conserved local romanian pig breeds using type 1 DNA markers. Genet Sel Evol. 2001;33(4):417-432
53. Groenen MA, Megens HJ, Crooijmans R. et al. Genetic Diversity in European and Chinese Pigs using SNP Markers. Personal communication. 2007
54. Ursing BM, Arnason U. The complete mitochondrial DNA sequence of the pig (sus scrofa). J Mol Evol. 1998;47(3):302-306
55. Watanobe T, Okumura N, Ishiguro N. et al. Genetic relationship and distribution of the japanese wild boar (sus scrofa leucomystax) and ryukyu wild boar (sus scrofa riukiuanus) analysed by mitochondrial DNA. Mol Ecol. 1999;8(9):1509-1512
56. Lin CS, Sun YL, Liu CY. et al. Complete nucleotide sequence of pig (sus scrofa) mitochondrial genome and dating evolutionary divergence within artiodactyla. Gene. 1999;236(1):107-114
57. Okumura N, Kurosawa Y, Kobayashi E. et al. Genetic relationship amongst the major non-coding regions of mitochondrial DNAs in wild boars and several breeds of domesticated pigs. Anim Genet. 2001;32(3):139-147
58. Kim KI, Lee JH, Li K. et al. Phylogenetic relationships of asian and european pig breeds determined by mitochondrial DNA D-loop sequence polymorphism. Anim Genet. 2002;33(1):19-25
59. Alves E, Ovilo C, Rodriguez MC, Silio L. Mitochondrial DNA sequence variation and phylogenetic relationships among iberian pigs and other domestic and wild pig populations. Anim Genet. 2003;34(5):319-324
60. Fang M, Berg F, Ducos A, Andersson L. Mitochondrial haplotypes of european wild boars with 2n = 36 are closely related to those of european domestic pigs with 2n = 38. Anim Genet. 2006;37(5):459-464
61. Fang M, Andersson L. Mitochondrial diversity in european and chinese pigs is consistent with population expansions that occurred prior to domestication. Proc Biol Sci. 2006;273(1595):1803-1810
62. Bruford MW, Bradley DG, Luikart G. DNA markers reveal the complexity of livestock domestication. Nat Rev Genet. 2003;4(11):900-910
63. Alexander LJ, Smith TP, Beattie CW, Broom MF. Construction and characterization of a large insert porcine YAC library. Mamm Genome. 1997;8(1):50-51
64. Rogel-Gaillard C, Bourgeaux N, Save JC. et al. Construction of a swine YAC library allowing an efficient recovery of unique and centromeric repeated sequences. Mamm Genome. 1997;8(3):186-192
65. Rogel-Gaillard C, Bourgeaux N, Billault A, Vaiman M, Chardon P. Construction of a swine BAC library: Application to the characterization and mapping of porcine type C endoviral elements. Cytogenet Cell Genet. 1999;85(3-4):205-211
66. Al-Bayati HK, Duscher S, Kollers S, Rettenberger G, Fries R, Brenig B. Construction and characterization of a porcine P1-derived artificial chromosome (PAC) library covering 3.2 genome equivalents and cytogenetical assignment of six type I and type II loci. Mamm Genome. 1999;10(6):569-572
67. Anderson SI, Lopez-Corrales NL, Gorick B, Archibald AL. A large-fragment porcine genomic library resource in a BAC vector. Mamm Genome. 2000;11(9):811-814
68. Suzuki K, Asakawa S, Iida M. et al. Construction and evaluation of a porcine bacterial artificial chromosome library. Anim Genet. 2000;31(1):8-12
69. Humphray S, Scott C, Clark R. et al. Sequencing the Pig Genome using a BAC by BAC Approach. Proceeding of the 30th international conference on animal genetics. 2006
70. Meyers SN, Rogatcheva MB, Larkin DM. et al. Piggy-BACing the human genome II. A high-resolution, physically anchored, comparative map of the porcine autosomes. Genomics. 2005;86(6):739-752
71. Yerle M, Echard G, Robic A. et al. A somatic cell hybrid panel for pig regional gene mapping characterized by molecular cytogenetics. Cytogenet Cell Genet. 1996;73(3):194-202
72. Yerle M, Pinton P, Robic A. et al. Construction of a whole-genome radiation hybrid panel for high-resolution gene mapping in pigs. Cytogenet Cell Genet. 1998;82(3-4):182-188
73. Yerle M, Pinton P, Delcros C, Arnal N, Milan D, Robic A. Generation and characterization of a 12,000-rad radiation hybrid panel for fine mapping in pig. Cytogenet Genome Res. 2002;97(3-4):219-228
74. Hawken RJ, Murtaugh J, Flickinger GH. et al. A first-generation porcine whole-genome radiation hybrid map. Mamm Genome. 1999;10(8):824-830
75. Milan D, Beever J, Lahbib Y, Schook L, Beattie C, Yerle M. An Integrated RH Map of the Porcine Genome with More than 5000 Anchoring Points on the Human Genome Provides a Framework for Sequencing of the Pig. Proceeding of the 30th international conference on animal genetics. 2006
76. Schook LB, Beever JE, Rogers J. et al. Swine genome sequencing consortium (SGSC): A strategic roadmap for sequencing the pig genome. Comparative and Functional Genomics. 2005;6(4):251-255
77. Wernersson R, Schierup MH, Jorgensen FG. et al. Pigs in sequence space: A 0.66X coverage pig genome survey based on shotgun sequencing. BMC Genomics. 2005;6(1):70
78. Chardon P. INRA, France. Personal communication. 2006
79. Gregory TR (ed). The Evolution of the Genome. New York, USA: Elsevier Academic Press. 2005
80. Kato H, Harada M, Tsuchiya K, Moriwaki K. Absence of correlation between DNA repair in ultraviolet irradiated mammalian cells and life span of the donor species. Jpn J Genet. 1980;55:99-108
81. Vinogradov AE. Genome size and GC-percent in vertebrates as determined by flow cytometry: The triangular relationship. Cytometry. 1998;31(2):100-109
82. Krishan A, Dandekar P, Nathan N, Hamelik R, Miller C, Shaw J. DNA index, genome size, and electronic nuclear volume of vertebrates from the miami metro zoo. Cytometry A. 2005;65(1):26-34
83. Levine M, Tjian R. Transcription regulation and animal diversity. Nature. 2003;424(6945):147-151
84. Graveley BR. Alternative splicing: Increasing diversity in the proteomic world. Trends Genet. 2001;17(2):100-107
85. Modrek B, Lee C. A genomic view of alternative splicing. Nat Genet. 2002;30(1):13-19
86. Bejerano G, Pheasant M, Makunin I. et al. Ultraconserved elements in the human genome. Science. 2004;304(5675):1321-1325
87. Nobrega MA, Ovcharenko I, Afzal V, Rubin EM. Scanning human gene deserts for long-range enhancers. Science. 2003;302(5644):413
88. Ovcharenko I, Loots GG, Nobrega MA, Hardison RC, Miller W, Stubbs L. Evolution and functional classification of vertebrate gene deserts. Genome Res. 2005;15(1):137-145
89. Dermitzakis ET, Reymond A, Antonarakis SE. Conserved non-genic sequences - an unexpected feature of mammalian genomes. Nat Rev Genet. 2005;6(2):151-157
90. Baltimore D. Our genome unveiled. Nature. 2001;409(6822):814-816
91. Murphy WJ, Stanyon R, O'Brien SJ. Evolution of mammalian genome organization inferred from comparative gene mapping. Genome Biol. 2001;2(6):S0005
92. Murphy WJ, Larkin DM, Everts-van der Wind A. et al. Dynamics of mammalian chromosome evolution inferred from multispecies comparative maps. Science. 2005;309(5734):613-617
93. Jiang Z, Michal JJ, Melville JS, Baltzer HL. Multi-alignment of orthologous genome regions in five species provides new insights into the evolutionary make-up of mammalian genomes. Chromosome Res. 2005;13(7):707-715
94. International Chicken Genome Sequencing Consortium, Hillier LW, Miller W. et al. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432(7018):695-716
95. Lindblad-Toh K, Wade CM, Mikkelsen TS. et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438(7069):803-819
96. Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437(7055):69-87
97. Murphy WJ, Pevzner PA, O'Brien SJ. Mammalian phylogenomics comes of age. Trends Genet. 2004;20(12):631-639
98. Coghlan A, Eichler EE, Oliver SG, Paterson AH, Stein L. Chromosome evolution in eukaryotes: A multi-kingdom perspective. Trends Genet. 2005;21(12):673-682
99. Pinton A, Ducos A, Berland H. et al. Chromosomal abnormalities in hypoprolific boars. Hereditas. 2000;132(1):55-62
100. Pinton A, Ducos A, Yerle M. Chromosomal rearrangements in cattle and pigs revealed by chromosome microdissection and chromosome painting. Genet Sel Evol. 2003;35(6):685-696
101. Gilbert W. The RNA world. Nature. 1986;319:618
102. Lander ES, Linton LM, Birren B. et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860-921
103. Waterston RH, Lindblad-Toh K, Birney E. et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420(6915):520-562
104. Doolittle WF, Sapienza C. Selfish genes, the phenotype paradigm and genome evolution. Nature. 1980;284(5757):601-603
105. Orgel LE, Crick FH. Selfish DNA: The ultimate parasite. Nature. 1980;284(5757):604-607
106. Brosius J. How significant is 98.5% 'junk' in mammalian genomes? Bioinformatics. 2003;19(Suppl 2):II35
107. Kazazian HH Jr. Mobile elements: Drivers of genome evolution. Science. 2004;303(5664):1626-1632
108. Volff JN. Turning junk into gold: Domestication of transposable elements and the creation of new genes in eukaryotes. Bioessays. 2006;28(9):913-922
109. Lyon MF. LINE-1 elements and X chromosome inactivation: A function for "junk" DNA? Proc Natl Acad Sci U S A. 2000;97(12):6248-6249
110. Bejerano G, Lowe CB, Ahituv N. et al. A distal enhancer and an ultraconserved exon are derived from a novel retroposon. Nature. 2006;441(7089):87-90
111. Kazazian HH Jr, Wong C, Youssoufian H, Scott AF, Phillips DG, Antonarakis SE. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature. 1988;332(6160):164-166
112. Sachidanandam R, Weissman D, Schmidt SC. et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409(6822):928-933
113. Shubitowski DM, Venta PJ, Douglass CL, Zhou RX, Ewart SL. Polymorphism identification within 50 equine gene-specific sequence tagged sites. Anim Genet. 2001;32(2):78-88
114. Brouillette JA, Andrew JR, Venta PJ. Estimate of nucleotide diversity in dogs with a pool-and-sequence method. Mamm Genome. 2000;11(12):1079-1086
115. Jungerius BJ, van Laere AS, Te Pas MF, van Oost BA, Andersson L, Groenen MA. The IGF2-intron3-G3072A substitution explains a major imprinted QTL effect on backfat thickness in a meishan x european white pig intercross. Genet Res. 2004;84(2):95-101
116. de Koning DJ, Rattink AP, Harlizius B, van Arendonk JA, Brascamp EW, Groenen MA. Genome-wide scan for body composition in pigs reveals important role of imprinting. Proc Natl Acad Sci U S A. 2000;97(14):7947-7950
117. Houston RD, Haley CS, Archibald AL, Cameron ND, Plastow GS, Rance KA. A polymorphism in the 5'-untranslated region of the porcine cholecystokinin type a receptor gene affects feed intake and growth. Genetics. 2006;174(3):1555-1563
118. Hiller M, Huse K, Platzer M, Backofen R. Creation and disruption of protein features by alternative splicing -- a novel mechanism to modulate function. Genome Biol. 2005;6(7):R58
119. Stamm S, Ben-Ari S, Rafalska I. et al. Function of alternative splicing. Gene. 2005;344:1-20
120. Brett D, Pospisil H, Valcarcel J, Reich J, Bork P. Alternative splicing and genome complexity. Nat Genet. 2002;30(1):29-30
121. Modrek B, Lee CJ. Alternative splicing in the human, mouse and rat genomes is associated with an increased frequency of exon creation and/or loss. Nat Genet. 2003;34(2):177-180
122. Johnson JM, Castle J, Garrett-Engele P. et al. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003;302(5653):2141-2144
123. Thanaraj TA, Clark F, Muilu J. Conservation of human alternative splice events in mouse. Nucleic Acids Res. 2003;31(10):2544-2552
124. Nurtdinov RN, Artamonova II, Mironov AA, Gelfand MS. Low conservation of alternative splicing patterns in the human and mouse genomes. Hum Mol Genet. 2003;12(11):1313-1320
125. Sorek R, Shamir R, Ast G. How prevalent is functional alternative splicing in the human genome? Trends Genet. 2004;20(2):68-71
126. Thanaraj TA, Stamm S, Clark F, Riethoven JJ, Le Texier V, Muilu J. ASD: The alternative splicing database. Nucleic Acids Res. 2004;32:D64-9
127. Pan Q, Bakowski MA, Morris Q. et al. Alternative splicing of conserved exons is frequently species-specific in human and mouse. Trends Genet. 2005;21(2):73-77
128. Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: Causes and consequences. Hum Genet. 1992;90(1-2):41-54
129. Lopez-Bigas N, Audit B, Ouzounis C, Parra G, Guigo R. Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett. 2005;579(9):1900-1903
130. Lopez AJ. Alternative splicing of pre-mRNA: Developmental consequences and mechanisms of regulation. Annu Rev Genet. 1998;32:279-305
131. Xing Y, Lee C. Alternative splicing and RNA selection pressure--evolutionary consequences for eukaryotic genomes. Nat Rev Genet. 2006;7(7):499-509
132. Keightley PD, Johnson T. MCALIGN: Stochastic alignment of noncoding DNA sequences based on an evolutionary model of sequence evolution. Genome Res. 2004;14(3):442-450
133. Chin CS, Chuang JH, Li H. Genome-wide regulatory complexity in yeast promoters: Separation of functionally conserved and neutral sequence. Genome Res. 2005;15(2):205-213
134. Dermitzakis ET, Kirkness E, Schwarz S, Birney E, Reymond A, Antonarakis SE. Comparison of human chromosome 21 conserved nongenic sequences (CNGs) with the mouse and dog genomes shows that their selective constraint is independent of their genic environment. Genome Res. 2004;14(5):852-859
Tables and Figures
Table 1
Total population size and number of global pig breeds (2006)
| Region | Africa | Asia | Europe | Lartin America | Near & Middle East | North America | Southwest Pacific | Total | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pig | Pop | Brd | Pop | Brd | Pop | Brd | Pop | Brd | Pop | Brd | Pop | Brd | Pop | Brd | Pop | Brd |
| No. | 23 | 49 | 594 | 229 | 192 | 165 | 72 | 67 | 0.3 | 1 | 75 | 18 | 3.8 | 12 | 960 | 541 |
| % | 2.4 | 9.1 | 61.9 | 42.3 | 20.0 | 30.5 | 7.5 | 12.4 | 0.0 | 0.2 | 7.8 | 3.3 | 0.4 | 2.22 | 100 | 100 |
Pop: population size in million heads; Brd: number of breeds; Per (%): share of the world total.
Source: The state of the world's animal genetic resources for food and agriculture (1st), 2006 [8].
Table 2
Characteristics of main classes of genetic markers
| mtDNA | Microsatellite | SNP | AFLP | |
|---|---|---|---|---|
| Type of loci (O'Brien) | I and II | II > I | I and II | I and II |
| Type of loci (Dodgson) | clone sequence-based | clone sequence-based | clone sequence-based | fingerprint |
| Distribution | mitochondria (less than 20 kb; 100-10,000 copies every cell) | nucleus (spaced every 5-50 kb; ubiquitous across the genome) | nucleus (spaced every 200-500 bp; millions of loci across the genome) | nucleus (ubiquitous across the genome) |
| PIC | high | high | low | moderate |
| Typical allele no. | hypervariable at the control region | 2 - 30 | 2 | 2 |
| Inheritance mode | maternally inherited | codominantedly inherited | codominantedly inherited | dominant inherited |
| Speed of assay | moderate | moderate | high | moderate |
| Development costs | moderate | high | moderate | low |
| Running costs | moderate | high | low | moderate |
| Major use | domestication; phylogeography | genome mapping; population genetics | phylogenomics; functional genomics; genetic diversity | population genetics; genome mapping |
| Major weakness | poor predictor of overall genomic diversity; loss of male-mediated gene flows | low abundance | ascertainment biases; biallelic | dominant mode of inheritance |
Table 3
Publicly available pig genomics and proteomics internet resources
| Resource type | Description | Resource name | URL |
|---|---|---|---|
| Genome | Pig genome sequencing by SGSC | Pig tales | http://www.piggenome.org |
| Genome | Pig PreEnsembl at Sanger | Pig PreEnsembl | http://pre.ensembl.org/Sus_scrofa |
| Genome | Pig genomics at UIUC | Swine genomics | http://www.swinegenomics.com |
| Genome | NAGRP pig genome program | U.S. pig genome mapping | http://www.animalgenome.org/pigs |
| Genome | Pig genome project at Japan | Animal genome program | http://animal.dna.affrc.go.jp |
| Genome | NCBI pig genome resources | Pig genome resources | http://www.ncbi.nlm.nih.gov/projects/genome/guide/pig |
| Genome | Pig genomic information system | PigGIS | http://www.piggis.org |
| Genome | 0.66X genome sequencing | Sino-Danish pig genome | http://piggenome.dk |
| Genome | 100 Mb genome sequencing | Korean pig genome project | http://www.nlri.go.kr |
| Genome | SSC7 and 14 genome sequencing | EU SABRE project | http://www.sabre-eu.eu |
| QTLs | Pig QTL database | Animal QTLdb | http://www.animalgenome.org/QTLdb/pig |
| Genes/markers | TIGR pig gene index | SsGI | http://www.tigr.org |
| Genes/markers | NCBI SNP database | Pig SNP database | http://www.ncbi.nlm.nih.gov/SNP |
| Linkage map | USDA MARC map | US MARC linkage map | http://www.marc.usda.gov/genome/swine |
| Linkage map | Pig genome map viewer | NCBI map viewer | http://web.ncbi.nlm.nih.gov/mapview |
| Linkage map | PIGMAP viewer at Roslin | ARKdb web | http://www.thearkdb.org |
| Physical map | Sanger porcine physical map | Sanger WebChrom | http://www.sanger.ac.uk/Projects/S_scrofa |
| Physical map | Pig FPC clones to linkage maps | BAC clone map | http://www.animalgenome.org/cgi-bin/QTLdb/SS |
| Physical map | Somatic cell hybrid panel | SCH map | http://www.toulouse.inra.fr/lgc/pig/hybrid.htm |
| Physical map | IMpRH maps | RH map | http://www.toulouse.inra.fr/lgc/pig/cyto/cyto |
| Physical map | UNR-1/UNR-2 | RH map | http://www.cabnr.unr.edu/beattie |
| Comparative map | Multispecies comparisons | Evolution highway | http://evolutionhighway.ncsa.uiuc.edu |
| Comparative map | Pig-human comparative map | Comparative map | http://www.toulouse.inra.fr/lgc/pig/compare/compare |
| Comparative map | Jackson labs | Mammalian maps | http://www.informatics.jax.org |
| Comparative map | Japan pig-mouse map | Pig mouse map | http://ws4.niai.affrc.go.jp/dbsearch2/java/mhomo/pig |
| Expression | Pig array from US pig genome project | Pig microarray | http://www.pigoligoarray.org |
| Expression | 98,988 pig ESTs database at Iowa | Pig EST database | http://pigest.genome.iastate.edu |
| Expression | Pig EST sequences at Denmark | Pig EST | http://pigest.kvl.dk |
| Expression | Full-length cDNA libraries and ESTs | PEDE at Japan | http://pede.dna.affrc.go.jp |
Table 4
Current chromosomal progress of the pig genome sequencing project (Nov 2006)
| Chr | Estimated length (bp) | No. of contigs | NoCs selected | NoCs sent | NoCs accessioned | NoCs finished | Total NoCs | Coverage (%) |
|---|---|---|---|---|---|---|---|---|
| SSC1 | 303,136,142 | 3 | 59 | 518 | 329 | 1 | 907 | 56.89 |
| SSC2 | 155,149,711 | 7 | 0 | 377 | 0 | 0 | 377 | 45.87 |
| SSC3 | 151,274,484 | 9 | 0 | 354 | 0 | 0 | 354 | 45.73 |
| SSC4 | 149,877,177 | 8 | 2 | 319 | 82 | 2 | 405 | 52.49 |
| SSC5 | 105,163,859 | 4 | 0 | 273 | 4 | 2 | 279 | 50.07 |
| SSC6 | 173,044,584 | 11 | 0 | 433 | 6 | 19 | 458 | 48.60 |
| SSC7 | 138,247,446 | 5 | 118 | 46 | 321 | 47 | 532 | 67.01 |
| SSC8 | 152,094,626 | 4 | 0 | 419 | 2 | 0 | 421 | 53.92 |
| SSC9 | 157,355,516 | 4 | 1 | 448 | 0 | 0 | 449 | 55.18 |
| SSC10 | 81,315,841 | 7 | 0 | 196 | 0 | 0 | 196 | 46.19 |
| SSC11 | 89,955,204 | 3 | 32 | 72 | 160 | 2 | 266 | 59.36 |
| SSC12 | 70,201,005 | 6 | 0 | 151 | 0 | 0 | 151 | 42.16 |
| SSC13 | 221,109,244 | 1 | 2 | 635 | 1 | 0 | 638 | 54.65 |
| SSC14 | 142,311,687 | 3 | 160 | 12 | 365 | 6 | 543 | 67.50 |
| SSC15 | 173,169,528 | 3 | 1 | 456 | 0 | 0 | 457 | 51.01 |
| SSC16 | 89,254,859 | 2 | 0 | 239 | 1 | 0 | 240 | 51.99 |
| SSC17 | 70,843,094 | 5 | 2 | 82 | 58 | 69 | 211 | 51.41 |
| SSC18 | 63,240,215 | 1 | 0 | 162 | 0 | 0 | 162 | 50.42 |
| SSCX | 135,575,825 | 21 | 0 | 275 | 0 | 0 | 275 | 39.29 |
| Total | 2,622,320,047 | 107 | 377 | 5,467 | 1,329 | 148 | 7,321 | |
| Average | 138,016,845 | 52.09 |
NoCs selected: number of clones selected for sequencing; NoCs sent: number of clones sent for sequencing; NoCs accessioned: number of accessioned sequence clones; NoCs finished: number of finished sequence clones; Total NoCs: total number of sequencing clones; Coverage (%): percentage of map covered by sequence clones. Source: Pig Pre-Ensembl: http://pre.ensembl.org/Sus_scrofa/
Figure 1
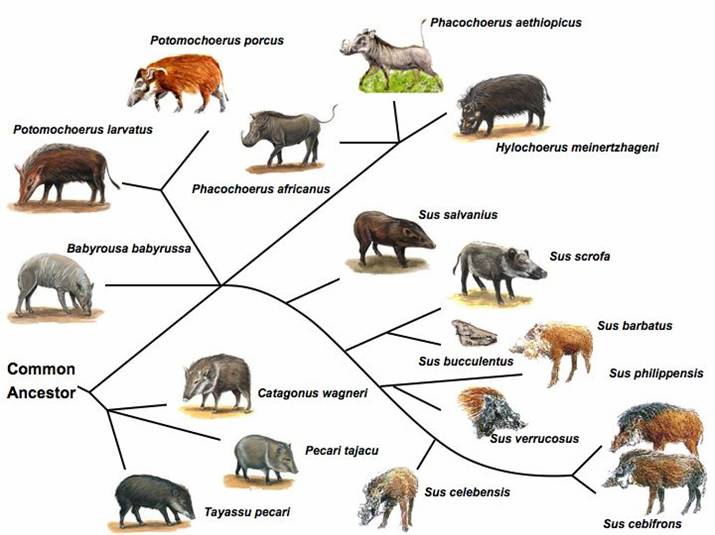
Suiforme diversity and phylogenetic relationship. Source: Randi et al (1996); Groves et al (1997); Fokkinga (2004); Robins et al (2006) [11-14]. Pig pictures were adapted from the animal diversity website at the University of Michigan Museum of Zoology (http://animaldiversity.ummz.umich.edu/); http://www.triplov.com/guinea_bissau/mammalia/suidae.htm.
Figure 2
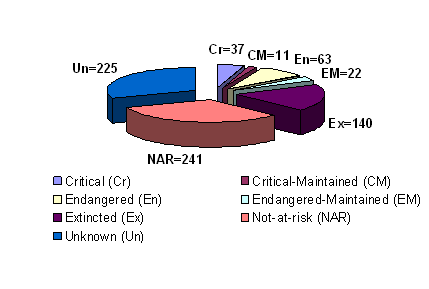
Global status of pig breeds. Source: The state of the world's animal genetic resources for food and agriculture (1st), 2006 [8]
Author contact
![]() Correspondence to: Dr. Lawrence B. Schook, University of Illinois at Urbana-Champaign, Department of Animal Sciences, Institute for Genomic Institute, 1201 W. Gregory Dr., Edward R. Madigan Laboratory 382, Urbana, IL 61801, USA. Phone: + 1-217-265-5326 Fax: + 1-217-244-5617 E-mail: schookedu
Correspondence to: Dr. Lawrence B. Schook, University of Illinois at Urbana-Champaign, Department of Animal Sciences, Institute for Genomic Institute, 1201 W. Gregory Dr., Edward R. Madigan Laboratory 382, Urbana, IL 61801, USA. Phone: + 1-217-265-5326 Fax: + 1-217-244-5617 E-mail: schookedu
Citation styles
APA
Chen, K., Baxter, T., Muir, W.M., Groenen, M.A., Schook, L.B. (2007). Genetic Resources, Genome Mapping and Evolutionary Genomics of the Pig (Sus scrofa). International Journal of Biological Sciences, 3(3), 153-165. https://doi.org/10.7150/ijbs.3.153.
ACS
Chen, K.; Baxter, T.; Muir, W.M.; Groenen, M.A.; Schook, L.B. Genetic Resources, Genome Mapping and Evolutionary Genomics of the Pig (Sus scrofa). Int. J. Biol. Sci. 2007, 3 (3), 153-165. DOI: 10.7150/ijbs.3.153.
NLM
Chen K, Baxter T, Muir WM, Groenen MA, Schook LB. Genetic Resources, Genome Mapping and Evolutionary Genomics of the Pig (Sus scrofa). Int J Biol Sci 2007; 3(3):153-165. doi:10.7150/ijbs.3.153. https://www.ijbs.com/v03p0153.htm
CSE
Chen K, Baxter T, Muir WM, Groenen MA, Schook LB. 2007. Genetic Resources, Genome Mapping and Evolutionary Genomics of the Pig (Sus scrofa). Int J Biol Sci. 3(3):153-165.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.