ISSN: 1449-2288International Journal of Biological Sciences

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(9):1267-1280. doi:10.7150/ijbs.4828 This issue Cite
Review
The Pathological and Physiological Roles of IL-6 Amplifier Activation
Masaaki Murakami1 ![]() , Toshio Hirano2
, Toshio Hirano2 ![]()
1. Laboratory of Developmental Immunology, JST-CREST, Graduate School of Frontier Biosciences, Graduate School of Medicine, and WPI Immunology Frontier Research Center, Osaka University, Osaka, Japan;
2. JST-CREST, Osaka University, Osaka, Japan.
Citation:
Murakami M, Hirano T. The Pathological and Physiological Roles of IL-6 Amplifier Activation. Int J Biol Sci 2012; 8(9):1267-1280. doi:10.7150/ijbs.4828. https://www.ijbs.com/v08p1267.htm
Other stylesAbstract
The NFκB-triggered positive feedback loop for IL-6 signaling in type 1 collagen+ non-immune cells (IL-6 amplifier) was first discovered to be a synergistic signal that is activated following IL-17A and IL-6 stimulation in type 1 collagen+ non-immune cells. Subsequent disease models have shown that it can also be stimulated by the simultaneous activation of NFκB and STAT3, functions as a local chemokine inducer, and acts as a mechanism for local inflammation, particularly chronic ones like rheumatoid arthritis and a multiple sclerosis. Moreover, we have recently shown that hyper activation of the IL-6 amplifier via regional neural activation establishes a gateway for immune cells including autoreactive T cells to pass the blood-brain barrier at dorsal vessels in 5th lumbar cord. Here we review how the IL-6 amplifier is activated by neural activation and the physiological relevance of the gateway to the central nervous system. Accumulating evidences continues to suggest that the IL-6 amplifier offers a potential molecular mechanism for the relationship between neural activation and the development of inflammatory diseases, which could establish a new interdisciplinary field that fuses neurology and immunology.
Keywords: IL-6, neural activation, inflammatory diseases
Introduction
After cloning IL-6, our research has been focusing on IL-6-mediated autoimmune diseases [1-4]. IL-6 is a pleiotropic cytokine that regulates multiple biological processes including the development of the nervous and hematopoietic systems, acute-phase responses, inflammation, and immune responses (Hirano, 1998). To date, ten IL-6 family cytokines have been identified: IL-6, oncostatin M, LIF, CNTF, CT-1, NNT-1, neuropoietin, IL-11, IL-27, and IL-31 [5-7]. All of these share gp130 as the signal transducer in their receptor complexes. Upon IL-6 stimulation, gp130 transduces two major signaling pathways: the JAK-signal transducer and activator of transcription 3 (STAT3) pathway, which is mediated by the YxxQ motif of gp130, and the SHP2-Gab-Ras-Erk-MAPK pathway, which is regulated by Y759, a cytoplasmic suppressor of cytokine signaling (SOCS3) binding residue in gp130 [7-9]. Additionally, a number of studies have suggested IL-6 has an important role in autoimmune diseases [3, 10-13]. Importantly, patients with RA showed high synovial concentrations of IL-6 [14], while anti-IL-6 receptor therapy is effective for some RA patients [15]. Additionally, it has been reported that IL-6 is expressed not only in immune cells but also in non-immune cells [16, 17].
The mutant mouse line F759, which is a mutant variant of gp130 where Y759 is substituted for phenylalanine (F), is a good model for studying how IL-6-mediated signals can trigger autoimmune diseases; in this case by enhancing STAT3 activation in the absence of SOCS3-mediated suppression [9, 18]. As these mice age, they spontaneously develop a rheumatoid arthritis-like tissue specific disease, indicating that constitutive activation of IL-6 signaling is involved in the development of autoimmune symptoms [19] (see Figure 1). We later found that the molecular mechanism of the pathogenesis involves type 1 collagen+ non-immune cells as well as major histocompatibility complex (MHC) class II-restricted CD4+ T cells. That non-immune cells express excessive IL-6 signaling serves two purposes for the disease pathogenesis in F759 mice. One is the production of excess IL-7 for the induction of homeostatic proliferation of CD4+ T cells including Th17 cells in a manner dependent on mouse age [18, 20]. The other is to activate a nuclear factor kappa B (NFκB)-triggered positive feedback loop of IL-6 signaling (IL-6 amplifier) in the presence of IL-17 from Th17 cells [20]. We have since shown in animal models for rheumatoid arthritis, multiple sclerosis (MS), and chronic rejection after transplantation that the IL-6 amplifier is stimulated by simultaneous activation of NFκB and STAT3, functions as a local inducer of chemokines, and acts as a mechanism for the local inflammation [13, 20-24].
However, mice from the MS model also developed experimental autoimmune encephalomyelitis (EAE). To induce MS-like symptoms, a passive transfer of myelin oligodendrocyte glycoprotein (MOG) specific CD4+ T cells was used. The intravenous transfer of the pathogenic CD4+ T cells developed the MS-like disease in the central nervous system (CNS) within two weeks after transfer; this despite the presence of the blood-brain barrier (BBB), which should prevent immune cell migration there. We later found that regional neural activation creates a gateway for immune cells including pathogenic CD4+ T cells to pass through the BBB and into the CNS by enhancing IL-6 amplifier activation in endothelial cells [23].
In this review, we explain the IL-6 amplifier in non-immune cells based on analysis of the rheumatoid arthritis model, F759 mice, and then describe how it acts as the connection point between neural and immune signals in endothelial cells from the 5th lumbar cord.
Figure 1
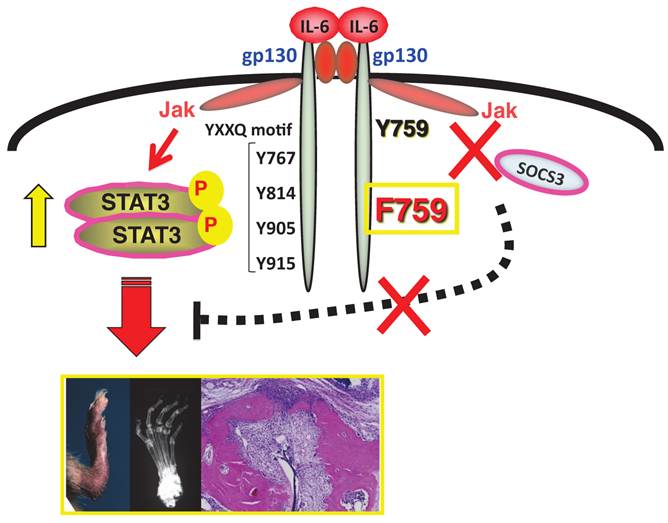
F759 mice have a point mutation at the SOCS3 binding tyrosine residue (Y759F) in gp130 and develop a rheumatoid arthritis like disease with age.
What is the IL-6 amplifier?
1. The establishment of an IL-6-dependent rheumatoid arthritis model, F759 arthritis
It has been reported that anti-IL-6 receptor antibodies can be used as medication for rheumatoid arthritis and Castleman's disease patients [25-27]. Although IL-6-mediated development of IL-17 expressing CD4+ T cells seems to play a role in these beneficial effects [28-30], how IL-6-mediated signaling or IL-17 develops such diseases remains unclear.
We have been studying intracellular signal events triggered by IL-6 stimulation since we cloned IL-6 cDNA. There exist two opposite signaling pathways via IL-6 receptor complexes after IL-6 ligation [13]. One is a positive signal via STAT3; the other is negative feedback signaling by SOCS3 [3]. We therefore hypothesized that deficient SOCS3-mediated signaling might offer a good arthritis model to investigate the roles of IL-6 in the pathogenesis. The result was the establishment of a knock-in mutant mouse line, F759, where a SOCS3 binding tyrosine reside in gp130, a signal transducer for IL-6, is changed to phenylalanine [9]. All F759 mice were found to have a rheumatoid arthritis-like disease at about 12-18 months after birth [19](Figure 1).
2. Molecular mechanism of arthritis development in F759 mice
(i) Roles of IL-6 signaling in hematopoietic cells
To identify important cell populations for rheumatoid arthritis development, F759 mice were crossed with mice deficient of CD4, CD8, or B cells. CD4-deficient F759 mice alone attenuated disease development [18]. It was confirmed that MHC class II-deficient F759 mice show only weak symptoms of the disease, while CD8-deficient and B cell (Igh6)-deficient F759 mice did not show these symptoms (Figure 2). In fact, CD4+ T cells were gradually activated as F759 mice aged. We hypothesized that excessive signaling of IL-6 in CD4+ T cells and/or dendritic cells induced the CD4+ T cell activation. The IL-6 signal in CD4+ T cells or dendritic cells inhibits key signals such as those mediated by T cell antigen receptors (TCR) or Toll-like receptors (TLR) [31, 32]. Consistent with these data, irradiated F759 recipients developed arthritis even after the transfer of healthy control bone marrow cells, which could be interpreted to mean that F759 arthritis is dependent on MHC class II-restricted CD4+ T cells and on excessive IL-6 signaling in non-immune cell populations (Figure 3). Thus, IL-6 signaling in hematopoietic cells is dispensable for the development of the arthritis in F759 mice.
(ii) Roles of IL-6 signaling in non-hematopoietic cells
Results of bone marrow transplantation above showed that IL-6 signaling in non-hematopoietic cells is dispensable for the development of the arthritis in F759 mice. One possible explanation for the development of the arthritis in F759 mice is that the excessive IL-6 signaling in non-immune cells converts naïve CD4+ T cells into activated ones, a phenomenon that accelerates with age. Indeed, homeostatic proliferation, which is an autonomous form of polyclonal CD4+ T cell proliferation, increased in F759 via the excessive expression of IL-7 from non-immune cells. Because blocking either homeostatic proliferation or IL-7 expression significantly suppressed the disease, it has been suggested that homeostatic proliferating CD4+ T cells via the IL-6-IL-7 axis in non-immune cells contributes to arthritis in F759 mice (Figure 4) [18], showing that the interaction between non-hematopoietic cells and immune cells plays roles in F759 arthritis [13].
Figure 2
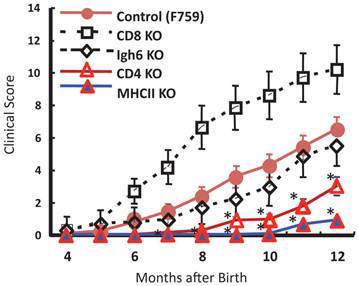
MHC class II-restricted CD4+ T cells enhance the development of arthritis in F759 mice.
Figure 3
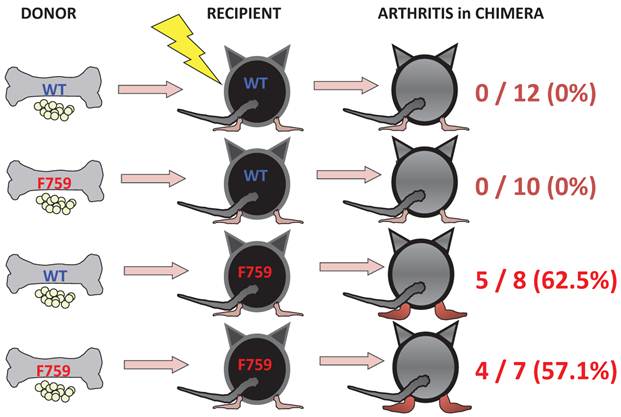
Enhanced IL-6 signaling in non-immune cells leads to arthritis in F759 mice.
Figure 4
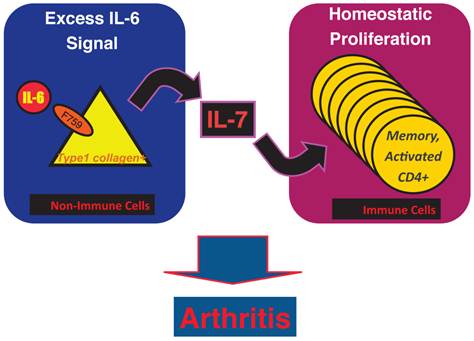
IL-7 induced by excessive IL-6 signaling in type 1 collagen+ non-immune cells increases the homeostatic proliferation of activated CD4+ T cells, which leads to the development of arthritis in F759 mice.
(iii) Discovery of the IL-6 amplifier in non-immune cells
How does the homeostatic proliferation of CD4+ T cells in aged F759 mice induce arthritis? We and others have shown that a new subset of activated CD4+ T cell differentiation is dependent on the IL-6-gp130-STAT3 pathway [28]. Indeed, polyclonal activated Th17 cells in spleen and superficial lymph nodes and serum IL-17 concentration increased in F759 mice with age (Figure 5). Additionally, a deficiency of IL-17 in F759 mice suppressed arthritis, while forced expression of IL-17 via a hydrodynamic method [33] enhanced it (Figure 6). It is possible, however, that the IL-17 effects are actually due to another cytokine, as following the forced expression of IL-17, IL-6 as well as some chemokines were found to be abnormally high in sera, which suggested a positive feedback mechanism for IL-6 signaling in the presence of IL-17 in F759 mice. Moreover, this mechanism was present in normal control animals too. We named this mechanism the NFκB-triggered positive feedback for IL-6 signaling, or IL-6 amplifier for short, in non-immune cells (Figure 7) [20]. In vivo and in vitro experiments have shown that the IL-6 amplifier is activated by simultaneous activation of NFκB and STAT3 and that it locally induces chemokines for inflammation [22].
Figure 5
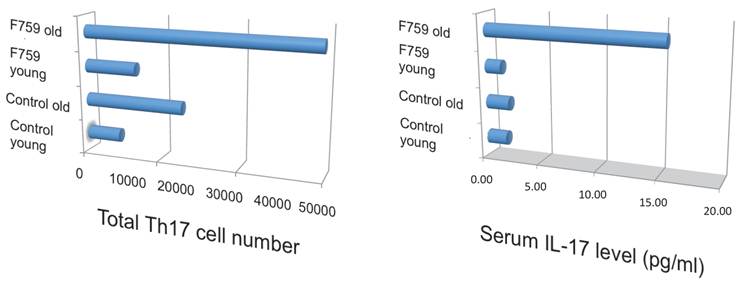
Th17 cells and serum IL-17 level increased in aged F759 mice.
Figure 6
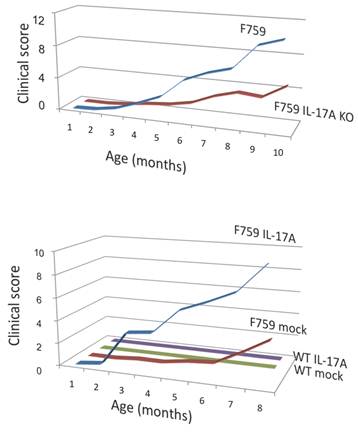
Deficiency of IL-17 decreased the development of arthritis, while forced expression of IL-17 via hydrodynamic method enhanced it in F759 mice comparing to mock control vector transduced F759 mice.
Figure 7
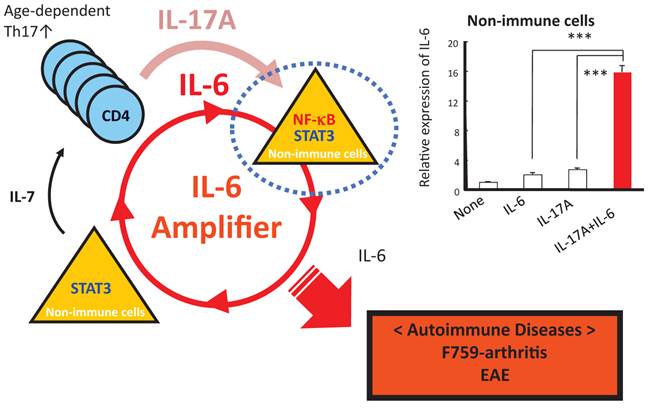
When in the presence of Th17 cells that hyper express IL-17, excessive IL-6 expression induced by a positive feedback loop for IL-6 signaling in non-immune cells plays a role for the development of arthritis in aged F759 mice.
(iv) A four-step model for tissue specific MHC class II associate autoimmune diseases
Standard dogma in immunology claims that the recognition of self-tissue specific antigens by autoreactive T cells leads to autoimmune diseases. Consistent with this notion, the use of Vβ by activated CD4+ T cells in old F759 mice was perturbed, suggesting the existence of tissue specific CD4+ T cells. To prove the importance of antigen recognition by CD4+ T cells, we established F759 mice that have one TCR which recognizes non-joint antigens such as a tuberculosis antigen, P25, or egg albumin, OVA. Even in these circumstances, the mice developed the disease (Figure 8), suggesting joint antigen recognition among polyclonal homeostatic proliferating CD4+ T cells including Th17 cells is not necessary for disease development in F759 mice.
We further showed that local events in joints such as microbleeding together with an accumulation of Th17 cells can induce arthritis in F759 mice in a manner independent of tissue antigen-recognition [13]. Specifically, a Th17 cell increase in F759 mice with age correlated with disease development, as joint microbleeding increased despite only normal activity. In other words, age increased the likelihood of Th17 cells entering the joints from the peripheral blood. This entry leads to a presence of IL-17, which can trigger IL-6 amplifier activation and corresponding chemokines [21](Figure 9). Thus, certain class II MHC-associated, tissue-specific autoimmune diseases may be induced by local events that cause an antigen-independent accumulation of effector CD4+ T cells, which induces the induction of the IL-6 amplifier via cytokines in the affected tissue. In other words, in certain cases, the target tissue itself may determine the specificity of the autoimmune disease via activation of the IL-6 amplifier. To explain this hypothesis, we have proposed a four-step model for MHC class II associated autoimmune diseases [13, 21]: 1) T cell activation regardless of the antigen specificity; 2) tissue-specific accumulation of activated T cells induced by local events; 3) transient activation of the IL-6 amplifier; and 4) enhanced sensitivity to cytokines in the target tissue. The result is chronic activation of the IL-6 amplifier and subsequent manifestation of autoimmune diseases. Thus, the IL-6 amplifier is a critical regulator of chromic inflammations in tissue specific MHC class II-associated autoimmune diseases.
Moreover, IL-6 amplifier activation in allogeneic grafts was responsible for transplantation rejection responses, which is another T cell-mediated inflammation type response [24](Figure 10). Thus, the IL-6 amplifier in non-immune cells is an important machinery for inflammation induction.
Figure 8
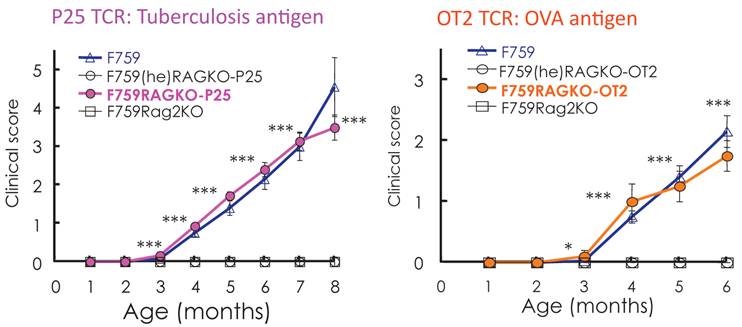
F759 mice that have TCR that recognize non-joint antigens develop arthritis.
Figure 9
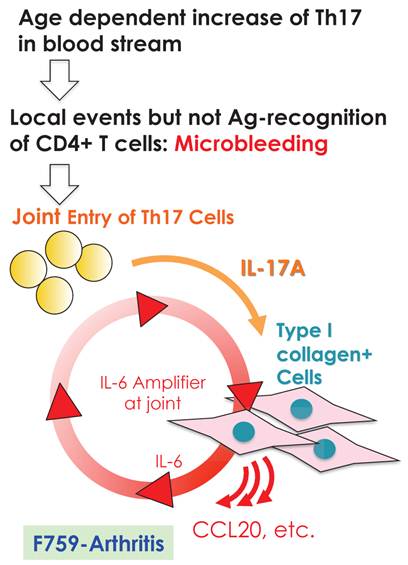
Local events such as microbleeding in the joints are trigger factors for IL-6 amplifier activation, which causes arthritis in aged F759 mice that have an excess number of Th17 cells in the blood stream.
Figure 10
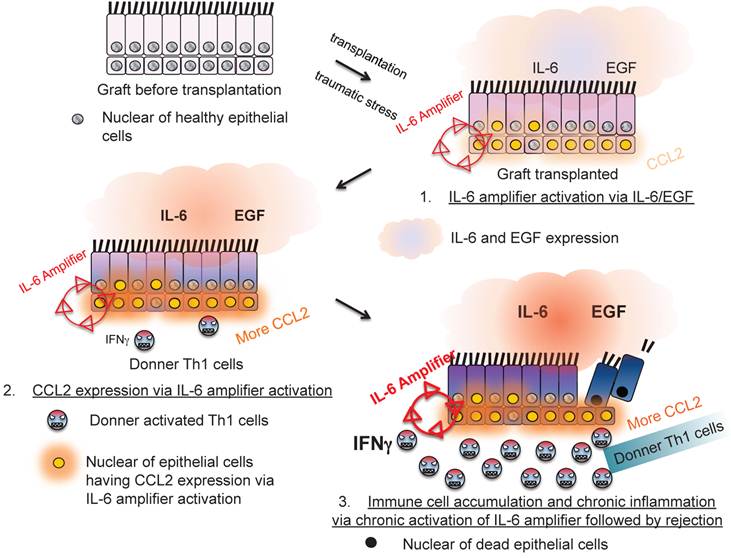
The IL-6 amplifier activation in grafts is a determinant factor for transplant rejection.
3. A gateway for immune cells in the CNS via IL-6 amplifier activation
(i) Pathogenic CD4+ T cells in the bloodstream induce a MS-like disease in the CNS
In the MS model, EAE, normal C57BL/6 mice are immunized with an epitope-peptide of CD4+ T cells from myelin oligodendrocyte glycoproteins (MOG) [20]. CD4+ T cells from these mice can be cultured with peptide-pulsed bone marrow derived dendritic cells to enrich an antigen-specific activated form of CD4+ T cells. These CD4+ T cells express IL-17 and IFNg, indicating that Th17 and Th1 cells are present. Furthermore, the CD4+ T cells are pathogenic, as their intravenous transfer into normal recipients led to paralysis. An essential contribution of the IL-6 amplifier to disease development was confirmed by four in vivo results using the EAE model: (i) IL-6 as well as chemokine concentrations increased after the pathogenic CD4+ T cell transfer, (ii) the pathogenic CD4+ T cell transfer in IL-6 deficient hosts significantly suppressed disease development, (iii) pathogenic CD4+ T cells deficient of IL-17 significantly suppressed disease development, and (iv) pathogenic CD4+ T cell transfer in hosts deficient of gp130 or STAT3 in type 1 collagen+ cells significantly suppressed disease development.
Even more interesting was that pathogenic CD4+ T cells in the blood stream could affect tissues of the central nervous system (CNS) including the brain and spinal cord. In other words, these cells were able to circumvent the BBB [34, 35]
(ii) Pathogenic CD4+ T cells accumulate in dorsal blood vessels of the 5th lumbar cord where various chemokines are expressed via IL-6 amplifier activation
Whole-mount frozen sections within 5 days after pathogenic CD4+ T cell transfer in C57BL/6 mice showed no T cells in the brain and upper spines including the cervical and thoracic spines, but did show an accumulation in the lumbar level [23]. In particular, pathogenic CD4+ T cells migrated to the 5th lumbar cord (L5) from the blood vessels of the dorsal side (Figure 11). Because IL-17 but not IFNg deficient pathogenic CD4+ T cells and deficient IL-6 signaling in type 1 collagen+ and endothelial cells suppressed the accumulation, it was suggested that the IL-6 amplifier was in part responsible for the migration (Figure 12).
Figure 11
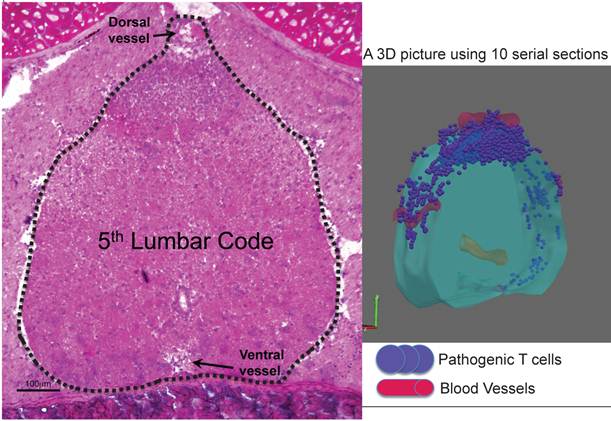
Dorsal vessels in the 5th lumbar cord (L5) create a gateway for autoreactive CD4+ T cells to pass through the brain-blood barrier. A section of L5 region 5 days after pathogenic CD4+ T cell transfer in C57BL/6 mice were made followed by hematoxylin eosin staining. 3D profile was made by using the serial sections.
Figure 12
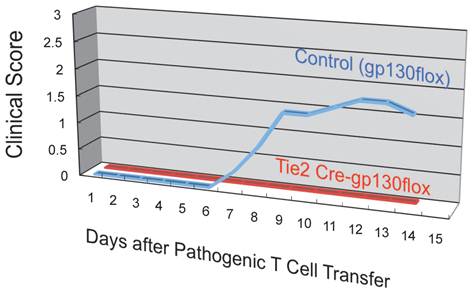
Endothelial specific depletion of IL-6 signaling suppressed the development of autoimmune disease in the EAE model.
Additionally, we have shown that CCL20, which binds to CCR6, induces the accumulation of Th17 cells [21]. Pathogenic CD4+ T cells prepared from conventional CCR6-deficient mice did not accumulate in the 5th lumbar cord (data not shown), and the dorsal blood vessels expressed an excessive CCL20 in a manner dependent on IL-6 signaling even at steady state (Figure 13). Moreover, anti-CCL20 neutralization antibody treatment suppressed pathogenic CD4+ T cell accumulation and disease development (Figure 14). Thus, it would seem CCL20 is a key chemokine for pathogenic CD4+ T cell accumulation in the 5th lumbar cord. Additionally, 10 other chemokines (CCL2, CCL3, CCL9, CCL21, CCL24, CX3CL1, CXCL1, CXCL2, CXCL9, and CXCL11) also significantly increased in the dorsal blood vessels of the 5th lumbar cord at steady state, suggesting that not only pathogenic Th17 cells but also many immune cell populations might migrate through these blood vessels and affect the CNS. In other words, dorsal blood vessels of the 5th lumbar cord could be a gateway for immune cells to the CNS, a phenomenon maintained by IL-6 amplifier activation.
Figure 13
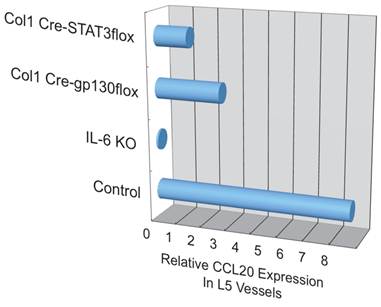
CCL20 expression in the dorsal vessels of the 5th lumbar cord decreased in mice that lacked IL-6 signaling in non-immune cells.
Figure 14
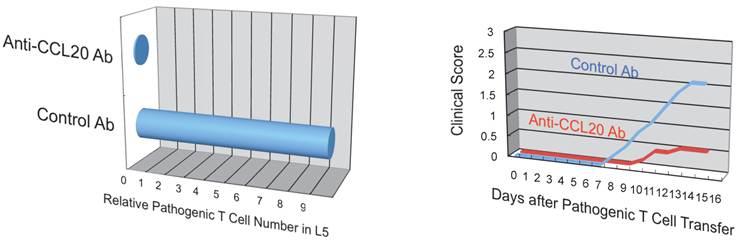
Anti-CCL20 antibody treatment suppressed the accumulation of autoreactive Th17 cells in the 5th lumbar cord and the development of autoimmune disease in the EAE model.
(iii) Chemokine expression in dorsal blood vessels of the 5th lumbar cord is dependent on IL-6 amplifier activation via regional neural activation in response to anti-gravity
Because the biggest dorsal root ganglion (DRG) is located near the 5th lumbar cord, and that sensory neurons from soleus muscles, which are major anti-gravity muscles, are present in the 5th lumbar DRG, it was considered whether continuous gravity stimulation could trigger pathogenic CD4+ T cell accumulation in the 5th lumbar cord by activating these muscles. Mouse experiments using the tail-suspension method, where anti-gravity responses from the soleus muscles are removed, caused pathogenic CD4+ T cell accumulation in the 5th lumbar cord and CCL20 expression in the dorsal blood vessels there, and suppressed disease development. Interestingly, electrical stimulations in the soleus muscles of mice suspended by their tail increased the pathogenic CD4+ T cell accumulation and CCL20 expression (Figure 15). Moreover, electrical stimulations in quadricep or tricep muscles increased CCL20 expression in the 3rd lumbar cord or lower cervical and upper thoracic cords. Thus, neural stimulation alters the status of the IL-6 amplifier in regional blood vessels such that chemokines are expressed andimmune cells can enter the CNS.
(iv) Sympathetic neural activation is involved in the accumulation of pathogenic CD4+ T cells in the 5th lumbar cord followed by the development of EAE
Blood flow speed in the dorsal vessels of the 5th lumbar cord decreased in mice tail-suspended, but increased when these mice received electrical stimulations in the soleus muscles (Figure 16). Furthermore, along with sensory neurons, sympathetic neurons were activated around the 5th lumbar cord. It is known that the status of blood vessels is mainly controlled by autonomic neurons like sympathetic and parasympathetic neurons. Consistently, noradrenalin, a sympathetic neurotransmitter, enhanced the activation of the IL-6 amplifier as monitored by CCL20 secretion at least in vitro (Figure 17). Moreover, inhibitors of noradrenalin receptors suppressed the pathogenic CD4+ T cell accumulation, or NFκB activation, and the CCL20 mRNA expression in the 5th lumbar cord or the dorsal vessels (Figure 18), which correlated with a suppression of disease development. Thus, regional neural activation can establish a gateway for immune cells including pathogenic T cells to pass through the BBB and into the CNS via the IL-6 amplifier [23](Figure 19).
Figure 15
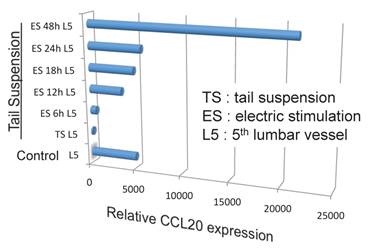
Anti-gravity stimulation in soleus muscles by tail suspension (TL) for 1 week decreased CCL20 expression in blood vessels in the 5th lumbar cord, while electric stimulation (ES) at 0.1 mA and 1-8 ms random intervals for 6 to 48 hours (6h, 12h, 18h, 24h, and 48h) in these muscles increased the expression in a manner dependent on the dose of the stimulation.
Figure 16
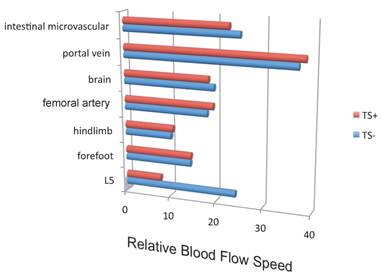
Blood flow speed in the vessels of the 5th lumbar cord alone decreased in mice suspended by their tails (TS+).
Figure 17
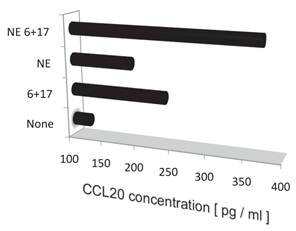
Noradrenalin (NE) enhances the IL-6 amplifier-dependent CCL20 secretion from endothelial cells.
Figure 18
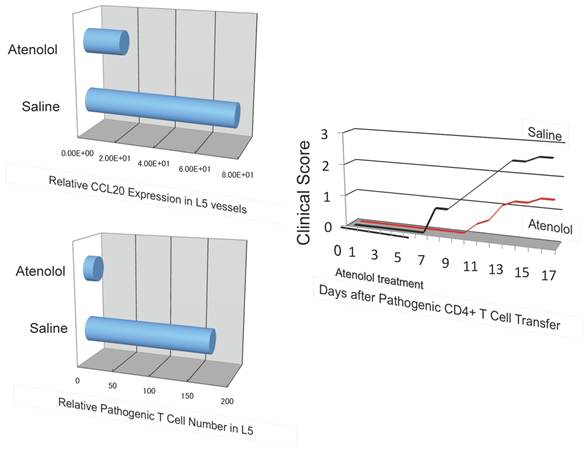
A beta 1 adrenalin receptor inhibitor, atenolol, decreased CCL20 expression in L5 vessels and accumulation of pathogenic T cells in the 5th lumbar cord followed by disease development.
Figure 19
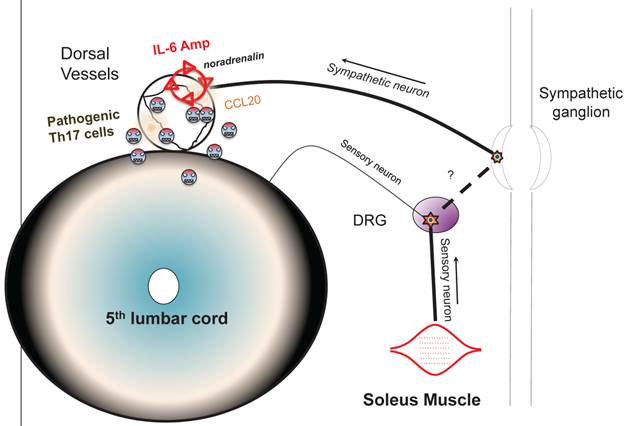
Schematic figure showing IL-6 amplifier activation via neural stimulation creates a gateway for immune cells to pass the brain-blood barrier.
Future direction
(i) Role of antigen recognition for the accumulation of pathogenic CD4+ T cells in the 5th lumbar cord
Because intravenously transferred, antigen non-specific Th17 cells or OVA-specific Th17 cells did not accumulate in the 5th lumbar cord (data not shown), but MOG-specific Th17 cells did, it is possible that antigen-recognition by transfused pathogenic CD4+ T cells also contributes to the accumulation of pathogenic CD4+ T cells. That is, pathogenic CD4+ T cell accumulation in the CNS is positively regulated by two factors: IL-6 amplifier activation via neural activation through soleus muscles and antigen recognition by pathogenic CD4+ T cells in the blood. Because endothelial cells sometimes express MHC class II molecules, one source for presenting the MOG antigen peptide to pathogenic CD4+ T cells might be endothelial cells in the vessels of the 5th lumbar cord. Another possibility is that dendritic cells in the CNS might play a role to reach their dendrites inside the vessels to present MOG peptides to the pathogenic CD4+ T cells in bloodstream like dendritic cells in gastrointestinal tract [36].
(ii) Relationship with human MS patients
Patients suffering from MS do not always display symptoms in their lower body. Instead, symptoms are generally isolated to the chest and cerebrum. This difference between MS patients and mouse models can be attributed to different sites of the pathogenic CD4+ T cell accumulation that follows local inflammation. Although neural activations in our EAE model were mainly induced by stimulating anti-gravity soleus muscles and electric stimulations in different muscles, and resulted in chemokine expression in the dorsal vessels of different spinal cords, excess neural activations commonly seen in the chest and/or cerebrum of MS patients may arise from various neural sources including the thoracic cords via nerves from the hands and fingers and/or the cerebrum via the optic-nerves or mental stress.
(iii) The physiological relevance of the gateway to the CNS
Although the reasons are unclear, a certain level of IL-6 amplifier activation occurs in the dorsal blood vessels of the 5th lumbar cord at steady state even in normal mice. One possibly purpose is that this activity acts as a surveillance system for immune cells to prevent pathogenic invasion or latent infectious states in the CNS [37, 38] The surveillance would elicit a reaction from various chemokines in dorsal vessels there. If so, neural activation-mediated IL-6 amplifier activation may have a critical role in maintaining CNS homeostasis by regulating various immune cells.
(iv) Future Directions
Current work is investigating the molecular mechanism that drives the synergy between NFκB and STAT3, which induces the excessive expression of chemokines and IL-6 and is a key regulator of the IL-6 amplifier. Our lab is now exploring genome-wide-screens to obtain positive-regulators, negative-regulators, and targets of the IL-6 amplifier activation using shRNA-based and DNA microarray-based screenings. At the moment, nearly 2000 candidates have been identified. These candidate genes could be targets for inflammatory medications much like anti-IL-6 receptor antibodies for rheumatoid arthritis. The IL-6 amplifier also provides a new model for bridging neurology and immunology. Neural stimulations beyond gravity-mediated ones, including mental-, stress-, and pain-mediated stimulations are now being considered as ways for immune cells including autoreactive ones to cross the BBB and compromise the CNS.
Acknowledgements
We thank Dr. P. Karagiannis (RIKEN, Osaka, Japan) for carefully reading the manuscript. This work was supported by KAKENHI (M. M., and T. H.), the JST-CREST program (M. M. and T. H.), and the Osaka Foundation for the Promotion of Clinical Immunology (M. M.).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hirano T, Yasukawa K, Harada H, Taga T, Watanabe Y, Matsuda T, Kashiwamura S, Nakajima K, Koyama K, Iwamatsu A. et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature. 1986;324:73-76
2. Hirano T. Interleukin-6 and its relation to inflammation and disease. Clin Immunol Immunopathol. 1992;62:S60-S65
3. Hirano T. Interleukin 6 and its receptor: ten years later. Int Rev Immunol. 1998;16(3-4):249-284
4. Hirano T, Kishimoto T. Interleukin-6. Handbook of Experimental Pharmacology. 1990;95:633-665
5. Suthaus J, Tillmann A, Lorenzen I, Bulanova E, Rose-John S, Scheller J. Forced homo- and heterodimerization of all gp130-type receptor complexes leads to constitutive ligand-independent signaling and cytokine-independent growth. Mol Biol Cell. 2010;21:2797-2807
6. Murakami M, Kamimura D, Hirano T. New IL-6 (gp130) family cytokine members, CLC/NNT1/BSF3 and IL-27. Growth Factors. 2004;2004(22):2
7. Kamimura D, Ishihara K, Hirano T. IL-6 signal transduction and its physiological roles: the signal orchestration model. Rev Physiol Biochem Pharmacol. 2003;149:1-38
8. Fukada T, Hibi M, Yamanaka Y, Takahashi-Tezuka M, Fujitani Y, Yamaguchi T, Nakajima K, Hirano T. Two signals are necessary for cell proliferation induced by a cytokine receptor gp130: involvement of STAT3 in anti-apoptosis. Immunity. 1996;5(5):449-460
9. Ohtani T, Ishihara K, Atsumi T, Nishida K, Kaneko Y, Miyata T, Itoh S, Narimatsu M, Maeda H, Fukada T. et al. Dissection of signaling cascades through gp130 in vivo: Reciprocal roles for STAT3- and SHP2-mediated signals in immune responses. Immunity. 2000;12:95-105
10. O'Shea JJ, Ma A, Lipsky P. Cytokines and autoimmunity. Nat Rev Immunol. 2002;2:37-45
11. Sakaguchi S, Sakaguchi N. Animal model of arthritis caused by systemic alteration of the immune system. Curr Opin Immunol. 2005;17:589-594
12. Awasthi A, Kuchroo VK. Th17 cells: from precursors to players in inflammation and infection. Int Immunol. 2009;21:489-498
13. Hirano T. Interleukin 6 in autoimmune and inflammatory diseases: a personal memoir. Proc Jpn Acad, Ser B. 2010;86:717-730
14. Hirano T, Matsuda T, Turne M, Miyasaka N, Buchan G, Tang B, Sato K, Shimizu M, Maini R, Feldmann M. et al. Excessive production of interleukin 6/B cell stimulatory factor-2 in rheumatoid arthritis. Eur J Immunol. 1988;18(11):1797-1801
15. Nakagawa T, Tsuruoka M, Ogura H, Okuyama Y, Arima Y, Hirano T, Murakami M. IL-6 positively regulates Foxp3+CD8+ T cells in vivo. Int Immunol. 2010;22:129-139
16. Kaplanski G, Teysseire N, Farnarier C, Kaplanski S, Lissitzky JC, Durand JM, Soubeyrand J, Dinarello CA, Bongrand P. IL-6 and IL-8 production from cultured human endothelial cells stimulated by infection with Rickettsia conorii via a cell-associated IL-1 alpha-dependent pathway. J Clin Invest. 1995;96:2839-2844
17. Van Wagoner NJ, Oh JW, Repovic P, Benveniste EN. Interleukin-6 (IL-6) production by astrocytes: autocrine regulation by IL-6 and the soluble IL-6 receptor. J Neurosci. 1999;19:5236-5244
18. Sawa S, Kamimura D, Jin GH, Morikawa H, Kamon H, Nishihara M, Ishihara K, Murakami M, Hirano T. Autoimmune arthritis associated with mutated interleukin (IL)-6 receptor gp130 is driven by STAT3/IL-7-dependent homeostatic proliferation of CD4+ T cells. J Exp Med. 2006;12:1459-1470
19. Atsumi T, Ishihara K, Kamimura D, Ikushima H, Ohtani T, Hirota S, Kobayashi H, Park S, Saeki Y, Kitamura Y. et al. A point mutation of Tyr-759 in interleukin 6 family cytokine receptor subunit gp130 causes autoimmune arthritis. J Exp Med. 2002;196(7):979-990
20. Ogura H, Murakami M, Okuyama Y, Tsuruoka M, Kitabayashi C, Kanamoto M, Nishihara M, Iwakura Y, Hirano T. Interleukin-17 Promotes Autoimmunity by Triggering a Positive-Feedback Loop via Interleukin-6 Induction. Immunity. 2008;29:628-636
21. Murakami M, Okuyama Y, Ogura H, Asano S, Arima Y, Tsuruoka M, Harada M, Kanamoto M, Iwakura Y, Takatsu K. et al. Local microbleeding facilitates IL-6- and IL-17-dependent arthritis in the absence of tissue antigen recognition by activated T cells. J Exp Med. 2011;208:103-114
22. Murakami M, Hirano T. A four step model for the IL-6 amplifier, a critical regulator of chromic inflammations in tissue specific MHC class II-associated autoimmune diseases. Fromtiers Immunology. 2011;2:22. doi: 10.3389/fimmu.2011.00022
23. Arima Y, Harada M, Kamimura D, Park J-H, Kawano F, Yull FE, Kawamoto T, Iwakaura Y, Betz HAK, Marquez G. et al. Regional neural activation defines a gateway for autoreactive T cells to cross the blood-brain barrier. Cell. 2012;148:447-457
24. Lee J, Nakagiri T, Oto T, Harada M, Morii E, Shintani Y, Inoue M, Takahashi Y, Iwakura Y, Miyoshi S. et al. NFκB-triggered positive-feedback for IL-6 signaling in grafts plays a role for allogeneic rejection responses. J Immunol. in press
25. Hirano T, Matsuda T, Turne M, Miyasaka N, Buchan G, Tang B, Sato K, Shimizu M, Maini R, Feldmann M. et al. Excessive production of interleukin 6/B cell stimulatory factor-2 in rheumatoid arthritis. Eur J Immunol. 1986;18(11):1797-1801
26. Tanaka T, Kishimoto T. Immunotherapeutic implication of IL-6 blockade. Immunotherapy. 2012;4:87-105
27. Nishimoto N, Kishimoto T, Yoshizaki K. Anti-interleukin 6 receptor antibody treatment in rheumatic disease. Ann Rheum Dis. 2000;59(Suppl 1):i21-27
28. Nishihara M, Ogura H, Ueda N, Tsuruoka M, Kitabayashi C, Tsuji F, Aono H, Ishihara K, Huseby E, Betz UA. et al. IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int Immunol. 2007;19:695-702
29. Bettelli E, Carrier Y, Gao W, Korn T, Strom T, Oukka M, Weiner H, Kuchroo V. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235-238
30. Veldhoen M, Hocking R, Atkins C, Locksley R, Stockinger B. TGF in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(179):179-189
31. Atsumi T, Sato M, Kamimura D, Moroi A, Iwakura Y, Betz UA, Yoshimura A, Nishihara M, Hirano T, Murakami M. IFN-gamma expression in CD8+ T cells regulated by IL-6 signal is involved in superantigen-mediated CD4+ T cell death. Int Immunol. 2009;21:73-80
32. Park S, Nakagawa T, Kitamura H, Atsumi T, Kamon H, Sawa S, Kamimura D, Ueda N, Iwakura Y, Ishihara K. et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J Immunol. 2004;173(6):3844-3854
33. Sawa Y, Arima Y, Ogura H, Kitabayashi C, Jiang JJ, Fukushima T, Kamimura D, Hirano T, Murakami M. Hepatic interleukin-7 expression regulates T cell responses. Immunity. 2009;30:447-457
34. Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol. 2007;28:12-18
35. Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Invest. 2010;120:1368-1379
36. Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, Vyas JM, Boes M, Ploegh HL, Fox JG. et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307:254-258
37. Salinas S, Schiavo G, Kremer EJ. A hitchhiker's guide to the nervous system: the complex journey of viruses and toxins. Nat Rev Microbiol. 2010;8:645-655
38. McGavern DB, Kang SS. Illuminating viral infections in the nervous system. Nat Rev Immunol. 2011;11:318-329
Author contact
![]() Corresponding author: murakamimed.osaka-u.ac.jp and hiranomed.osaka-u.ac.jp.
Corresponding author: murakamimed.osaka-u.ac.jp and hiranomed.osaka-u.ac.jp.
Received 2012-7-6
Accepted 2012-8-14
Published 2012-10-25
Citation styles
APA
Murakami, M., Hirano, T. (2012). The Pathological and Physiological Roles of IL-6 Amplifier Activation. International Journal of Biological Sciences, 8(9), 1267-1280. https://doi.org/10.7150/ijbs.4828.
ACS
Murakami, M.; Hirano, T. The Pathological and Physiological Roles of IL-6 Amplifier Activation. Int. J. Biol. Sci. 2012, 8 (9), 1267-1280. DOI: 10.7150/ijbs.4828.
NLM
Murakami M, Hirano T. The Pathological and Physiological Roles of IL-6 Amplifier Activation. Int J Biol Sci 2012; 8(9):1267-1280. doi:10.7150/ijbs.4828. https://www.ijbs.com/v08p1267.htm
CSE
Murakami M, Hirano T. 2012. The Pathological and Physiological Roles of IL-6 Amplifier Activation. Int J Biol Sci. 8(9):1267-1280.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.