Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Embryonic development of the...
3. Post-natal epidermis:...
4. The E2F family
5. The E2F/pRB growth regulatory...
6. Role and regulation of E2F...
7. Role of the Rb family...
8. Role of cyclin-dependent...
9. The E2F/pRb pathway in...
10. Conclusions
Acknowledgements
References
Figures
Author biography

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2005; 1(2):87-95. doi:10.7150/ijbs.1.87 This issue Cite
Review
Signalling In The Epidermis: The E2f Cell Cycle Regulatory Pathway In Epidermal Morphogenesis, Regeneration And Transformation
Iordanka A. Ivanova1, Sudhir J. A. D'Souza1, Lina Dagnino1 2 3 ![]()
1 Dept. of Physiology & Pharmacology, University of Western Ontario, London, ON, Canada
2 Dept. of Paediatrics, University of Western Ontario, London, ON, Canada,
3 Child Health Research Institute and Lawson Health Research Institute, London, ON, Canada
Received 2004-10-1; Accepted 2005-2-1; Published 2005-4-1
Abstract
The epidermis is the outermost layer in the skin, and it is the first line of defence against the environment. The epidermis also provides a barrier against loss of fluids and electrolytes, which is crucial for life. Essential in the maintenance of this tissue is its ability to continually self-renew and regenerate after injury. These two characteristics are critically dependent on the ability of the principal epidermal cell type, the keratinocyte, to proliferate and to respond to differentiation cues. Indeed, the epidermis is a multilayered tissue composed of keratinocyte stem cells and their differentiated progeny. Central for the control of cell proliferation is the E2F transcription factor regulatory network. This signaling network also includes cyclins, cdk, cdk inhibitors and the retinoblastoma (pRb) family of proteins. The biological importance of the E2F/pRb pathway is emphasized by the fact that a majority of human tumours exhibit alterations that disrupt the ability of pRb proteins to inhibit E2F, leading to permanent activation of the latter. Further, E2F is essential for normal epidermal regeneration after injury. Other member of the E2F signaling pathway are also involved in epidermal development and pathophysiology. Thus, whereas the pRb family of proteins is essential for epidermal morphogenesis, abnormal regulation of cyclins and E2F proteins results in tumorgenesis in this tissue. In this review, we discuss the role of each member of this important growth regulatory network in epidermal formation, homeostasis and carcinogenesis.
Keywords: E2F, epidermis, proliferation, keratinocytes, tissue regeneration, stem cells
1. Introduction
Proper regulation of cell proliferation and differentiation is critical for the maintenance of self-renewing tissues, such as the epidermis, and is centrally involved in their pathogenesis. In mammalian cells, the E2F family of transcription factors is an important downstream effector in a pathway activated by multiple signaling events, which controls the expression of genes involved in cell cycle progression, G1/S transition and DNA replication. The E2F proteins are downstream components of a growth regulatory pathway that also includes members of the retinoblastoma (pRB) family, cyclins, cyclin-dependent kinases (cdk), as well as cdk inhibitors (reviewed in [1, 2, 3]). More recently, other E2F targets have been identified, which include genes involved in apoptosis, DNA repair and damage checkpoint.
A substantial body of work has been conducted to decipher how the E2F/pRB network functions, mostly using tumour cell lines or serum-starved fibroblasts, and largely in the context of progression from the G0/G1 to the S phases of the cell cycle. Several excellent recent reviews discuss these functions of E2F [1, 2, 3]. This review will focus on the regulation and role of the E2F growth regulatory network on homeostasis and pathogenesis of the epidermis, a complex tissue composed of stem cells and their undifferentiated progeny, as well as keratinocytes at various stages of differentiation and varied proliferative potential.
2. Embryonic development of the mammalian epidermis
The skin is the largest organ in the body and is the first line of defence against environmental insults, and microbial infection, as well as water and electrolyte loss. The skin is formed by two layers: The outer epidermis and the inner dermis. The epidermis is a complex stratified squamous epithelium formed by one basal and several suprabasal layers of keratinocytes, which provide barrier functions to the skin (Figure 1). Each epidermal layer contains keratinocytes at various stages of differentiation and proliferative potential (reviewed in [4]). In addition, the mammalian epidermis contains appendages, such as hair follicles, sebaceous glands, mammary and eccrine sweat glands, teeth and nails.
Architecture of the Skin. In the skin, the dermis and the epidermis are separated by a basement membrane. The epidermis is formed by a lower basal layer, and several suprabasal layers. the uppermost suprabasal layer is the cornified envelope (ce).

During early embryogenesis, the surface epithelium consists of a single-layered, highly proliferative ectoderm, present in embryonic day (E) 9-10 murine embryos [5, 6]. Between E9 and E12, the ectoderm undergoes stratification to produce a transitory peridermal layer, which is shed later on. The embryonic ectoderm is highly proliferative, as it must quickly expand to cover the growing embryo. Additional stratification of the ectoderm between E12 and E15 produces a basal and an intermediate layer. The cells in the intermediate layer undergo early terminal differentiation at E15-E16, ultimately giving rise to the upper (suprabasal) spinous and granular layers of the epidermis. Formation of epidermal appendages involves fate decisions in ectodermal stem cell lineages partly directed by signals originating in the underlying mesenchyme (reviewed in [7]). One of the last events during embryonic epidermal development is the acquisition of barrier function, which occurs shortly prior to birth, between E16 and E17 [8].
3. Post-natal epidermis: Architecture, self-renewal and regeneration after injury
The postnatal epidermis is formed by a lower, basal layer directly in contact with a basement membrane, and composed by epidermal stem cells and their committed progeny, termed transit amplifying cells (Fig. 1). As transit amplifying cells differentiate, they migrate upwards, sequentially giving rise to the suprabasal spinous, granular and cornified layers. The epidermis is continuous with appendages such as hair follicles and sebaceous glands, and undergoes constant renewal and remodeling, as cells on the cornified layers are shed and are replenished. In humans the epidermis is completely renewed every three weeks [9] . Critical for the self-renewal properties of the epidermis is the existence of multipotent stem cells, which can give rise to all lineages of skin epithelia, including interfollicular epidermis, hair follicles, and sebaceous glands. Epidermal stem cells are found at various sites, including in or near the bulge region of the hair follicle and in interfollicular epidermis [7, 9, 10, 11].
Wounding of the skin triggers extensive proliferation of epidermal stem cells. Clinically, grafting of cultured autologous epidermal stem cells efficiently reconstitutes the epidermis, although appendages are not regenerated, indicating that other signals, including those originating in the underlying mesenchyme are necessary for appendage formation (reviewed in [12, 13]). Re-epithelialization after wounding is a complex process that requires signals from inflammatory cells, dermal fibroblasts and keratinocytes themselves. The epidermal cells at the wound margin undergo pronounced changes, including the acquisition of migratory properties. In the keratinocytes just behind the actively migrating cells, a pronounced activation of proliferative pathways takes place, inducing extensive cell division which provides new keratinocytes needed to effectively cover the wound. Once this occurs, epidermal cells re-establish the basement membrane and initiate stratification, reverting to their resting phenotype. All of these changes involve tight regulation of expression and activity of growth regulatory proteins, especially those in the E2F pathway.
4. The E2F family
The E2F family of transcription factors consists of two subgroups termed E2F and DP (reviewed in [1, 2, 3]). In mammals, seven E2F and two DP genes have been identified. The functional E2F unit is generally a heterodimer containing one E2F and one DP protein, associated through their respective dimerization domains. The exception is E2F7, which does not interact with DP proteins, but functions as a homodimer [14, 15, 16]. Structurally, E2F1 through –5 contain a DNA binding domain, a leucine-zipper dimerization domain for interactions with DP proteins, and a C-terminal transcriptional activation domain that also mediates interactions with the pRb family of proteins (Fig. 2). E2F1, -2 and –3 also contain a cyclin A-binding domain and interact exclusively with pRb, whereas E2F4 and –5 do not bind cyclin A and interact preferentially with the two other pRb-related proteins, p107 and p130. E2F6 contains DNA-binding and dimerization domains very similar to the other E2F proteins, but lacks pRB-binding and transcriptional activation regions. There are two DNA-binding domains in E2F-7, which, remarkably, lacks dimerization and pRB-binding regions. DP proteins have a DNA-binding and a dimerization domain very similar to those found in the E2F subgroup, but they lack transcriptional activation and pRB-binding regions. The complexity of the E2F family is increased by the existence of multiple splice forms of DP2, E2Fs –3, -6 and –7, although the exact role of each splice form is unclear at present.
Schematic structure of the E2F family of transcription factors. All member of the E2F subgroup contain a DNA-binding domain (DNA). E2F1 through –6 contain a leucine-zipper dimerization domain that mediates interactions with DP proteins (DP), and E2F1 through 3 also have a cyclin A-binding region (CycA). C-terminal transactivation domains that mediate binding of E2F1 through –5 to pRb family proteins are also indicated. E2F7 has two DNA-binding domains and lacks DP- and pRb-binding regions. DP proteins only have DNA and E2F-binding domains.
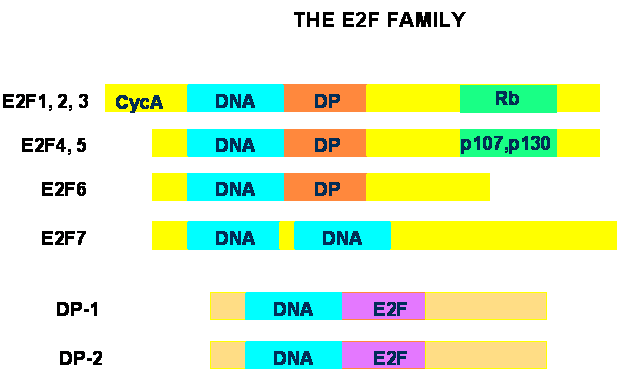
A large body of work has characterized the role of E2F in G1/S transition and DNA replication in mammalian cells. More recently, a broader function of some E2F proteins in mediating apoptosis, DNA damage and repair has been established (reviewed in [1, 17, 18]). Clearly, the breadth of biological activities of E2F factors is accompanied by a large variety of gene targets [19, 20], and the presence in E2F complexes of chromatin-modifying factors has emerged as a common mechanism involved in transcriptional regulation by E2F [2]. Such association is likely central for the ability of E2F-containing complexes to either activate or repress transcription of target genes.
The generation of mutant mouse models with targeted inactivation of E2F genes has shed light into some of their biological roles. Inactivation of E2F1 results in tissue-specific impairment of several functions in epidermal keratinocytes, including a marked reduction in proliferation rates, adhesion, migration and chemotaxis in response to serum or to transforming growth factor-β (TGF-β). In vivo, these defects result in significantly delayed wound healing [21]. In addition, E2F1-/- mice exhibit defective thymocyte apoptosis during normal thymic selection [22, 23], and abnormal pancreatic growth and function [24]. E2F2-/- mice develop autoimmune disorders consequent to enhanced T-lymphocyte proliferation [25], whereas E2F3-null animals exhibit abnormal cardiac function and develop congestive heart failure [26], and fibroblasts from these animals show abnormal centrosome amplification [27]. E2F4 is necessary for proper haematopoietic and intestinal epithelium maturation [28, 29], whereas E2F5 is indispensable for normal development and function of the differentiated choroid plexus epithelium [30].
5. The E2F/pRB growth regulatory pathway
E2F is a downstream component of a signaling network that regulates cell growth, as well as the transition between cell proliferation and terminal differentiation. This signaling network also includes cyclins, cdk, cdk inhibitors and the pRb family of proteins [1, 2, 3, 31]. In quiescent or terminally differentiated cells, E2Fs form complexes that generally containing p130 and, in some cases, pRb. These complexes repress transcription of genes necessary for DNA replication. Mitogenic stimulation of quiescent cells induces activation of cyclin D-cdk4(6) complexes, which phosphorylate pRb and p130 (Fig. 3). Phosphorylated pRb family proteins dissociate from E2F, and this “free” E2F becomes a transcriptional activator, directing the synthesis of factors involved in cell cycle progression and in DNA replication (E. g. cyclins E and A, cdk2, DNA polymerase). The biological importance of this pathway is emphasized by the fact that a majority of human tumours exhibit alterations that disrupt the ability of pRb proteins to inhibit E2F, leading to permanent activation of the latter [32, 33].
Regulation of G1/S transition by the E2F/pRb pathway. A. During G0 and early G1 phases, hypophosphorylated pRb family proteins form transcriptional repressor complexes that also contain histone deacetylace (HDAC) activity. B. Mitogenic stimulation activates cyclin D/cdk4 and/or cyclin D/cdk6 activity, resulting in pRb protein phosphorylation. Cyclin D/cdk complexes can be negatively regulated by cdk inhibitors (p15, p16, p18 and p19). C. Phosphorylation of pRb family proteins results in their dissociation from E2F factors, which, in turn allows activation of transcription by E2F. Some activator E2F complexes also contain hitone acetylase activitiy. E2F activation results in transcription of a variety of genes, including those encoding enzymes necessary for DNA replication and repair.
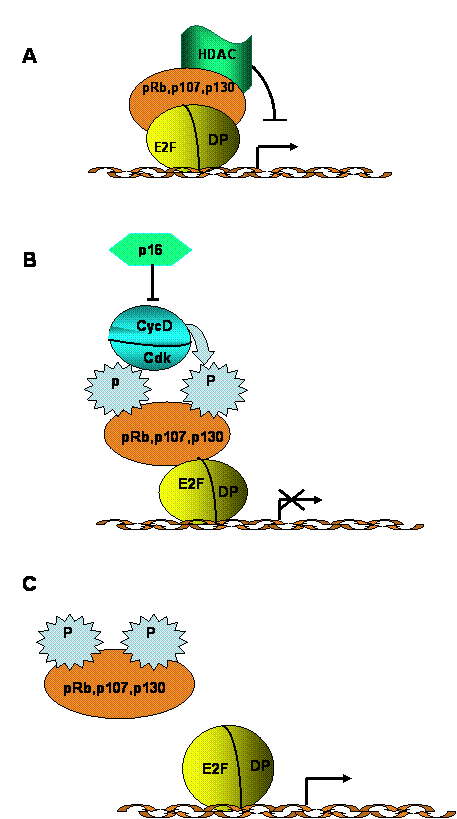
The E2F pathway is a target of many signaling cascades associated with entry into or exit from quiescence [31]. For example, activation of the ras and MAPK pathway induces cyclin D expression, pRb (p107 or p130) phosphorylation and E2F activation. Senescence and mitogenic inhibitors, such as TGF-β, induce cyclin D/cdk inhibitors, which results in the production of hypophosphorylated pRb proteins, which then associate with E2F, forming repressor complexes. DNA damage can trigger cell cycle arrest or apoptosis mediated through ATM and E2F1. Finally, signaling pathways activated by differentiation can activate E2F-mediated induction of key homeobox and Polycomb group proteins involved in development.
6. Role and regulation of E2F proteins in epidermal morphogenesis and homeostasis
The epidermis expresses at least 6 of the seven E2F forms isolated to-date (L. Dagnino, unpublished). During murine epidermal development, E2F4 mRNA is first detected in the E12.5 ectoderm, followed by appearance of E2F2 in basal cells underneath the intermediate layer in E14.5 embryos. Epidermal stratification at E15.5 triggers differential expression of E2F transcripts. E2F2 and E2F4 mRNA are detected in the basal keratinocyte layer and follicle epithelium around the dermal papilla, whereas E2F5 mRNA is abundant in the terminally differentiated suprabasal layers [34].
In primary cultured mouse keratinocytes, E2F1, 2, and 3 protein levels decrease during differentiation induced by Ca2+ or bone morphogenetic protein (BMP)-6 [35], although Ca2+-induced differentiation does not decrease E2F1 mRNA levels. Ca2+ activates multiple signaling pathways, including protein kinase C (PKC) [36], and the reduction in E2F1 protein levels in differentiating mouse keratinocytes involves activation of PKC δ and η (Ivanova and Dagnino, unpublished observations). E2F1 mRNA levels and stability decrease in primary human keratinocytes induced to differentiate by treatment with interferon γ or phorbol esters, and during senescence [37, 38, 39, 40]. In contrast, E2F1 protein levels did not decrease in immortalized human HaCaT keratinocytes induced to differentiate. Other differences have been reported in the regulation of E2F during differentiation of mouse keratinocytes relative to HaCaT cells. Specifically, whereas E2F4 and E2F5 proteins decrease during differentiation induced by serum withdrawal in HaCaT cells, induction of differentiation by Ca2+ or BMP-6 in murine keratinocytes does not significantly alter E2F4 levels, but results in E2F5 protein upregulation, consistent with the abundant E2F-5 transcripts in the suprabasal epidermal layers [35, 41]. The reason for these differences is not clear, and may be related to the distinct nature of the cell models used.
E2F1 plays a central role in maintaining keratinocytes in a proliferative, undifferentiated state. Specifically, primary cultured undifferentiated E2F1-/- keratinocytes exhibit doubling times about seven-fold longer than normal cells due to lengthening of the G1 and S phases. These cells also exhibit reduced signaling through integrins, which may partially account for their abnormal proliferation phenotype [21]. In contrast, exogenous E2F1 expression induces DNA synthesis in terminally differentiated mouse keratinocytes [35], and suppresses expression of the differentiation markers keratin K10, loricrin and involucrin in human cells induced to differentiate with Ca2+ treatment [41, 42, 43]. E2F1 also plays a role in the expression of the tumor suppressor gene BRCA1. Transgenic mice that specifically express E2F1 in the basal layer exhibit BRCA1 levels about 4-fold higher than wild type littermates [44]. Although the biological significance of this finding is unclear at present, as BRCA1 mutations have not been associated with epidermal disorders, E2F1 and BRCA1 may modulate DNA repair processes in UV-irradiated keratinocytes.
Signaling through TGF-β receptors in epidermal keratinocytes has multiple effects, including inhibition of proliferation and increased migration, but not induction of differentiation [45]. One mechanism for the antiproliferative effect of TGF-β involves nuclear translocation of pre-existing cytoplasmic complexes that contain Smad3 and E2F4 or E2F5, which bind to the c-myc promoter, thus repressing its transcription. TGF-β also induces down-regulation of E2F1, -2 and -3 [35, 46] and signaling through Smad7, which results in E2F1 mRNA down-regulation in keratinocytes [47].
7. Role of the Rb family proteins in the epidermis
Accumulating evidence from cultured cells and transgenic models shows the importance of the pRb family of proteins in epidermal morphogenesis. During late mouse embryogenesis, pRb, p107 and p130 transcripts are present the epidermis [48]. pRb is present in all mouse post-natal epidermal layers [49], and p130 is expressed in undifferentiated and differentiated murine cultured keratinocytes at similar levels [35]. In human epidermis, pRb and p107 proteins are expressed in all layers, whereas p130 is restricted to suprabasal keratinocytes [50]. Conflicting data exist for human keratinocytes induced to differentiate by culture in suspension. In one report, suspension reduced pRb levels and increased p130 abundance relative to undifferentiated cells [51], whereas another study reported little change in the abundance of these proteins [52].
The roles of the pRb family of proteins have been explored using exogenous expression in cultured cells and transgenic mouse models. In HaCaT cells, simultaneous exogenous expression of pRb and p107, or pRb, p107 and p130 induced growth arrest and expression of the differentiation markers keratin 10 and involucrin [50]. pRb is essential for epidermal differentiation and exit from the cell cycle, as mice with inactivation of Rb in the epidermis exhibited hyperplasia, DNA synthesis and keratin 14 expression in suprabasal layers normally negative for both, abnormal responses to radiation and centrosome synthesis, as well as reduced keratin 10 abundance in suprabasal keratinocytes [49]. One of the targets of the E2F/pRb pathway during differentiation is the cdk inhibitor p21, the transcription of which is activated directly by pRb [53].
Although mice with targeted inactivation of P107 or P130 do not show epidermal alterations, p107-/-/p130-/- double mutant animals exhibit impaired differentiation of the interfollicular epidermis and abnormalities in hair follicles [54]. This is indicative of a fundamental role for these two proteins in epidermal development, and suggests a degree functional redundancy between them. Taken together, the data indicate that pRb family proteins play central and not totally overlapping roles in the epidermis, as evidenced by the increased severity in epidermal abnormalities in p107-/-/p130-/- and pRb-/-/p107-/- double mutant mice, relative to the single null animals [54, 55].
A functional link between the E2F/pRb and the BMP signaling pathways has recently been described. The decrease in hair follicles and delayed follicle formation observed in p107-/-/p130-/- mice is accompanied by impaired expression of BMP-4, an essential follicle morphogen, and of other modulators of BMP signaling, including noggin [54]. Although the direct involvement of the E2F/pRb network in BMP4 expression has yet to be explored, clearly there is a genetic interaction between these two pathways. Curiously, although these double mutant mice also exhibit increased levels of nuclear β-catenin [56], their follicular phenotype contrasts with that of transgenic mice with increased levels of a stable, nuclear β-catenin mutant, which show greater hair follicle formation [57]. Folliculogenesis requires multiple interactions among signaling pathways and it is possible that in p107-/-/p130-/- mice additional signaling components are abnormal, resulting in reduced follicle formation in spite of the apparent increase in nuclear β-catenin.
Treatment of keratinocytes with TGF-β results in the formation of multiple pRb family complexes, including formation of E2F/p130 species associated with repression of cdc2 and cdc25 expression, the latter two proteins being indispensable for cell division [58, 59].
8. Role of cyclin-dependent kinases (cdk) in epidermal keratinocytes
The balance of active (hypophosphorylated) and inactive (hyperphosphorylated) pRb family proteins is partially regulated by cdk and their associated cyclins and inhibitors and, consequently, cdk activity is one of the upstream regulators of the E2F/pRb pathway. Many of these upstream regulators of E2F and pRb proteins are ubiquitously expressed in the epidermis, including cyclins D1, D3, E and A, cdk2, cdk4 and cdk6, as well as the cdk inhibitors p21, p27, p57, p15, p16 and p18 [60]. In cultured keratinocytes induced to differentiate by Ca2+ treatment, there is a pronounced downregulation of cyclins D1, D2, D3, and E, as well as cdk2, together with an increased abundance of cdk4, cdk6 and p21. These changes also reflect reduction in cdk2-associated kinase activity, normally present in proliferating cells [61], which generates hypophosphorylated pRb.
Keratinocytes isolated from p21-/- or p27-/- mice display increased proliferative capacity relative to wild type cells. Although the capacity of these mutant cells to exit the cell cycle upon differentiation is unaffected, expression of late differentiation markers is also impaired in p21-/- keratinocytes [62]. The epidermis of double mutant mice carrying an inactivating mutation in the INK/Arf (which encodes p16 and p19) and p21 loci is morphologically normal. However, expression of the differentiation markers keratin 10, filaggrin and loricrin is decreased, and keratin 6 is abnormally expressed in the suprabasal layers. Although basal keratinocyte proliferation was enhanced in these mutant cells, their ability to exit the cell cycle upon treatment with TGF-β or Ca2+ was unaffected [63].
Several mediators crucial for epidermal development act directly upstream from the E2F/pRb pathway. For example, sonic hedgehog (Shh) activation of the transcription factors Gli2 and Gli3 is indispensable for folliculogenesis [64]. Although, individually, cyclin D1 and cdk4 are dispensable for epidermal formation [65, 66], Shh- or Gli2-null mice show decreased cell proliferation in hair follicles due, in part, to impaired cyclin D1 and D2 expression [67]. Conversely, exogenous Shh expression in human keratinocytes induces epidermal hyperplasia, proliferation in suprabasal keratinocytes, and impaired cell cycle exit upon Ca2+-induced differentiation [68].
The epidermal phenotype of transgenic mouse models has confirmed a positive regulatory effect of D-type cyclins and cdk in the epidermis. Mice with targeted cyclin D1, D2 or D3 expression to the basal layer exhibit expansion of proliferating layers, and moderate acanthosis and hyperkeratosis, but retain normal differentiation responses [69, 70]. Similarly targeted cdk4 expression resulted in increased keratinocyte proliferation and epidermal thickness characterized by expansion of the differentiated spinous and granular layers, indicating that differentiation responses are operative in these animals. Sequestration of p27 by cdk4 with concomitant activation of cdk2/cyclin E complexes appears to mediate these effects [71].
Other pathways regulate keratinocyte proliferation and differentiation through modulation of cdk. For example, Ca2+-induced differentiation activates PKCη, which associates with complexes containing p21/cyclin E/cdk2, resulting in cdk2 inactivation and generation of hypophosphorylated pRb [72]. In addition, inhibition of keratinocyte proliferation by interferon-γ, tumor necrosis factor-α, TGF-β and phorbol esters involves activation of p16 [73].
9. The E2F/pRb pathway in epidermal regeneration and pathogenesis
Epidermal regeneration after wounding requires a proliferative burst and acquisition of migratory properties in keratinocytes [12]. Skin injury in humans triggers a marked up-regulation of E2F1 and E2F2, but not of E2F3 or E2F4 transcripts. E2F1 and E2F2 are expressed in migrating keratinocytes at the wound margin, as well as in all the layers of proliferative keratinocytes in the regenerating epidermis, just behind the migrating cell front [21].
Although the hyperproliferative phenotype of keratinocytes in inflammatory disorders such as psoriasis has been well documented, the role and regulation of the E2F/pRb pathway has received little attention. Chaturvedi et al. [73] have reported heterogenous regulation of the cdk inhibitors p12, p14ARF and p16, with elevated levels of these proteins in 33% to 88% of psoriatic samples, suggesting that none of these proteins may play a universal causative role in the keratinocyte phenotype observed in this disorder. Cyclosporin treatment of psoriatic plaques induces reduction in epidermal proliferation characterized by a reduction in overexpressed cyclins D1, A and B [74], although, paradoxically, cyclosporine also interferes with p21 expression in keratinocytes [75]. The role of the E2F/pRb pathway in psoriasis is unclear, but pharmacological intervention with agents that regulate cell cycle proteins could aid in the therapy of this disorder.
Several lines of evidence implicate the E2F/pRb pathway in carcinogenesis. In pre-cancerous actinic keratoses and in seborrhoeic keratoses, a condition caused by excessive sunlight exposure, but not in normal epidermis, weak to moderate p16 expression has been detected. In contrast, progression to squamous cell carcinoma is associated with high p16 expression [76, 77]. Correlative studies between E2F expression and clinical outcomes in human tumors indicate that the consequences of changes in E2F expression depend on multiple factors, including tissue type and status of other cell growth regulators, such as pRb, p53 and p16 [78]. E2F1 amplification associated with poor prognosis occurs in esophageal squamous cell carcinomas, suggesting association between abnormal E2F regulation and tumorigenesis in squamous epithelia [79, 80]. However, the status of E2F1 in epidermal squamous cell carcinomas has yet to be investigated.
Experimental tumorigenic treatments in mouse epidermis result in up-regulation of E2F1, -2, -4 and –5, p16 and p57, as well as increased abundance of cdk4-containing complexes during pre-malignant progression [81, 82]. Overexpression of E2F1 targeted to basal keratinocytes in transgenic mice induces epidermal hyperplasia and tumor formation with long latency, and cooperates with Ha-ras or p53 inactivation in tumor induction [83, 84, 85]. These E2F1 transgenic animals were resistant to experimental carcinogenesis, likely due to increased levels of apoptosis, unlike transgenic mice overexpressing E2F4 in basal keratinocytes, which developed larger tumors [86]. Finally, transgenic mice expressing DP1 targeted to the basal layer exhibited increased tumor size and numbers when subjected to experimental carcinogenesis protocols, but not spontaneously, indicating that DP1 deregulation may facilitate tumor formation subsequent to other insults [87]. Cyclin D1 is necessary for epidermal tumor formation, as in similar carcinogenesis models, cyclin D1-null mice develop lesions with reduced frequency compared with wild type animals [88], whereas transgenic mice expressing cdk4 in basal keratinocytes present high rates of squamous cell carcinoma formation [89].
Exposure to ultraviolet (UV) radiation is a major risk factor for the development of epidermal tumors. Following epidermal exposure to UV-B, p16 and p21 protein levels increase [90], possibly to mediate growth arrest associated with UV-induced DNA damage. Notably, absence of E2F1 increases UV-induced keratinocyte apoptosis, in stark contrast with the well established role of this protein in induction of apoptosis in other systems [91].
10. Conclusions
Impressive progress has been made in elucidating the functions and mechanisms of action of the various components of the E2F/pRb pathway. This pathway is now recognized as a crucial network in the regulation of tissue development, remodeling and homeostasis. The involvement of alterations in the E2F/pRb network in the formation of epidermal tumors has been suggested by recent experimental evidence, and modulation of this pathway may represent an effective therapeutic tool for a variety of epidermal disorders.
Acknowledgements
Work in our laboratory has been supported by grants from the Canadian Institutes of Health Research (CIHR), the National Science and Engineering Research Council of Canada, and the National Cancer Institute of Canada (NCIC) with funds from the Canadian Cancer Society and the Terry Fox Foundation raised through the Terry Fox Run. IAI is a Research Student of the Terry Fox Foundation, through an award from the NCIC. SJAD is a CIHR New Investigator, and LD is a CIHR/Cancer Research Society Inc. New Investigator.
Conflict of interest
The authors declare that no conflict of interest exists.
References
1. Cam H, Dynlacht BD. Emerging roles for E2Fs: Beyond the G1/S transition and DNA replication. Cancer Cell. 2003 ;3:311-316
2. Frolov MV, Dyson NJ. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J Cell Science. 2004 ;117:2173-2181
3. Stevens C, La Thangue NB. E2F and cell cycle control: a double-edge sword. Arch Biochem Biophys. 2003 ;412:157-169
4. Fuchs E, Byrne C. The epidermis: rising to the surface. Curr Opin Genet Dev. 1994 ;4:725-36
5. Byrne C, Hardman MJ, Nield K. Covering the limb - formation of the integument. J Anat. 2003 ;202:113-124
6. M'Boneko V, Merker HJ. Development and morphology of the periderm of mouse embryos (days 9-12 of gestation). Acta Anat. 1988 ;133:325-336
7. Niemann C, Watt FM. Designer skin: lineage committment in postnatal epidermis. Trends Cell Biol. 2002 ;12:185-192
8. Hardman MJ. et al. Patterned acquisition of skin barrier function during development. Development. 1998 ;125:1541-1552
9. Brouard M, Barrandon Y. Controlling skin morphogenesis: hope and despair. Curr Opin Biotech. 2003 ;14:520-525
10. Alonso L, Fuchs E. Stem cells of the skin epithelium. Proc Natl Acad Sci USA. 2003 ;100:11830-11835
11. Gambardella L, Barrandon Y. The multifaceted adult epidermal stem cell. Curr Opin Cell Biol. 2003 ;15:771-777
12. Martin P. Wound healing -aiming for perfect skin regeneration. Science. 1997 ;276:75-81
13. Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999 ;341:738-746
14. De Bruin A. et al. Identification and characterization of E2F7, a novel mammalian E2F family member capable of blocking cellular proliferation. J Biol Chem. 2003 ;278:42041-42049
15. DiStefano L, Jensen MR, Helin K. E2F7, a novel E2f featuring DP-independent repression of a subset of E2F-regulated genes. EMBO J. 2003 ;22:6289-6298
16. Logan N. et al. E2F-7: a distinctive E2F family member with an unusual organization of DNA-binding domains. Oncogene. 2004 ;23:5138-5150
17. Bell LA, Ryan KM. Life and death decisions by E2F-1. Cell Death Differ. 2004 ;11:137-142
18. Hitchens MR, Robbins PD. The role of the transcription factor DP in apoptosis. Apoptosis. 2003 ;8:461-468
19. Muller H. et al. E2Fs regulate the expression of genes involved in differentiation, development, proliferation and apoptosis. Genes Dev. 2001 ;15:267-285
20. Young AP, Nagarajan R, Longmore GD. Mechanisms of transcriptional regulation by Rb-E2F segregate by biological pathway. Oncogene. 2003 ;22:7209-7217
21. D'Souza SJA. et al. E2F-1 is essential for normal epidermal wound repair. J Biol Chem. 2002 ;277:10626-10632
22. Field SJ. et al. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell. 1996 ;85:549-561
23. Yamasaki L. et al. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell. 1996 ;85:537-548
24. Fajas L. et al. Impaired pancreatic growth, beta cell mass and beta cell function in E2F1(-/-) mice. J Clin Invest. 2004 ;113:1288-1295
25. Murga M. et al. Mutation of E2F2 in mice causes enhanced T lymphocyte proliferation, leading to the development of autoimmunity. Immunity. 2001 ;15:959-970
26. Rogers JE. et al. Mutant mouse models reveal the relative roles of E2F1 and E2F3 in vivo. Molec Cell Biol. 2002 ;22:2663-2672
27. Saavedra HI. et al. Inactivation of E2F3 results in centrosome amplification. Cancer Cell. 2003 ;3:333-346
28. Humbert PO. et al. E2F4 is essential for normal erythrocyte maturation and neonatal viability. Mol Cell. 2000 ;6:281-291
29. Rempel RE. et al. Loss of E2F4 activity leads to abnormal development of multiple cellular lineages. Mol Cell. 2000 ;6:293-306
30. Lindeman GJ. et al. A specific, nonproliferative role for E2F-5 in choroid plexus function revealed by gene targeting. Genes Dev. 1998 ;12:1092-1098
31. Stevaux O, Dyson NJ. A revised picture of the E2F transcriptional network and RB function. Curr Opin Cell Biol. 2002 ;14:684-691
32. Hunter T. Oncoprotein networks. Cell. 1997 ;88:333-346
33. Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995 ;81:323-330
34. Dagnino L. et al. Expression patterns of the E2F family of transcription factors during murine epithelial development. Cell Growth Differ. 1997 ;8:553-563
35. D'Souza SJA. et al. Ca+2 and BMP-6 signalling regulate E2F during epidermal keratinocyte differentiation. J Biol Chem. 2001 ;276:23531-23538
36. Denning MF. Epidermal keratinocytes: regulation of multiple cell phenotypes by multiple protein kinase C isoforms. Int J Biochem Cell Biol. 2004 ;36:1141-1146
37. Jones SJ. et al. E2F as a regulator of keratinocyte proliferation: implications for skin tumor development. J Invest Dermatol. 1997 ;109:187-193
38. Saunders NA. et al. E2F1 messenger RNA is destabilized in response to a growth inhibitor in normal human keratinocytes but not in a squamous carcinoma cell line. Cancer Res. 1998 ;58:1646-1649
39. Saunders NA, Jetten AM. Control of growth regulatory and differentiation-specific genes in human epidermal keratinocytes by interferon gamma. Antagonism by retinoic acid and transforming growth factor beta 1. J Biol Chem. 1994 ;269:2016-2022
40. Saunders NA, Smith RJ, Jetten AM. Regulation of proliferation-specific and differentiation-specific genes during senescence of human epidermal keratinocyte and mammary epithelial cells. Biochem Biophys Res Commun. 1993 ;197:46-54
41. Paramio JM. et al. Opposite functions for E2F1 and E2F4 in human epidermal keratinocyte differentiation. J Biol Chem. 2000 ;275:41219-41226
42. Dicker AJ. et al. E2F-1 induces proliferation-specific genes and suppresses squamous differentiation-specific genes in human epidermal keratinocytes. Oncogene. 2000 ;19:2887-2894
43. Wong CF. et al. E2F modulates keratinocyte squamous differentiation: Implications for E2F inhibition in squamous cell carcinoma. J Biol Chem. 2003 ;278:28516-28522
44. Wang A. et al. Regulation of BRCA1 expression by the Rb-E2F pathway. J Biol Chem. 2000 ;275:4532-4536
45. Dagnino L. et al. Transforming growth factor regulation of keratinocyte growth. Recent Results Cancer Res. 1993 ;128:15-29
46. Chen CR. et al. E2F4/5 and p107 as Smad cofactors linking the TGFβ receptor to c-myc repression. Cell. 2002 ;110:19-32
47. Smith L. et al. Modulation of proliferation-specific and differentiation-specific markers in human keratinocytes by SMAD7. Exp Cell Res. 2004 ;294:356-365
48. Jiang Z. et al. The retinoblastoma gene family is differentially expressed during embryogenesis. Oncogene. 1997 ;14:1789-1797
49. Balsitis SJ. et al. Recapitulation of the effects of the human papillomavirus type 16 E7 oncogene on mouse epithelium by somatic Rb deletion and detection of pRb-independent effects of E7 in vivo. Molec Cell Biol. 2003 ;23:9094-9103
50. Paramio JM. et al. Differential expression and functionally co-operative roles for the retinoblastoma family of proteins in epidermal differentiation. Oncogene. 1998 ;17:949-957
51. Harvat BL. et al. Up-regulation of p27Kip1, p21WAF1/Cip1 and p16Ink4a is associated with, but not sufficient for, induction of squamous differentiation. J Cell Sci. 1998 ;111:1185-1196
52. Tibudan SS, Wang Y, Denning MF. Activation of protein kinase C triggers irreversible cell cycle withdrawal in human keratinocytes. J Invest Dermatol. 2002 ;119:1282-1289
53. Decesse JT. et al. RB regulates transcription of the p21/WAF1/CIP1 gene. Oncogene. 2001 ;20:962-971
54. Ruiz S. et al. Abnormal epidermal differentiation and impaired epithelial-mesenchymal tissue interactions in mice lacking the retinoblastoma relatives p107 and p130. Development. 2003 ;130:2341-2353
55. Ruiz S. et al. Unique and overlapping functions of pRb and p107 in the control of proliferation and differentiation in epidermis. Development. 2004 ;131:2737-2748
56. Ruiz S. et al. Functional link between retinoblastoma family proteins and the Wnt signaling pathway in mouse epidermis. Dev Dyn. 2004 ;230:410-418
57. Gat U. et al. De Novo hair follicle morphogenesis and hair tumors in mice expressing a truncated beta-catenin in skin. Cell. 1998 ;95:605-614
58. Herzinger T. et al. The pRb-related protein p130 is a possible effector of transforming growth factor beta 1 induced cell cycle arrest in keratinocytes. Oncogene. 1995 ;10:2079-2084
59. Iavarone A, Massague J. E2F and histone deacetylase mediate transforming growth factor beta repression of cdc25A during keratinocyte cell cycle arrest. Mol Cell Biol. 1999 ;19:916-922
60. Bartkova J. et al. Cell cycle regulatory proteins in human wound healing. Arch Oral Biol. 2003 ;48:125-132
61. Martinez LA. et al. Coordinated changes in cell cycle machinery occur during keratinocyte terminal differentiation. Oncogene. 1999 ;18:397-406
62. Missero C. et al. The absence of p21Cip1/WAF1 alters keratinocyte growth and differentiation and promotes ras-tumor progression. Genes Dev. 1996 ;10:3065-3075
63. Paramio JM. et al. The ink4/arf tumor suppressors cooperate with p21cip/waf in the processes of mouse epidermal differentiation, senescence and carcinogenesis. J Biol Chem. 2001 ;276:44203-44211
64. St-Jacques B. et al. Sonic hedgehog signaling is essential for hair developmment. Curr Biol. 1998 ;8:1058-1068
65. Fantl V. et al. Mice lacking cyclin D1 are small and show defects in eye and mammary gland development. Genes Dev. 1995 ;9:2364-2372
66. Rodriguez-Puebla ML. et al. Ckd4 deficiency inhibits skin tumor development but does not affect normal keratinocyte proliferation. Am J Phatol. 2002 ;161:405-411
67. Mill P. et al. Sonic hedgehog-dependent activation of Gli2 is essential for embryonic hair follicle development. Genes Dev. 2003 ;17:282-294
68. Fan H, Khavri PA. Sonic Hedgehog opposes epithelial cell cycle arrest. J Cell Biol. 1999 ;147:71-76
69. Robles AI. et al. Expression of cyclin D1 in epithelial tissues of transgenic mice results in epidermal hyperproliferation and severe thymic hyperplasia. Proc Natl Acad Sci USA. 1996 ;93:7634-7638
70. Rodriguez-Puebla ML. et al. Cyclin D2 overexpression in transgenic mice induces thymic and epidermal hyperplasia whereas cyclin D3 3expression results only in epidermal hyperplasia. Am J Phatol. 2000 ;157:1039-1050
71. Miliani de Marval PL. et al. Transgenic expression of cyclin-dependent kinase 4 results in epidermal hyperplasia, hypertrophy and severe dermal fibrosis. Am J Phatol. 2001 ;159:369-379
72. Kashiwagi M. et al. PKCη associates with cyclinE/dck2/p21 complex, phosphorylates p21 and inhibits cdk2 kinase in keratinocytes. Oncogene. 2000 ;19:6334-6341
73. Chaturvedi V. et al. Role of INK4a/Arf locus-encoded senescent checkpoints activated in normal and psoriatic keratinocytes. Am J Phatol. 2003 ;162:161-170
74. Miracco C. et al. Cyclin D1, B and A expression and cell turnover in psoriatic skin lesions before and after cyclosporin treatment. Br J Dermatol. 2000 ;143:950-956
75. Santini MP. et al. Cross talk among calcineurin, Sp1/Sp3 and NFAT in control of p21(WAF/CIP1) expression in keratinocye differentiation. Proc Natl Acad Sci USA. 2001 ;98:9575-9580
76. Hodges A, Smoller BR. Immunohistochemical comparison of p16 expression in actinic keratoses and squamous cell carcinomas of the skin. Mod Pathol. 2002 ;15:1121-1125
77. Nakamura S, Nishioka K. Enhanced expression of p16 in seborrhoeic keratosis: a lesion of accumulated senescent epidermal cells in G1 arrest. Br J Dermatol. 2003 ;19:175-181
78. Zacharatos P. et al. Distinct expression patterns of the transcription factor E2F-1 in relation to tumor growth parameters in common human carcinomas. J Pathol. 2004 ;203:744-753
79. Ebihara Y. et al. Over-expression of E2F-1 in esophageal squamous cell carcinoma correlates with tumor progression. Dis Esophagus. 2004 ;17:150-154
80. Fujita Y. et al. Chromosome arm 20q gains and other genomic alterations in esophageal squamous cell carcinoma, as analyzed by comparative genomic hybridization and fluorescence in situ hybridization. Hepatogastroenterology. 2003 ;50:1857-1863
81. Balasubramanian S, Ahmad N, Mukhtar H. Upregulation of E2F transcription factors in chemically induced mouse skin tumors. Int J Oncol. 1999 ;15:387-390
82. Rodriguez-Puebla ML. et al. Deregulated expression of cell-cycle proteins during premalignant progression in SENCAR mouse skin. Oncogene. 1998 ;17:2251-2258
83. Pierce AM. et al. Increased E2F1 activity induces skin tumors in mice heterozygous and nullizygous for p53. Proc Natl Acad Sci USA. 1998 ;95:8858-8863
84. Pierce AM. et al. Deregulated expression of E2F1 induces hyperplasia and cooperates with ras in skin tumor development. Oncogene. 1998 ;16:1267-1276
85. Pierce AM. et al. E2F1 has both oncogenic and tumor-suppressive properties in a transgenic model. Mol Cell Biol. 1999 ;19:6408-6414
86. Wang D, Russell JL, Johnson DG. E2F4 and E2F1 have similar proliferative properties but different apoptotic and oncogenic properties in vivo. Mol Cell Biol. 2000 ;20:3417-3424
87. Wang D. et al. Deregulated expression of DP1 induces epidermal proliferation and enhances skin carcinogenesis. Mol Carcinog. 2001 ;31:90-100
88. Robles AI. et al. Reduced skin tumor development in cyclin D1-deficient mice highlights the oncogenic ras pathway in vivo. Genes Dev. 1998 ;12:2469-2474
89. Miliani de Marval PL. et al. Enhanced malignant tumorigenesis in Cdk4 transgenic mice. Oncogene. 2004 ;23:1863-1873
90. Ahmed NU, Ueda M, Ichihashi M. Induced expression of p16 and p21 proteins in UVB-irradiated human epidermis and cultured keratinocytes. J Dermatol Sci. 1999 ;19:175-181
91. Wikonkal NM. et al. Inactivating E2f1 reverts apoptosis resistance and cancer sensitivity in Trp53-deficient mice. Nat Cell Biol. 2003 ;5:655-660
Figures
Author biography
Lina Dagnino received Ph.D. in Pharmacology at the University of Alberta, Canada. Currently she is an assistant Professor at Dept .of Physiology & Pharmacology and Dept. of Paediatrics of University of Western Ontario. She is also a Scientist of the Lawson Health Research Institute and the Child Health Research Institute, Canada. Previously she was an investigator at the Ottawa Hospital Research Institute. Her research interests include the transcriptional control of growth regulatory networks and the mechanisms that modulate epidermal cell fate, differentiation, regeneration and transformation. She has been the recipient of a Canadian Foundation for Innovation New Investigator Award, and currently holds a Canadian Institutes of Health Research/Cancer Research Society New Investigator Award, and a Premier's Research Excellence Award from the Ontario Ministry of Health.
Iordanka A. Ivanova is a doctoral student at the University of Western Ontario. She has received graduate awards from the Ontario Graduate Studentship Programme, the National Science and Engineering Council of Canada and the Terry Fox Foundation through the National Cancer Institute of Canada.
Sudhir J. A. D'Souza received a Ph.D. in Pharmacology from the University of Alberta, Canada and an M. D. C. M. degree from McGill University, Canada. He trained at Vanderbilt University, USA, where he did a residence in Paediatrics, followed by a fellowship in Paediatric Nephrology at the Hospital for Sick Children in Toronto, Canada. He is currently an attending Paediatric Nephrologist at St. Josephs's Health Center, Toronto, and Assistant Professor in the Dept. of Physiology and Pharmacology at the University of Western Ontario. Dr. D'Souza's research interests focus on the mechanisms of maintenance of the epithelial phenotype and the role of proteins in the “Regulators of G protein Signalling” (RGS) family in mitosis and microtubule dynamics. He has been recipient of multiple awards, including a Canadian Foundation for Innovation New Investigator Award, a Kidney Foundation Scholarship, and is currently a Canadian Institutes of Health Research New Investigator.
![]() Corresponding address:
Corresponding address:
L. Dagnino, Dept. Physiology & Pharmacology, University of Western Ontario, London, ON, Canada, N6A 5C1. Fax: (519) 661-3827, email: ldagninoca