Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2005; 1(3):114-122. doi:10.7150/ijbs.1.114 This issue Cite
Research Paper
Purification, properties and alternate substrate specificities of arginase from two different sources: Vigna catjang cotyledon and buffalo liver
Snehal Dabir1, Pankaj Dabir2, Baburao Somvanshi1 ![]()
1 Department of Biochemistry, Dr BA Marathwada University, Aurangabad – 400 001 (MS), India
2 Department of Biochemistry and JB Tropical Disease Research Centre, Mahatma Gandhi Institute of Medical Sciences, Sevagram - 442 102, (MS), India
Received 2005-3-10; Accepted 2005-7-8; Published 2005-8-1
Abstract
Arginase was purified from Vigna catjang cotyledons and buffalo liver by chromatographic separations using Bio-Gel P-150, DEAE-cellulose and arginine AH Sepharose 4B affinity columns. The native molecular weight of an enzyme estimated on Bio-Gel P-300 column for Vigna catjang was 210 kDa and 120 kDa of buffalo liver, while SDS-PAGE showed a single band of molecular weight 52 kDa for cotyledon and 43 kDa for buffalo liver arginase. The kinetic properties determined for the purified cotyledon and liver arginase showed an optimum pH of 10.0 and pH 9.2 respectively. Optimal cofactor Mn++ ion concentration was found to be 0.6 mM for cotyledon and 2 mM for liver arginase. The Michaelis-Menten constant for cotyledon arginase and hepatic arginase were found to be 42 mM and 2 mM respectively. The activity of guanidino compounds as alternate substrates for Vigna catjang cotyledon and buffalo liver arginase is critically dependent on the length of the amino acid side chain and the number of carbon atoms. In addition to L-arginine cotyledon arginase showed substrate specificity towards agmatine and L-canavanine, whereas the liver arginase showed substrate specificity towards only L-canavanine.
Keywords: Purification, kinetic properties, substrate specificity arginine amidino hydrolase, Vigna catjang cotyledon, buffalo liver
1. Introduction
L-Arginase (L-arginine amidino hydrolase, EC 3.5.3.1.) is a binuclear manganese metalloenzyme that catalyzes the hydrolysis of L-arginine to form L-ornithine and urea. Arginase is the terminal enzyme of the urea cycle among the six enzymes. The enzyme was found to exist in two forms and has a broad tissue distribution. A cytosolic form, AI is highly expressed in the liver or hepatic cells and is important in ureogenesis. Extra-hepatic AII form is thought to be involved in the biosynthesis of polyamines, the amino acids ornithine, proline and glutamate and in the inflammatory process, among others [1]. Recently studies have shown that increased stimulation of arginase expression in animal systems leads to production of polyamines that promote tumor cell proliferation [2] and wound healing [3].
Most of the studies on plant arginase have focused on its role in mobilizing arginine during early seedling germination. Storage proteins are mobilized to provide amino acids for protein synthesis in the expanding axis. Arginase activity increases sharply during germination in several species including soybean [4, 5], arabidopsis and loblolly pines [6, 7]. In higher plants urea and ornithine formed as products of arginase reaction are used in the assimilation of nitrogen into amino acids and the synthesis of polyamines through urease and ornithine decarboxylase respectively [8, 9]. The enzyme has been isolated and characterized from cotyledons as well as axis of soybean [5, 8]. But no properties of the purified enzyme from the cotyledons of Vigna catjang and buffalo liver have been reported. In animal's arginase is purified and characterized from a variety of sources having different characteristic properties with regard to molecular weight, subunit structure and its regulation especially in the urea cycle [10].
In contrast far less is known about the purification and properties of the enzyme in higher plants [11, 12]. Hence the present study was undertaken for the purification and properties of arginase from Vigna catjang cotyledon and buffalo liver, and also reports physicochemical and kinetic properties of the purified enzyme.
This purified enzyme can be used to study the effect of L-amino acids on the inhibition of arginases, to know the composition of amino acids at the active site of enzyme and may be useful in investigating its role in mobilizing the storage proteins.
2. Materials and methods
Source of enzyme
Seeds of cow pea Vigna catjang were obtained from the local market; surface sterilized by treating with Lysol, soaked in water for 6 hrs and then allowed to germinate for 24 hrs on moist filter paper at 25oC.
Buffalo liver sample was collected from the local slaughterhouse. A portion of liver tissue was removed within 15 min after sacrificing the animal. It was placed in ice bath and brought to the laboratory as early as possible. The liver sample was immediately used for the isolation and purification of arginase.
Cotyledon arginase activity assay
Arginase activity was measured according to the Roman and Ruy's method [13]. Briefly the reaction mixture consisting of 10 mM carbonate bicarbonate buffer (pH 10), 2 mM MnCl2, 130 mM L-arginine and enzyme solution in a total volume of 1 ml was incubated for 30 minutes at 37 oC. The reaction was terminated by adding 10% TCA. Protein was removed by centrifugation; 1 ml of the supernatant was mixed with 1ml of anhydrous glacial acetic acid and ninhydrin reagent. The mixture was kept in boiling water bath for 20 min. The intense red colour developed was diluted with 2 mls of glacial acetic acid. The reagent blank was run in a parallel way and the results were compared with control assay. The optical density was read at 515 nm on spectrophotometer (UV 160A Shimatzu, Japan.) against reagent blank. The activity of arginase was expressed in units. A unit of an enzyme activity is defined as that amount of an enzyme, which produces one µmole of L-ornithine per min at 37oC.
Liver arginase activity assay
Enzyme assay and experimental conditions used for buffalo liver arginase were same as described above for Vigna catjang cotyledon arginase, except millimolar concentrations of MnCl2 (6 mM), L-arginine (25 mM) and carbonate bicarbonate buffer (50 mM, pH 9.2).
Protein determination
The protein content of each fraction was determined by the method of Lowry et al. [14] with the bovine serum albumin (Sigma, USA) as standard.
Purification of cotyledon arginase
Purification of the enzyme was carried out at 0-4 oC. The cotyledons (100 gm) were homogenized in a chilled mixer with 300 ml of 50 mM Tris-HCl buffer (pH 7.5) containing 0.6 mM MnCl2, 100 mM KCl and 10% Glycerol. The homogenate was filtered through four layers of gauze and clarified by centrifugation at 13,000 rpm for 30 min at 4 ºC.
Ammonium sulphate precipitation
The supernatant was adjusted to 30% saturation with solid ammonium sulphate and stirred at 4ºC for 5 hours. The solution was then centrifuged and the pellet was discarded. The supernatant was brought to 70% saturation with solid (NH4) 2 SO4 and treated as above except that the pellet was retained. The pellet containing the enzyme was resuspended with 30 mls of 10 mM Tris-HCl buffer (pH 7.5) containing 0.6 mM MnCl2 and 10% glycerol. The extract was purified by centrifugation.
Bio-Gel P-150 chromatography
The clear supernatant was applied to a Bio-Gel P-150 column (2 X 80 cm) equilibrated with Tris-HCl buffer (10 mM, pH 7.5) containing 0.6 mM MnCl2 and 10% glycerol. The enzyme was eluted using the same buffer with a flow rate of 12 ml / hour.
DEAE-cellulose chromatography
Active fractions obtained from above step were applied to DEAE-cellulose (Sigma) column (2.5 X 12 cm) previously equilibrated with Tris-HCl buffer (10 mM, pH 7.5) containing 10% glycerol. The column was washed with the same buffer and the enzyme was eluted using a linear gradient of sodium chloride (0-0.5 M) prepared in column-equilibrated buffer.
Affinity chromatography
Active fractions from above step were pooled and applied on a hydroxyapatite column (1.2 X 10 cm) (Bio-Rad, USA) previously equilibrated with Tris-HCl buffer (10 mM, pH 7.5) containing 0.6 mM MnCl2 and 10% glycerol followed by 15 ml of 0.5 M KCl prepared in column equilibrating buffer. The flow rate of column was adjusted to 12 ml / hour, each 1.0 ml fractions were collected and its protein content and arginase activity were assayed.
Purification of liver arginase
Liver sample was weighed (30 gm) homogenized with 120 ml of chilled Tris-HCl buffer (100 mM, pH 7.5) containing 5 mM MnCl2 and 100 mM KCl. The homogenate was centrifuged at 13,000 rpm, 30 minutes and the supernatant was collected.
Heat treatment
The heat treatment was given to the above supernatant at 60 oC, 20 min with swirling. It was then immediately cooled in ice bath and denatured proteins were removed by centrifugation.
Ammonium sulphate precipitation
The resulting supernatant was adjusted to 30% saturation with solid (NH4) 2 SO4 and stirred at 4 ºC for 1 hour. The solution was then centrifuged and the pellet was discarded. The supernatant was brought to 70% saturation with solid ammonium sulphate and treated as above except that the pellet was retained. The pellet was resuspended in 15 mM Tris-HCl buffer pH 8.0 containing 5 mM MnCl2 and kept for 2 hours. The unresolved proteins were removed by centrifugation and dialyzed against the Tris-HCl buffer used for homogenization.
DEAE-cellulose chromatography
The dialyzed material was applied to a 2.5 X 12 cm column of DEAE-cellulose (Sigma) at 4 oC, equilibrated with the Tris-HCl buffer (15 mM, pH 8.0) containing 2 mM MnCl2. Elution was carried out with a linear gradient of NaCl from 0 to 0.5 M in same buffer containing 2 mM MnCl2 at a flow rate of 20 ml / hour. The active fractions containing arginase activity were pooled together.
Bio-Gel P-150 chromatography
Above pooled fractions were dialyzed against Tris-HCl buffer (15 mM, pH 8.0) containing 2 mM MnCl2 and applied as a concentrated zone on Bio-Gel P-150 (2 X 80 cm) column equilibrated with Tris-HCl buffer (15 mM, pH 8.0) containing 2 mM MnCl2. Each 2 mls of fractions were collected at 12 ml / hour and active fractions containing arginase activity were pooled.
Affinity chromatography
The pooled fractions from above step were dialyzed at 4 oC in 3 X 3 L of Tris-HCl buffer (15 mM, pH 8.0) containing 2 mM MnCl2 and passed through the arginine AH-Sepharose 4B (1.2 X 10 cm) column equilibrated with 15 mM Tris-HCl buffer pH 8.0 containing 1 mM MnCl2.The column was washed with 20 mls of equilibrating buffer without MnCl2. The adsorbed proteins were eluted by passing 20 mls of 10 mM Tris-HCl (pH 8.0), 10 mls of 50 mM Tris-HCl (pH 7.5), 10 mls of 50 mM Tris-HCl (pH 8.5) and finally with 1.0 M sodium chloride in column equilibrating buffer. The fractions of 2 ml were collected with a flow rate of 12 ml / hour and arginase activity and protein content were assayed.
SDS-PAGE electrophoresis
SDS-polyacrylamide gel electrophoresis (10%) was carried out according to Laemmli et al (15). The purified enzyme was treated with 1% SDS and β-mercaptoethanol for 10 minutes at 100 oC and loaded in wells. Sample stacking was done at 10 mM and resolution was carried out at 15 mA constant current. Molecular weight standards were bovine serum albumin (66,000), Egg albumin (45,000), Glyceraldehydes triphosphate dehydrogenase (35,000), Carbonic anhydrase (29,000) and β-lactoglobuline (18,400) (Sigma). After electrophoresis gel was stained in 0.25% w/v Coomassie brilliant blue (R-250) prepared in 50% v/v methanol and 10% v/v acetic acid. The gels were destained by passive diffusion of dye in 50% v/v methanol and 1% acetic acid after changing the destaining solution for 2-3 times.
Native molecular weight determination
The molecular weight of the purified enzyme was estimated by gel filtration with a Bio-Gel P-300 column (2.2 X 78 cm). Blue dextran was used to measure the void volume. Catalase (bovine) (2, 40,000), Aldolase (rabbit muscle) (147,000), Albumin (bovine serum) (67,000), Albumin (egg) (45,000), Chymotrypsinogen A (25,000) and Cytochrome C (12,300) were used as a molecular weight marker. The elution volume of arginase was determined by estimating enzymatic activity whereas the protein content of each fraction was determined by Lowry's method [14]. The position of each marker protein was defined by plotting the elution volume against protein concentration. The molecular weight of an arginase was estimated by plotting log of molecular weight versus Kav for the standards. Kav = (Ve – Vo)/ (Vt Vo), where Vo is the void volume of the column; Vt is the total volume of the column and Ve is the elution volume of a protein.
Effect of pH on arginase activity
The pH dependence of the enzymatic activity of arginase from cotyledon and liver were determined by using three buffer systems: 10 mM Potassium phosphate (pH 6.0 to 6.5), Tris-HCl (pH 7.0 to 9.0) and carbonate bicarbonate buffer (pH 9.0 to 11.0).
Effect of divalent cations
To know the effect of divalent cations on the activity of arginase, the purified cotyledon arginase was diluted (1:2) with distilled water. The enzyme was dialyzed extensively with distilled water and further with 10 mM Tris-HCl buffer (pH 7.5) in the absence of manganese and then assayed the purified enzyme with the various metals.
The same procedure was followed to study the effect of different cations on the activity of buffalo liver arginase under the suitable enzyme assay conditions.
Estimation of urea
Arginase activity assay of the Vigna catjang cotyledon and hepatic tissue was carried out according to the procedure of Marsh WH et al. [16] by estimating urea. The reaction mixture consisted of 10 mM carbonate bicarbonate buffer (pH 10), 2 mM Mncl2, 130 mM L-arginine and enzyme solution in a total of 1 ml was incubated for 30 minutes at room temperature. This enzymatic reaction was terminated by adding 10% TCA. Two mls of reaction mixture was taken for urea estimation. The control tube was run simultaneously by inactivating the enzyme with 10% TCA prior to the addition of compound tested as alternate substrate (s) for arginase. The tubes were centrifuged, 2 ml of the clear supernatant was transferred from each tube and the enzyme activity was recorded by spectrophotometric detection of urea at 520 nm by using diacetyl monoxime as described by Marsh WH et al. Enzyme assay and experimental conditions used for buffalo liver arginase were the same as described above for the Vigna catjang arginase, except millimolar concentrations of MnCl2 (6 mM), L-arginine (25 mM) and carbonate bicarbonate buffer (50 mM, pH 9.2).
Substrate specificity
The substrate specificity of Vigna catjang cotyledon and buffalo liver arginase was investigated by testing its activity towards structurally similar compounds with guanidino group. The agmatine, d-arginine, L-arginine, L-canavanine, L-argininic acid, β-guanidino propionic acid, γ-guanidino butyric acid, L-homoarginine and streptomycin (Sigma, USA.) tested as a potential alternate substrate for arginase. The solutions of above compounds (30 mM) were prepared in 10 mM carbonate bicarbonate buffer. Enzyme activity was measured by spectrophotometric detection of urea as described above.
3. Results and Discussion
Purification
The purification of arginase from Vigna catjang cotyledon and buffalo liver was carried out as described earlier 'Materials and Methods'. The results from the purification of arginase from cotyledons and liver are summarized in Table 1. The fractional precipitation of arginase with chilled acetone (-10 ºC) and heat treatment results in reduced stability of the cotyledon arginase, but fractional precipitation with ammonium sulphate yielded good results. It showed 2.2-fold purification corresponding to a specific activity of 70.15. The enzyme fractions were freed of ammonium sulphate and low molecular weight protein by passing the solution through a Bio-Gel P-150 column. Arginase was adsorbed on a DEAE-cellulose column and eluted at about 300 mM sodium chloride in column equilibrating buffer (Tris-HCl, 10 mM, pH 7.5) containing 0.6 mM MnCl2 and 10% glycerol. Inclusion of 10% glycerol in the column buffers has given stability to arginase activity during chromatographic purification. The enzyme was specifically bound on an arginine Sepharose 4B column and was eluted at a high molarity of Tris-HCl buffer (Figure 1). The specific activity of the arginase obtained from affinity column was about five fold higher than DEAE-cellulose chromatography. Probably arginine AH-Sepharose 4B minimized non-specific adsorption. The purification of Vigna catjang cotyledon arginase was achieved about 64 fold with 1.64% yield. This is about two and half fold lower than that reported for the axis of Soybean [5].
Purification of Arginase from Vigna catjang cotyledon and buffalo liver
Elution profiles of Vigna catjang arginase activity and protein content. A) Bio-Gel P-150 B) DEAE-cellulose and C) Arginine AH-Sepharose 4B chromatography
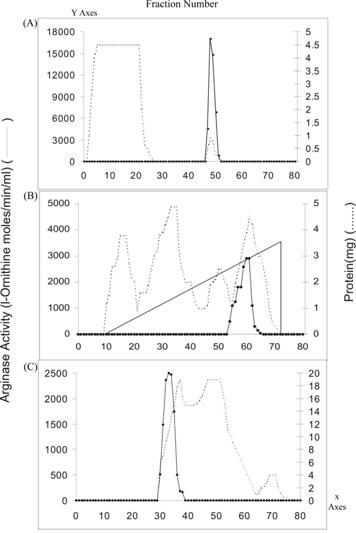
Arginase isozymes have not been reported in Iris bulbus and Pisum sativum and Jerusalem artichoke tubers [11, 12, 17]. We could not find any evidence of the presence of arginase isozymes in purification steps.
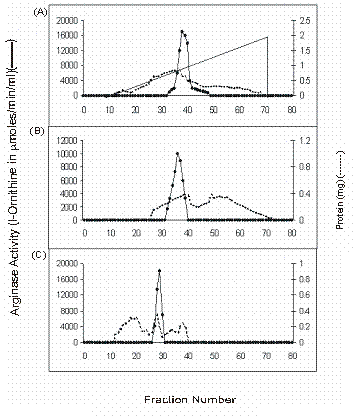
Heat treatment was given to the crude extract of buffalo liver to enhance the coagulation of thermolabile proteins, which were further removed by centrifugation and supernatant was collected to test the enzymatic activity. It was found that arginase from buffalo liver was stable at 60 oC. Such thermal stability of arginase has also been reported from livers of beef [18] and rabbit [19]. 70% saturation of arginase with solid ammonium sulphate has resulted in increased specific activity of liver arginase. We did not observe any isozyme for buffalo liver arginase on DEAE-cellulose chromatography. The enzyme was eluted in a single symmetrical peak, with linear gradient of sodium chloride (Figure 2). The above active enzyme fractions were passed through Bio-Gel P-150 chromatography and a single homogenous peak of arginase activity was appeared. We have obtained about 2028.97 fold purification from arginine AH-Sepharose 4B affinity adsorbent (Table 1). The overall recovery was about 18 %. From the purification data it is clear that the protocol which we have used for the purification of Vigna catjang cotyledon and buffalo liver arginase is simple and effective.
Elution profiles of Buffalo liver arginase activity and protein content. A) DEAE-cellulose B) Bio-Gel P-150 and C) Arginine AH-Sepharose 4B chromatography
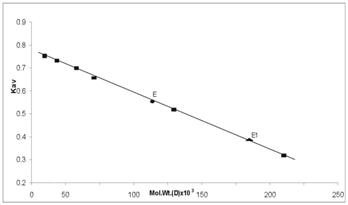
Molecular weight
The molecular weight of the native enzyme determined by gel filtration was found to be 210 kDa for Vigna catjang and 120 kDa for buffalo liver arginase (Figure 3). The molecular weight on SDS-PAGE was found to be 52 kDa and 43 kDa for Vigna catjang and buffalo liver arginase respectively (Figure 4).
Determination of molecular weight of Vigna catjang cotyledon (E) and Buffalo liver (E1) arginase by gel filtration technique on Bio-Gel P-300
SDS-PAGE (10%) analysis of purified Vigna catjang cotyledon and buffalo liver arginase. M: Molecular weight standard, size in kilodalton is shown on the left. A: Purified Vigna catjang cotyledon arginase and B: Purified buffalo liver arginase
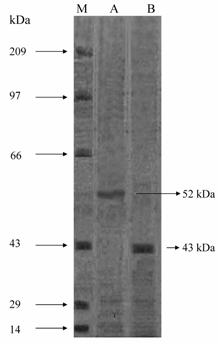
The molecular weight obtained for cotyledon arginase is smaller, compared with those of soybean axes 220 kDa [5] and E. prunastri 330 kDa [20]. Reports obtained for buffalo hepatic arginase agrees with the reported molecular weights of arginases from beef 115 kDa [18] and rabbit liver 110 kDa [19].
Kinetic properties
The effect of incubation time on the activities of cotyledon and liver arginase was studied up to 75 min. There was a direct relationship although not linear, between the product formed and incubation time for the enzymes. From the observations common incubation time selected for the studies was 30 min.
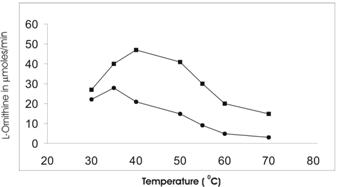
Buffalo liver arginase is stable at 55-60 oC as in rat mammary gland [21] and rabbit liver [19], but in case of cotyledon arginase it is found to be less stable at the same temperature. The maximum activity of cotyledon and liver arginase was found at 35 oC and 42 oC respectively (Figure 5).
Determination of optimal temperature for the activity of Vigna catjang (●) cotyledon and buffalo liver (■) arginase. The data represents an average of multiple experiments
Effect of pH
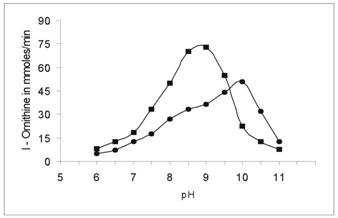
The activity of cotyledon and liver arginase was determined in the buffer pH range from 6.0 to 11 at 37 oC. The optimum pH of cotyledon arginase was found to be 10.0. This is found to be slightly higher than Evernia prunastri and Soybean axes [5, 20]. Liver arginase showed high activity at pH 9.2 (Figure 6). Generally mammalian arginases appear to have basic pH optima of 9.5-10.5, although some exceptions have been noted. Gasiorowska [22] have found four different isoenzymes of arginase in rat tissues, one of which had a pH optimum of 7.5. On the other hand, Patil et al [23] noted optimum pH 11.5 for Ox-erythrocyte arginase at 55 oC. The variation of activity with pH suggests that an ionisable group may function at the catalytic site [24].
Determination of the optimal pH for Vigna catjang (●) cotyledon and buffalo liver (■) arginase. The enzymatic activity of arginase was determined by replacing the buffer in the pH range of 6.0-11.0. The data represents an average of multiple experiments
Michaelis-Menten constant (Km)
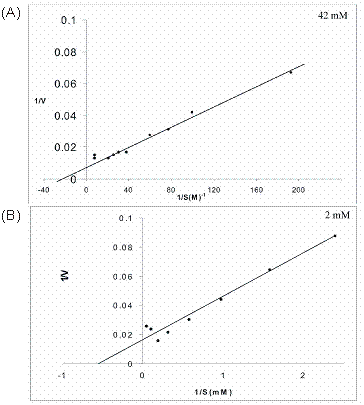
The effect of substrate concentration on the activity of cotyledon and liver arginase was determined by Lineweaver-Burk, double reciprocal (1/V Vs 1/S) plots. The Km values for cotyledon and liver arginase were found to be 42 mM and 2 mM respectively as shown in figure 7 (A) and (B). The Km value of Vigna catjang cotyledon arginase is in the range of other reported Km (40-83 mM) for plant arginases [5, 11, 25]. The Km value of buffalo liver arginase is in the range reported for type I arginase isolated from human 6.3 mM [26] and rabbit liver [19]. A variation in millimolar concentration of arginine is may be due to the variety of assays used and non-physiological conditions generally used [27, 28]. It is interesting to note that at a higher concentration of substrate, double reciprocal plot is non-linear and seems to indicate excess substrate inhibition in both Vigna catjang cotyledon and buffalo liver arginase.
Linewaver-Burk plots of initial reaction velocities (µmoles/min) for (A) Vigna catjang cotyledon and (B) buffalo liver arginase measured at various concentrations of arginine
Effect of metal ions
Arginase is a metallo enzyme in which manganese acts as a cofactor as well as activator in almost all reported arginases [20, 29]. However the amount required for optimal activity of the enzyme varies. About 0.6 mM and 2 mM Mncl2 in the reaction mixture were found to be optimal for the activity of Vigna catjang and buffalo liver arginase respectively.
Replacement of Mn++ ion with other metal ions such as Mg++ and Co++ restored more than 60 % while Ni++ and Fe++ restored about 50 % activity, whereas Ca++ and Zn++ restored about 35 to 38 % and Cd++ restored only 10 % of the original activity of Vigna catjang cotyledon arginase. In case of buffalo liver arginase, Mg++ ions restored almost the original activity, whereas Ca++, Ni++, Co++ and Cd++ restored about 40-50 % of the original activity of the enzyme and Fe++ and Zn++ has completely inactivated the arginase activity (Table 2).
Effect of divalent cations on the activity of Vigna catjang cotyledon and buffalo liver arginase
| Metal ion 2 mM | Residual activity (%) | |
|---|---|---|
| Cotyledon arginase | Liver arginase | |
| Mn++ | 100 | 100 |
| Mg++ | 61 | 93 |
| Ca++ | 35 | 52 |
| Co++ | 66 | 45 |
| Ni++ | 55 | 51 |
| Cd++ | 11 | 43 |
| Fe++ | 52 | 00 |
| Zn++ | 38 | 00 |
The amount of Mn++ required for optimal activity of the soybean axes enzyme was reported as 1 mM [5], 1-2 mM for human liver [30], 8-10 mM for human erythrocytes [31], 40 mM for mammalian liver arginase [32] and 50 mM for beef liver arginase [18]. Maggini et al [33] have measured arginase activity by reassessment of assay conditions using near physiological steady-state concentrations of arginine (5-35 mM) and excess Mn++ (30 mM). They obtained specific activity values for rat liver arginase of 1.2 U / g of tissue, compared with activity values of 70 U / g from initial velocity experiments. This modified activity values suggest that arginase could play a role in the regulation of urea cycle activity, its activity being regulated by the concentrations of arginine and Mn++ ions. Increased streptozotoein-induced in diabetic rat's resulted in activation of hepatic arginase [34]. Similarly, the concentration of manganese in the diet has been shown to regulate the activity of hepatic arginase in the rat [35].
Substrate specificity
Hepatic arginase of ureotelic and urecotelic animals have nearly same substrate specificity towards L-arginine and to some extent L-canavanine [36]. The leupine arginase hydrolyzes L-homoarginine more efficiently than L-arginine [37]. With this background it was interesting to investigate the substrate specificity of Vigna catjang cotyledon and buffalo liver arginase. Different substrates containing guanidino group common in their structure were tested as potential substrates for Vigna catjang cotyledon among these only agmatine, and L-canavanine were hydrolyzed significantly in addition to L-arginine. The ratio of the reaction product, urea of L-arginine to agmatine and L-arginine to L-canavanine is six and fifteen respectively Table 3. Arginase/canavanase high ratios (>10) have been reported from arginase from rat liver [35] and silk moth [38] Table 3.
Substrate specificities of Vigna catjang cotyledon and buffalo liver arginase
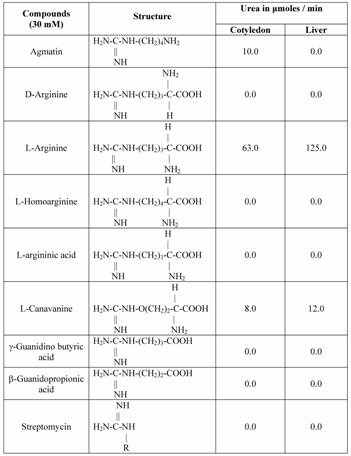
Buffalo liver arginase hydrolyzed only L-arginine and L-canavanine under the standard assay conditions and the results are shown in (Table 3). The ratio of L-arginine to L-canavanine is more than ten. The enzyme does not hydrolyze the isomer of L-arginine, the D-arginine. Agmatine has no α-carboxyl group, whereas γ-guanidino butyric acid and β-guanidino propionic acid lack α-amino group. Streptomycin does not posses α- NH2 as well as α-COOH groups. Therefore buffalo liver arginase does not utilize these guanidine compounds as its substrate to liberate urea as one of the product.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Cederbaum SD, Yu H, Grody WW. et al. Arginase I and II: do their functions overlap? Med Genet Metab. 2004 ;81(suppl 1):S38-44
2. Chang CI. et al. Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer Res. 2001 ;61:1100-1106
3. Satriano J. Agmatine: At the Crossroads of the Arginine Pathways. Ann N Y Acad Sci. 2003 ;1009:34-43
4. Kang JH, Cho YD. Purification and properties of arginase from soybean (Glycine max). Plant physiol. 1990 ;93:1230-1234
5. Matsubara S, Suzuki Y. Arginase activity in the cotyledons of soybean seedlings. Plant physiol. 1984 ;62:309-314
6. Zonia LE. et al. Essential role of arginase in germination of nitrogen limited Arabidopsis thaliana seeds. Plant physiol. 1995 ;107:1097-1103
7. King JE, Gifford DJ. Amino acid utilization in seeds of Loblolly pine during germination and early seedlings growth L-Arginine and arginase activity. Plant physiol. 1997 ;113:1125-1135
8. Dawnun KR, Rosenthal GA, Cohen WS. L-Arginine and L-canavanine metabolism in jack bean, Canavalia ensiformis (L) DC and soybean, Glycine max (L) mere. Plant physiol. 1983 ;73:965-968
9. Polacco JC, Holland MA. Roles of urease in plant cells. Int Rev Cytol. 1993 ;146:65-103
10. Carvajal N, Ascoria M, Rodriguez JP. et al. Evidence of cooperative effects in human liver arginase. Biochim Biophys Acta. 1982 ;701:146-148
11. Butin JP. Purification, properties and subunit structure of arginase from Iris bulbus. Eur J Biochem. 1982 ;127:237-243
12. Wright LC, Brady GJ, Hinde RW. Purification and properties of arginase from Jerusalem artichoke tubers. Phytochem. 1981 ;20:2641-2645
13. Roman W, Ruys J. Simple method for the estimation of l-ornithine. J Clin. Enzyme. 1969 ;2:121-128
14. Lowry OH, Roserbrough NJ, Farr AL. et al. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951 ;193:265-275
15. Laemmli UK. Cleavage of structural proteins during the assembly of the head of the bacteriophage T4. Nature. 1970 ;227:680-685
16. Marsh WH, Fingerhut B, Miller H. Automated and manual direct methods for determination of urea. Clin Chem. 1965 ;11:624-627
17. Talor AA, Stevart GR. Tissue and Subcellular localization of enzymes to arginine metabolism in Pisum sativum. Biochem Biophys Res Commun. 1981 ;101:1281-1289
18. Harell D, Sokolovsky M. Beef-liver arginase. Isolation and molecular properties. Eur J Biochem. 1972 ;25:102-108
19. George AK Harold JS. Isolation and characterization of arginase from rabbit liver. Biochem J. 1973 ;133:779-788
20. Angels MF, Legaz ME. Purification and properties of the constitutive arginase of Evernia prunastri. Plant Physiol. 1984 ;76:1065-1069
21. Christopher PJ, Murray RG. Rat mammary arginase: Isolation and characterization. Biochemical Med and Meta Biol. 1994 ;51:156-165
22. Gasiorowska I, Porembaska Z, Jochimowicz J. et al. Isozymes of arginase in rat tissue. Acta Biochim Polon. 1970 ;17:19-30
23. Patil NB, Somvanshi BS, Kothari RM. Isolation and characterization of arginase from Ox-erythrocyte. Biotech Tech. 1990 ;4(2):133-136
24. Kuhn NJ, Talbor J, Ward S. pH-sensitive control of arginase by Mn (II) ions at submicromolar concentrations. Arch Biochem Biophys. 1991 ;286:217-221
25. Kollofel C, Van Dijke HD. Mitochondrial arginase activity form cotyledons of developing and germinating seeds of Vicia faba. Plant physiol. 1975 ;55:507-510
26. Buscur L, Cabellow J, Veliz M. et al. Molecular forms of human liver arginase. Biochem Biophys Acta. 1966 ;128:149-154
27. Ganganta CL, Bond JS. Assay and kinetics of arginase. Anal Biochem. 1986 ;154:388-394
28. Pace CN, Buonanno A, Simmons - Hansen J. Steady-state kinetic studies of arginase with an improved direct spectrophotometric assay. Anal Biochem. 1980 ;109:261-265
29. Helga HK, Greenberg DM. Molecular characteristics of rat liver arginase. J Biol Chem. 1968 ;243:6123-
30. Carvajal N, Venegas A, Ostreicher G. et al. The effect of manganese on the quaternary structure of human liver arginase. Biochim Biophys Acta. 1971 ;250:437- 442
31. Nishibe H. Isolation and characterization of arginase in human erythrocyte. Physiol Chem Physics. 1973 ;54:53- 462
32. Hirsch-Kolb H, Kolb HJ, Greenberg DM. Comparative physical and chemical studies of mammalian arginase. Comp Biochem Physiol. 1970 ;37:345-359
33. Maggini S, Stockin-Tschan FB, Morikofer-Zwez S. et al. New kinetic parameters for rat liver arginase measured at near physiological steady-state concentrations of arginine and Mn2+. Biochem J. 1992 ;283:53-660
34. Bond JS, Failla ML, Unger DF. Elevated manganese concentration and arginase activity in livers of streptozotoein - induced diabetic rats. J Biol Chem. 1983 ;258:8004-8009
35. Brock AA, Chapman SA, Ulman EA. et al. Dietary manganese deficiency decreases rat hepatic arginase activity. Nutrition. 1994 ;124:340-344
36. Mora J, Tarrab R, Maratuscelli L. et al. Characteristics of arginases from ureotelic and non-ureotelic animals. Biochem J. 1965 ;96:588-594
37. Muszynska G, Reifer I. Purification, properties and inhibition of plant arginase. Acta Biochim Pol. 1968 ;15:55-66
38. Reddy SRR, Campbell JW. Arginine metabolism in insects: properties of insect fat body arginase. Comp Biochem Physiol. 1969 ;28:515-534
Tables & Figures
Author contact
![]() Corresponding address:
Corresponding address:
Dr. Snehal Dabir, Phone: (+91)9822562943, E-mail: snehal_214com