Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Materials and methods
3. Results
4. Discussion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2006; 2(1):1-9. doi:10.7150/ijbs.2.1 This issue Cite
Research Paper
Dephosphorylation specificities of protein phosphatase for cardiac troponin I, troponin T, and sites within troponin T
Nathan M. Jideama, Brian H. Crawford, AKM A. Hussain, Robert L. Raynor ![]()
Department of Biological Sciences, Clark Atlanta University, Atlanta, GA 30314, USA
Received 2006-2-4; Accepted 2006-3-1; Published 2006-3-1
Abstract
Protein dephosphorylation by protein phosphatase 1 (PP1), acting in concert with protein kinase C (PKC) and protein kinase A (PKA), is a pivotal regulatory mechanism of protein phosphorylation. Isolated rat cardiac myofibrils phosphorylated by PKC/PKA and dephosphorylated by PP1 were used in determining dephosphorylation specificities, Ca2+-stimulated Mg2+ATPase activities, and Ca2+ sensitivities. In reconstituted troponin (Tn) complex, PP1 displayed distinct substrate specificity in dephosphorylation of TnT preferentially to TnI, in vitro. In situ phosphorylation of cardiomyocytes with calyculin A, a protein phosphatase inhibitor, resulted in an increase in the phosphorylation stiochiometry of TnT (0.3 to 0.5 (67%)), TnI (2.6 to 3.6 (38%)), and MLC2 (0.4 to 1.7 (325%)). These results further confirmed that though MLC2 is the preferred target substrate for protein phosphatase in the thick filament, the Tn complex (TnI and TnT) from thin filament and C-protein in the thick filament are also protein phosphatase substrates. Our in vitro dephosphorylation experiments revealed that while PP1 differentially dephosphorylated within TnT at multiple sites, TnI was uniformly dephosphorylated. Phosphopeptide maps from the in vitro experiments show that TnT phosphopeptides at spots 4A and 4B are much more resistant to PP1 dephosphorylation than other TnT phosphopeptides. Mg2+ATPase assays of myofibrils phosphorylated by PKC/PKA and dephosphorylated by PP1 delineated that while PKC and PKA phosphorylation decreased the Ca2+-stimulated Mg2+ATPase activities, dephosphorylation antagonistically restored it. PKC and PKA phosphorylation decreased Ca2+ sensitivity to 3.6 µM and 5.0 µM respectively. However, dephosphorylation restored the Mg2+ATPase activity of PKC (99%) and PKA (95%), along with the Ca2+ sensitivities (3.3 µM and 3.0 µM, respectively).
Keywords: Protein phosphatase, protein kinase C, protein kinase A, troponin complex
1. Introduction
Protein dephosphorylation, acting in concert with protein phosphorylation has become increasingly appreciated as a fundamental mechanism in controlling phosphorylation states of proteins, which play a cardinal role in the regulation of biological activities such as cardiac muscle contraction and relaxation [1]. Evidence exists that there is a significant increase of PP2A activity in the myocardium of transgenic mice [2]. It has been shown that the subcellular localization of two B subunits (B56α and B56γ1) of PP2A were assessed in cultured neonatal and adult rat ventricular myocytes [3]. One of the roles of protein phosphatases in cardiac muscle physiology is its ability to stimulate calcium release in cardiac myocytes [4, 5]. Of several serine/threonine protein phosphatases identified to date, PP1 is unique in its particulate localizations and substrate specificities. All forms of PP1 holoenzymes (110 kD) appear to share a similar 37-kD catalytic subunit (PP1C), but differ in the associated regulatory subunits. It has been shown that these different regulatory subunits are crucial for targeting the holoenzymes to specific subcellular locations and hence enabling them to act preferentially on specific substrates in these locations [6-8]. Accordingly, PP1 holoenzymes have been classified into the following subspecies: PP1G, which is associated with glycogen particle or sarcoplasmic reticulum; PP1, with myofibrils; and PP1N, with the nucleus [1]. PP1 has been purified from skeletal and cardiac muscle [6-7] and smooth muscle [8], and their properties characterized [6-8]. Because PP1 represents about 60 to 90% of the phosphatase activity toward native myosin in rabbit skeletal muscle and bovine cardiac muscle, respectively, it has been suggested that this enzyme is primarily responsible for dephosphorylating MLC2 in vivo [6-7]. PP1/PP2A have also been shown to play a role in regulation of cardiac inotropy and relaxation [9] and that inhibiting these enzymes with calyculin A (CCA) affects the excitation-contraction coupling cascade in murine ventricular myocytes [9-10].
Dephosphorylation of native myosin or isolated MLC2 has been used for assaying the PP1 activity in previous studies [6-8]. In addition to MLC2 in the thick filament, reversible phosphorylation also occurs in other myofibrillar proteins. These proteins include, notably, C- protein in the thick filament, and Tnl and TnT in the thin filament [11, 12]. As others and we have reported, PKC phosphorylates cardiac TnI and TnT in vitro [12-19] and in situ in cardiomyocytes [12, 20] at multiple sites, leading to decreases in maximal activity and/or Ca2+ sensitivity of Ca2+-stimulated Mg2+ATPase of reconstituted actomyosin [15-19] or native myofibrils [12, 17]. With the use of a number of phosphorylation site- substitution and -deletion mutants of cardiac TnI, we found that PKC phosphorylation of Ser-43/Ser-45 and Ser-23/Ser-24 resulted in decreased activity and Ca2+ sensitivity, respectively, of Ca2+ -stimulated Mg2+ATPase of reconstituted actomyosin S-1 [19]. Others and we also reported that PKC isozymes has shown to be expressed in adult rat cardiomyocytes [21-25], exhibited distinct intermolecular specificities in differentially phosphorylating Tnl and TnT subunits in the Tn complex, as well as intermolecular specificities in differentially phosphorylating multiple sites within TnI and TnT [25]. PKC-δ was unique among the isozymes in its ability to favorably phosphorylate Ser-23/Ser-24 in Tnl, the bonafide phosphorylation sites for PKA [25], and hence functioned as a hybrid of typical PKC and PKA in the regulation of myofilament properties [25]. It was recently reported by others that Ca2+-/calmodulin-dependent protein kinase phosphatase dephosphorylates and regulates multifunctional Ca2+/calmodulin-dependent protein kinase [27]. Incubation of isolated adult rat cardiomyocytes with CCA, a potent inhibitor of PPI and PP2A [1], resulted in elevated phosphorylation of Tnl, TnT, and MLC2 [12, 20], indicating that dephosphorylation of contractile proteins was an active process.
We suspected that PP1 could not only effectively act on Tnl and TnT in the thin filament as it does on MLC2 in the thick filament [6-8] but could differentially dephosphorylate TnI and TnT at multiple sites within them that are phosphorylated by PKC and/or PKA. Also, we suspected that PP1 could antagonistically restore myofibrillar Mg2+ATPase activity and Ca2+sensitivity to normal following PKC/PKA phosphorylation. In the present study, we have examined these issues, as well as, the functional consequences of dephosphorylation of Tnl and/or TnT.
2. Materials and methods
Preparation of protein phosphates
PP1 was purified from adult male Sprauge-Dawley rat hearts. Crude extract was obtained by mincing and grinding of rat fat-free cardiac ventricular muscles in buffer A (containing 50 mM imidazole chloride, 5 mM EDTA, and 0.5 mM DTT (pH 7.5)) and centrifuging at 3300 x g for 30 min. The supernatant containing the crude extract was filtered through glass wool, brought to neutral pH by adding solid potassium bicarbonate, and brought to 70% saturation with ammonium sulfate by the addition of solid salt (43.6 g/100ml) and stirring. Ammonium sulfate was precipitated by allowing the extract to stand on ice for 1h and centrifuged at 3300 x g for 30 min. The precipitate was resuspended in buffer A (room temperature) and mixed with 95% ethanol. The resulting suspension was centrifuged at 3300 x g for 5 min and the precipitate was resuspended in buffer A, followed by centrifugation at 10,000 x g for 20 min. The extract was resuspended in buffer A (4 fold diluted buffer A) and dialyzed for 3 h, followed by dialysis in buffer C (2 fold dilution of buffer A) for 3 h, and finally dialyzed overnight in buffer A. The dialyzed enzyme solution was loaded onto a DEAE-Sepharose column (5 × 9 cm) previously equilibrated with buffer A. The column was washed with 0.1 M NaCl/buffer A and the enzyme eluted with 0.5 M NaCl/buffer A. Fractions were assayed for phosphorylase phosphatase activity. Fractions with phosphorylase phosphatase activity were dialyzed for 2-3 h against buffer A and loaded onto a DEAE -Sepharose column (2.5 × 90 cm) previously equilibrated with buffer A. The column was washed with 0.1 M NaCl/buffer A and then eluted with a linear gradient of 0.1 to 0.5M NaCl. Fractions were assayed for PP1 and PP2 (usually fractions in peak 1 contained PP1 and peak 2 contained PP2). Fractions of PP1 were pooled, dialyzed, and loaded onto a haperin-Sepharose column previously equilibrated with buffer A/20% glycerol. The column was washed with 0.1 M NaCl/buffer A/20% glycerol and PP1 eluted with 0.5 M NaCl/buffer A/20% glycerol. Fractions with PP1 activity was pooled and chromatographed on a Sephacryl S-200 column equilibrated with 0.3 M NaCl/buffer A/20% glycerol. Active fractions were dialyzed against 50% buffer/50% glycerol and stored at -20°C.
Preparation of rat cardiac myofibrils
Myofibrils were extracted from rat ventricular myocardia by detergent extraction as described by Murphy and Solaro [28]. Briefly, rat ventricles were minced and homogenized in cold standard buffer (60 mM KCl, 30 mM imidazole (pH 7.0) and 2 mM MgCl2) and centrifuged at 12,000 x g for 15 min. The pellet was resuspended in 1 volume of solution (10 mM EGTA, 8.2 mM MgCl2, 14.4 mM KCl, 60 mM imidazole (pH 7.0), 5.5 mM ATP, 0.01mg/ml protease inhibitors (leupeptin, antipain, aprotinin and pepstatin), and 1% triton X – 100) and was allowed to incubate on ice for 30 min. The suspension was washed three times with standard buffer and stored in -70 °C.
Reconstituion of rat cardiac Tn complex
Reconstituted rat cardiac Tn complex was prepared as we previously reported [19, 25, 29]. Briefly, TnI and TnT were mixed in 10 mM imidazole HCl (pH 7.0) containing 6 M urea, 1 mM CaCl2, and 1mM DTT at a molar ratio of 1:1. The mixture was dialyzed for 48 h at 4°C against 10 mM imidazole HCl (pH 7.0) containing 1 mM magnesium acetate and 1 mM DTT with a KCl concentration of 1.0, 0.7, 0.3, and 0.1 M. This mixture was then concentrated using ultrafiltration. Formation of the Tn complex was verified by gel filtration chromatography on Bio-Gel A-0.5 M (Bio-Rad®).
Phosphorylation of rat cardiac reconstituted Tn complex
Reconstituted rat cardiac Tn complex was phosphorylated, as described elsewhere, using PKC [19, 25, 29]. Briefly, reaction mixture (0.4ml) containing 50 mM Tris-HCl (pH 7.5) 10 mM MgCl2, 1.0mM DTT, 0.9 mM CaCl2, 0.5 mM EGTA, 0.3 M KCl, 5 µM [γ-32 P] ATP (2-4 × 106 cpm), phosphotidylserine (38µg/ml), diolein (5µg/ml), 100 nM TPA, and 4 µM reconstituted Tn. The reaction was carried out for 60 min. at 30°C. The γ-32 P labeled Tn was anaylyzed by SDS- polyacrylamide gel electrphoresis.
Phosphorylation of cardiac myofibrils
Rat cardiac myofibrils were phosphorylated using brain PKC as we previously reported [19, 25, 29]. The γ-32P labeled myofibrils were then analyzed by direct precipitation with 5% trichloroacetic acid containing tungstate. Bovine serum albumin was used as a carrier protein during precipitation of the myofibrils. Phosphorylation of myofibrils was visualized using sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE) followed by autoradiography. Phosphorylation by PKA was carried out as in brain PKC except that 10 mM cAMP replaced PS/Ca2+/diolein as an activator.
In situ phosphorylation of rat ventricular myofibrils
Isolated cardiomyocytes were incubated (1.5 hour at 37° C) in phosphate free Dulbecco's modified eagle's medium (DMEM) and metabolically labeled using phosphorus-32 (32P). Samples were pelleted and washed in buffer A (1 mM PMSF, 100 µg/ml leupeptin, 100 µg/ml antipain, 100 µg/ml pepstatin A, 0.1 mM NaOV, 100 nM CCA, and 5 mM EGTA), sonicated in buffer B (50 mM KH2PO4, 70 mM NaF, 5 mM EDTA, 1% Triton X-100, protease inhibitors, and phosphatase inhibitors), iced for 30 min., and washed in modified buffer B (without Triton X-100). The phosphorylations of samples were visualized by SDS-PAGE and autoradiography.
Dephosphorylation of phosphorylated rat cardiac reconstituted Tn and rat ventricular myofibrils
Previously, γ-32 P labeled reconstituted troponin phosphorylated by PKC was dephosphorylated by PP1 according to a modified method of Venema et al. [20]. 32P-labeled Tn was incubated with PP1 (10 units/ml) in 20 mM Hepes buffer (0.2 mM MnCl2, 1 mM DDT, 0.5 mM CaCl2, 0.04 mM EDTA, and 200 mM KCl) (pH 7.5). A parallel experiment with myelin base protein (MBP) was used as a dephosphorylation control, since MBP is a very well known substrate for PP1. Samples from the incubation mixture were removed for dephosphorylation analysis at 0, 2, 10, 30, 60, and 120 min. intervals. Dephosphorylation of PKC and PKA phosphorylated myofibrils was performed as described above except that samples from the incubating mixture were removed for dephosphorylation analysis by SDS-PAGE at 0, 10, 30, and 60 min intervals.The dephosphorylation analysis was carried out by SDS-PAGE, followed by autoradiography.
Two dimensional phosphopeptide mapping
Two dimensional phosphopeptide mapping of phosphorylated and dephosphorylated TnI, TnT, and MLC2 was performed as we previously described [19, 25, 29]. γ-32P labeled TnI, TnT, and MLC2 were excised from the gel and extracted using 0.2 M ammonium bicarbonate buffer (pH 8.6). Extracts were exhaustively digested with 50µl of 2 mg /ml trypsin at 37°C for 17 h. 25 µl of trypsin was added for an additional 7 hours. The digested phosphopeptide supernatant was separated from the gel by centrifugation. The supernatant was degassed by addition of 5µl of glacial acetic acid and vortexed. The phosphopeptides were lyophilized, and spotted on precoated thin layer chromatography (TLC) sheets. Electrophoresis in the first dimension was carried out for 2 h at 450 V in pyridine: acetic acid: water (1:10:89, v/v). Column chromatography in the second dimension was performed in butanol:pyridine:acetic acid:water (125:100:3:125, v/v). The phosphopeptide spots were visualized by autoradiography.
Determination of Ca2+- stimulated Mg2+ATPase activity of phosphorylated myofibrils
Ca2+-stimulated Mg2+ATPase activity was assayed according to the methods of Noland and Kuo [18] using previously phosphorylated/dephosphorylated cardiac myofibrils. The assay was carried out at 30°C for 10 min in standard reaction mixtures (0.5 ml) containing 50 µg of rat cardiac myofibrils, 10 mM MOPS-KOH (PH 7.0), 4.1 mM Mg acetate, 2.1mM [γ-32P] ATP (5-8 x 106 CPM), 1 mM DTT, 1 mM EGTA, and 1.05 mM CaCl2 (the free Ca2+ was calculated to be 40 mM). CaCl2, when present, was added as a mixture with EGTA and MOPS-KOH to give calculated free Ca2+ concentration of 0.01-100 mM in the assay mixture. KCl was added to bring the final ionic strength to 18 mM. The reactions were initiated by addition of radioactive ATP, terminated by addition of 10% charcoal suspension, and the released 32Pi was determined as we recently reported [25, 29].
3. Results
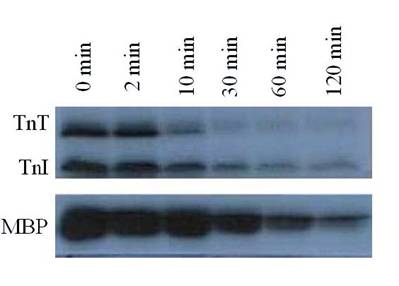
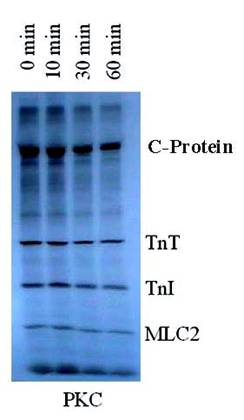
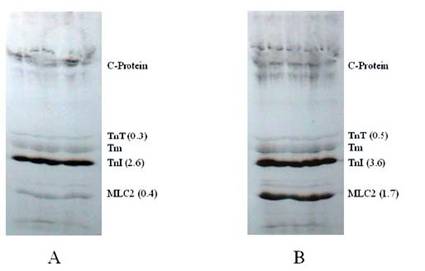
It was observed that PP1, a serine/threonine protein phosphatase effectively dephosphorylated C-protein, TnT and Tnl in the thin filament, as suspected, and in MLC2 in the thick filament, as reported by others [6-8]. PP1 catalyzed dephosphorylation acted in concert with PKC catalyzed phosphorylation of myofibrillar proteins. PP1 substrate specificity in the dephosphorylation was confirmed by the result obtained from dephosphorylating PKC phosphorylated reconstituted Tn complex (Fig. 1). In this experiment, PP1 displayed distinct substrate specificity in dephosphorylation of TnT preferentially to TnI. It is observed that TnT was almost completely dephosphorylated within the first 30 min of the dephosphorylation experiment, whereas TnI was still resistant to PP1 dephosphorylation even after 120 min of the experiment. Myelin basic protein (MBP) dephosphorylation by PP1 used as a control because it is a well-known substrate for PP1 confirmed this result that Tn complex is also an excellent PP1 substrate. PP1 substrate specificity in the dephosphorylation was also confirmed by the result obtained from dephosphorylating PKC and PKA phosphorylated myofibrils, in vitro (Fig. 2). This time-dependent dephosphorylation of the myofibrils revealed that PP1 dephosphorylation of PKC phosphorylated myofibrils were uniformly dephosphorylated starting between 0-10 min. of the dephosphorylation experiment. In situ phosphorylation of cardiomyocytes incubated with CCA, a PP1 and PP2A inhibitor, was used to determine which myofibrillar proteins are dephosphorylated by PP1/PP2A in situ (Fig. 3). Phosphorylation stiochiometry of TnT from the in situ experiment increased from 0.3 to 0.5 (67%), TnI from 2.6 to 3.6 (38%), and MLC2 0.4 to 1.7 (325%). From this experiment, it is therefore concluded that though MLC2 is the preferred target substrate for PP1 in the thick filament [6-8] the Tn complex (TnI and TnT) is also a PP1 and PP2A substrate in the thin filament, as we suspected.
Autoradiogram of PP1 mediated time dependent dephosphorylation patterns of PKC phosphorylated TnT and TnI. 32P-labeled reconstituted Tn complex was incubated with PP1 (10 units/ml) in 20 mM Hepes buffer (0.2 mM MnCl2, 1 mM DDT, 0.5 mM CaCl2, 0.04 mM EDTA, and 150-300 mM KCl) (pH 7.5) for 5-120 min at 22°C. Dephosphorylation of myelin basic protein (MBP) was used as a control. The findings were confirmed by three other experiments
Autoradiogram of PKC in vitro phosphorylated rat cardiac myofibrils time dependent dephosphorylation by PP1. 32P-labeled myofibrils were incubated with PP1 (10 units/ml) in 20 mM Hepes buffer (0.2 mM MnCl2, 1 mM DDT, 0.5 mM CaCl2, 0.04 mM EDTA, and 150-300 mM KCl) (pH 7.5) for 0-60 min at 22°C. The findings were confirmed by three other experiments
Autoradiogram showing the effects of protein phosphatase in situ dephosphorylation on rat cardiomyocytes without (A) and with (B) calyculin A. Isolated cardiomyocytes were incubated (1.5 hour at 37° C) in phosphate free DMEM and metabolically labeled using phosphorus-32 (32P). Further incubations were conducted with or without CCA. The phosphorylation of samples was evaluated by SDS-PAGE, autoradiography, densitometry, and liquid scintillation counts. The findings were confirmed by three other experiments
It was suspected that PP1 and PP2A might exhibit further discrete substrate specificities in terms of dephosphorylating multiple sites on TnI (Fig. 4) and TnT (Fig. 5), and MLC2 (Fig. 6). We discovered that it was true for TnT but not true for TnI and MLC2 in in vitro phosphorylation/dephosphorylation experiments. Phosphopeptide maps generated from these experiments showed that (a) TnI phosphopeptides are uniformly dephosphorylated by PP1 (Fig. 4) and (b) TnT phosphopeptides in spots 4A and 4B are much more resistant to PP1 dephosphorylation than the other TnT phosphopeptides (Fig. 5). The phosphopeptide in spot 4B is most resistant to PP1 followed by spot 4A. The most integral finding is that, although the phosphopeptide in spot 5 had the highest 32P incorporation, it was the fastest to be dephosphorylated by PP1. On the other hand, spot 4B, which was one of the least phosphorylated, became the most resistant to PP1 dephosphorylation. In vitro dephosphorylation experiments with PP2A did not show any preferential dephosphorylation of TnI and TnT (result not shown). Phosphopeptide maps generated from in situ phosphorylation experiment with and without CCA revealed that phosphopeptides in spots 2 and 4 (Fig. 7) of TnI are much more resistant to PP1 and PP2A than much larger phosphopeptides in spot 5.
Autoradiogram of 2-D peptide maps showing dephosphorylation of phosphopetides generated from TnI excised from PKC phosphorylated rat cardiac myofibrils and dephosphorylated by PP1, in vitro. γ-32P labeled TnI was excised from the gel, extracted, and exhaustively trypsin digested at 37°C for 17 h. The digested phosphopeptide supernatant was separated and spotted on TLC sheets. Electrophoresis was carried out in the first dimension and chromatography in the second dimension. The phosphopeptide spots were visualized by autoradiography. The findings were confirmed by three other experiments

Autoradiogram of tryptic phosphopeptide maps showing in vitro dephosphoryltion patterns of cardiac TnT maximally phosphorylated by PKC. γ-32P labeled TnT was excised from the gel, extracted, and exhaustively trypsin digested at 37°C for 17 h. The digested phosphopeptide supernatant was spotted on TLC sheets. Electrophoresis was carried out in the first dimension and chromatography in the second dimension. The phosphopeptide spots were visualized by autoradiography. The findings were confirmed by three other experiments

Autoradiogram of tryptic phosphopeptide maps of rat cardiac MLC2 in situ dephosphorylated by protein phosphatase in the presence and absence of CCA. γ-32P labeled MLC2 was excised from the gel, extracted, and exhaustively trypsin digested at 37°C for 17 h. The digested phosphopeptide supernatant was spotted on TLC sheets. Electrophoresis was carried out in the first dimension and chromatography in the second dimension. The phosphopeptide spots were visualized by autoradiography. The findings were confirmed by three other experiments
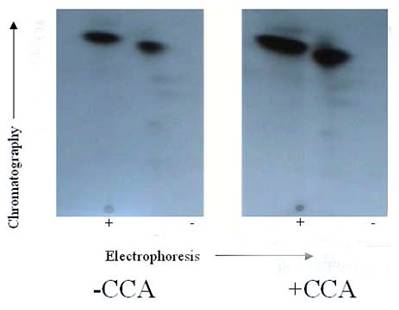
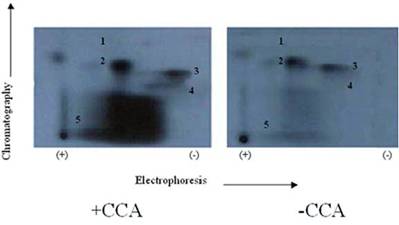
Autoradiogram of tryptic phosphopeptide maps 2-D phosphopeptide map of in situ protein phosphatase dephosphoylation of PKC phosphorylated TnI with and without CCA. γ-32P labeled TnI was excised from the gel, extracted, and exhaustively trypsin digested at 37°C for 17 h. The digested phosphopeptide supernatant was spotted on TLC sheets. Electrophoresis was carried out in the first dimension and chromatography in the second dimension. The phosphopeptide spots were visualized by autoradiography. The findings were confirmed by three other experiments
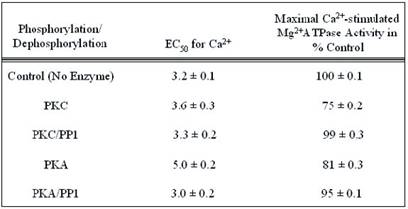
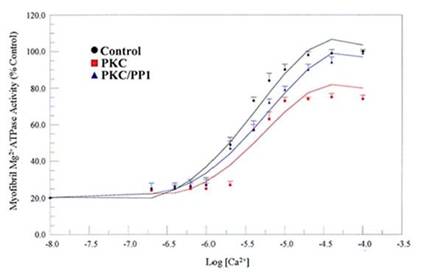
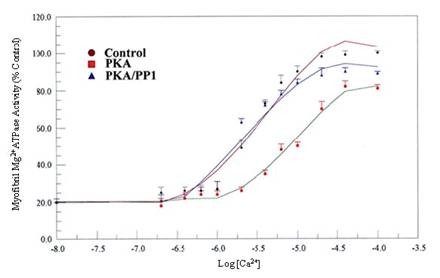
We examined the physiological relevance of dephosphorylation of myofibrillar proteins in intact myofibrils. PP1, acting in concert with PKC and PKA phosphorylation, conferred substrate specificities in dephosphorylating contractile proteins in the thick and thin filaments of the cardiac myofibrils (Figs. 8, and 9; Table 1). The data obtained delineated that while PKC and PKA phosphorylation decreased Ca2+-stimulated Mg2+ATPase activities of the rat cardiac myofibrils by 25%, PP1 dephosphorylation restored it by 24%. Also, PKA phosphorylation of the myofibrils decreased the Ca2+-stimulated Mg2+ATPase activities of the rat cardiac myofibrils by 18% while PP1 dephosphorylation restored it by 14%. PKC and PKA phosphorylation decreased myofibrillar Mg2+ATPase activity to 75% and 81% and decreased Ca2+ sensitivity to 3.6 µM and 5.0 µM respectively. However, dephosphorylation restored myofibrillar Mg2+ATPase activity to 99% for PKC and 95% for PKA and restored Ca2+ sensitivity to (3.3 µM and 3.0 µM, respectively) values nearly equal to or better than that of the control (Figs. 8, and 9; Table 1). This present study demonstrates for the first time, distinct substrate specificity of PP1 in dephosphorylating rat intact myofibrillar proteins.
Summary of the kinetic data for Maximal Ca2+-stimulated Mg2+ATPase activity and calcium sensitivity of phosphorylated and dephosphorylated rat cardiac myofibrils. The values were calculated from the data presented in Figs. 8 and 9, and are displayed as the means ± S.E. of three experiments. The maximal enzymatic activity is expressed as percentages of the values obtained from PKC or PKA phosphorylated and PP1 dephosphorylated rat cardiac myofibrils compared to the unphosphorylated control, which is taken as 100%
Effects of in vitro phosphorylation (PKC) and dephosphorylation (PP1) of rat cardiac myofibrils on Ca2+-stimulated Mg2+ATPase activity and Ca2+sensitivity. Ca2+-stimulated Mg2+ATPase activity assay was carried out at 30°C for 10 min in standard reaction mixtures. Kinetic data for Ca2+ stimulation of Mg2+ATPase (as a function of Ca2+) was calculated. The data shown are the means ± S.E. of three experiments, and the curves drawn are the polynomial “best fits” of the data. The maximal activity obtained for the unphosphorylated (Control) myofibrils was taken as 100%
Effects of in vitro phosphorylation (PKA) and dephosphorylation (PP1) of rat cardiac myofibrils on Ca2+-stimulated Mg2+ATPase activity and Ca2+sensitivity. Ca2+-stimulated Mg2+ATPase activity assay was carried out at 30°C for 10 min in standard reaction mixtures. Kinetic data for Ca2+ stimulation of Mg2+ATPase (as a function of Ca2+) was calculated. The data shown are the means ± S.E. of three experiments, and the curves drawn are the polynomial “best fits” of the data. The maximal activity obtained for the unphosphorylated (Control) myofibrils was taken as 100%
4. Discussion
In this report, we provided unique evidence that PP1, a serine/threonine protein phosphatase, effectively dephosphorylated TnT and TnI in the thin filament and C-protein (as we suspected) and in MLC2 in the thick filament, as reported by others [6-8]. We also reported that the in situ phosphorylation of rat cardiomyocytes with and without PP1 and PP2A inhibitor, CCA, increased the phosphorylation stiochiometry of TnT from 0.3 to 0.5 (67%), TnI from 2.6 to 3.6 (38%), and MLC2 0.4 to 1.7 (325%). It was therefore concluded that though MLC2 is the preferred target substrate for PP1 in the thick filament [6-8], the Tn complex (TnI and TnT) is also a PP1 substrate. It was observed that although the in situ phosphorylation of TnT, with and without CCA, appeared to be the minimal (Fig. 3) compared with TnI, its phosphorylation stochiometry increased approximately two times more than that of TnI (67% for TnT and 38% for TnI). The poor phosphorylation of TnT could be due to the fact that TnT is the preferred substrate of PKC-ζ, in situ, which phosphorylated TnT very poorly in vitro, as we have reported elsewhere [19, 25, 29]. On the other hand, TnI is a favored substrate for PKA and other PKC isozymes (PKC-α, -δ, and –ε) found to be expressed in the heart [21-25] and these isozymes phosphorylate TnI very well. In 1995, we reported that PKC isozymes (PKC-α, -δ, –ε, and –ζ) and PKA have specificities in phosphorylating specific sites within TnI [19]. It was clearly shown that the phosphopeptide in spot 1 contained Ser-78, spot 2 contained Ser-43/Ser-45, spot 3 contained Thr-144, and spot 5 contained Ser-23/Ser-24.
The phosphopeptide maps generated from the in situ phosphorylation of cardiomyocytes with and without CCA revealed that the TnI phosphopeptides in spots 2 and 3 are most resistant to PP1 dephosphorylation (Fig. 7), whereas those in spot 5 (Ser-23/Ser-24 (PKA and PKC-δ sites)) was most vulnerable to PP1 dephosphorylation since CCA failed in effectively protecting this site from PP1. Since phosphorylation sites in spots 2 and 3 are preferred phosphorylation sites for PKC-α, -δ and –ε, and PKC-δ acts as a hybrid of PKC and PKA [25] (which preferably share the phosphorylation of the phosphopeptide in spot 5), it appears that PP1 and PP2A dephosphorylation of these sites act in concert with catalyzed phosphorylation by PKC-δ and PKA.
We reported previously that TnT phosphopeptide maps from in vitro phosphorylation contained Thr-190 in spot 1, Ser-194 in spot 6, Thr-199 in spot 4A, and Thr-280 in spots 3 and 5 [19, 25, 29]. In this study, the TnT phosphopeptide map from phosphorylation/dephosphorylation revealed that the Thr-199 in spot 4A and the phosphopeptide in spot 4B were the least affected by PP1 dephosphorylation. These phosphorylation sites are located in the C-terminal half of the TnT where binding to tropomyosin (Tm) and troponin C (TnC) [27, 30] occur, and most likely where TnT may interact with actin. These results suggest that it is essential to look at the physiological implications of PP1 dephosphorylation of the cardiac myofibrils.
We hypothesized that PP1 will antagonistically affect PKC and PKA induced changes in Ca2+stimulated Mg2+ATPase and calcium sensitivity of the myofibrils. Our hypothesis was proven correct. Our data (Figs. 8, and 9; Table 1) reveal that while PKC and PKA phosphorylation decreased the Ca2+-stimulated Mg2+ATPase activities of the rat cardiac myofibrils, PP1 dephosphorylation restored it close to that of the control values. Ca2+ sensitivity was also restored to values close to that of the control, suggesting that PP1 acts as a reset regulator of cardiac muscle phosphorylation. This data agrees to that previously reported by others indicating that PP1/PP2A stimulate Ca2+ release [4, 5].
As we and others have earlier reported, different PKC isozymes (PKC-α, -δ, - ε and - ζ) and PKA conferred changes in Ca2+-stimulated Mg2+ATPase activities and Ca2+ sensitivities of the rat cardiac myofibrils (Figs. 8, and 9; Table 1) due to their ability to incorporate γ-32P into specific site on TnT and TnI [29, 31]. PP1, on the other hand, dephosphorylated the same phosphopeptides phosphorylated by PKC and PKA. It is, therefore, expected that PP1 will impose an opposite effect on myofibrillar Ca2+-stimulated Mg2+ATPase activities and Ca2+ sensitivities. It was observed that in most cases, there are differences in the extent of restoration of Ca2+-stimulated Mg2+ATPase activities and Ca2+ sensitivities due to dephosphorylation. This discrepancy may be due to the dephosphorylation resistance observed on phosphopeptide spots 2 and 3 on TnI and 4A and 4B on TnT.
In conclusion, this investigation has proven that protein phosphatase has substrate specificities in dephosphorylating myofibrillar proteins and the dephosphorylation sites within them. It has also shown that dephosphorylation affects Ca2+-stimulated Mg2+ATPase activities and Ca2+ ion sensitivities not exactly equivalent to the extent incurred on them by PKC and PKA phosphorylation.
Abbreviations
PP2A and PP1: protein phosphatase 2A and 1, respectively; CCA: calyculin A; MLC2: myosin light chain 2; TnI and TnT: troponin I and T subunits, respectively; PKC: protein kinase C; PKA: protein kinase A; DTT: dithiothreitol; Tn: troponin; Tm: tropomyosin; MBP: myelin basic protein.
Acknowledgements
This work was supported by NIH/NHLBI Grant # KO1HL03835, MBRS/RISE Grant # R25GM60414 and P20CA91366.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Cohen P. The structure and regulation of protein phosphatases. Ann Rev Biochem. 1989 ;58:453-508
2. Gergs U, Boknik P, Buchwallow I, Fabritz L, Matus M, Justus I, Hanske G, Schmitz W, Neumann J. Overexpression of the catalytic subunit of protein phosphatase 2A impairs cardiac function. J Biochem. 2004 ;279(39):40827-40834
3. Gigena MS, Ito A, Nojima H, Rogers TB. A B56 regulatory subunit of protein phosphatase 2A localizes to nuclear speckles in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2005 ;289:H285-H294
4. Terentyev D, Viatchenko-Karpinski S, Gyorke I, Terentyeva R, Gyorke S. Protein phosphatases decrease sarcoplasmic reticulum calcium content by stimulating calcium release in cardiac myocytes. J Physiol. 2003 ;552:109-118
5. DuBell WH, Rogers TB. Protein phosphatase 1 and an opposing protein kinase regulate steady-state L-type Ca2+ current in mouse cardiac myocytes. J Physiol. 2004 ;556(1):79-93
6. Chisholm AAK, Cohen P. Identification of a third form of protein phosphatase 1 in rabbit skeletal muscle that is associated with myosin. Biochem Biophys Acta. 1988 ;968:392-400
7. Chisholm AAK, Cohen P. The myosin-bound form of protein phosphatase 1 (PP-1M) is the enzyme that dephosphorylates native myosin in skeletal and cardiac muscles. Biochem Biophys Acta. 1988 ;971(2):163-169
8. Pato MD, Kerc E. Purification and characterization of a smooth muscle myosin phosphatase from turkey gizzards. J Biol Chem. 1985 ;260:12359-12366
9. Boknik P, Khorchidi S, Bodor GS, Huke S, Knapp J, Linck B, Luss H, Muller FU, Schmitz W, Neumann J. Role of protein phosphatases in regulation of cardiac inotropy and relaxation. Am J Physiol Heart Circ Physiol. 2001 ;280:H786-H794
10. DuBell WH, Gigena MS, Guatimosim S, Long X, Lederer WJ, Rogers TB. Effects of PP1/PP2A inhibitor calyculin A on the E-C coupling cascade in murine ventricular myocytes. Am J Physiol Heart Circ Physiol. 2002 ;282:H38-H48
11. Solaro RJ. Protein phosphorylation in heart muscle. Boca Raton, FL: CRC Press. 1986
12. Venema RC, Kuo JF. Protein kinase C-mediated phosphorylation of troponin I and C-protein in isolated myocardial cells is associated with inhibition of myofibrillar actomyosin MgATPase. J Biol Chem. 1993 ;268:2705-2711
13. Katoh N, Wise BC, Kuo JF. Phosphorylation of cardiac troponin inhibitory subunit (troponin I) and tropomyosin-binding subunit (troponin T) by cardiac phospholipid-sensitive Ca2+-dependent protein kinase. Biochem J. 1983 ;209:189-195
14. Noland TA Jr, Raynor RL, Kuo JF. Identification of sites phosphorylated in bovine cardiac troponin I and troponin T by protein kinase C and comparative substrate activity of synthetic peptides containing the phosphorylation sites. J Biol Chem. 1989 ;264:20778-20786
15. Noland TA Jr, Kuo JF. Protein kinase C phosphorylation of cardiac troponin I or troponin T inhibits Ca2(+)-stimulated actomyosin MgATPase activity. J Biol Chem. 1991 ;266:4974-4978
16. Noland TA Jr, Kuo JF. Protein kinase C phosphorylation of cardiac troponin T decreases Ca(2+)-dependent actomyosin MgATPase activity and troponin T binding to tropomyosin-F-actin complex. Biochem J. 1992 ;288:123-129
17. Noland TA Jr, Kuo J.F. Protein kinase C phosphorylation of cardiac troponin I and troponin T inhibits Ca(2+)-stimulated MgATPase activity in reconstituted actomyosin and isolated myofibrils, and decreases actin-myosin interactions. J Mol Cel Cardiol. 1993 ;25:53-65
18. Noland TA Jr, Kuo JF. Phosphorylation of cardiac myosin light chain 2 by protein kinase C and myosin light chain kinase increases Ca(2+)-stimulated actomyosin MgATPase activity. Biochem Biophys Res Commun. 1993 ;193:254-260
19. Noland TA Jr, Guo X, Raynor RL, Jideama NM, Averyhart-Fullard V, Solaro RJ, Kuo JF. Cardiac Troponin I Mutants. J Biol Chem. 1995 ;270:25445-25454
20. Venema RC, Raynor RL, Noland TA Jr, Kuo JF. Role of protein kinase C in the phosphorylation of cardiac myosin light chain 2. Biochem J. 1993 ;294:401-406
21. Bogoyevitch MA, Parker PJ, Sugden PH. Characterization of protein kinase C isotype expression in adult rat heart. Protein kinase C-epsilon is a major isotype present, and it is activated by phorbol esters, epinephrine, and endothelin. Circ Res. 1993 ;72:757-767
22. Rybin VO, Steinberg SF. Protein kinase C isoform expression and regulation in the developing rat heart. Circ Res. 1994 ;74:299-309
23. Disatnik MH, Buraggi G, Mochly-Rosen D. Localization of protein kinase C isozymes in cardiac myocytes. Exp Cell Res. 1994 ;210:287-297
24. Puceat M, Hilal-Dandan R, Strulovici B, Brunton LL, Brown JH. Differential regulation of protein kinase C isoforms in isolated neonatal and adult rat cardiomyocytes. J Biol Chem. 1994 ;269:16938-16944
25. Jideama N, Noland T Jr, Raynor R, Blode G, Fabbro D, Hannun Y, Kuo J. Phosphorylation Specificities of Protein Kinase C Isozymes for Bovine Cardiac Troponin I and Troponin T and Sites within These Proteins and Regulation of Myofilament Properties. J Biol Chem. 1996 ;271(38):23277-23289
26. Murphy AM, Jones L, Sims HF, Strauss AW. Molecular cloning of rat cardiac troponin I and analysis of troponin I isoform expression in developing rat heart. Biochemistry. 1991 ;30:707- 712
27. Zot AS, Potter JD. Structural aspects of troponin-tropomyosin regulation of skeletal muscle contraction. Annu Rev Biophys Biophys Chem. 1987 ;16:535-559
28. Murphy AM, Solaro R.J. Developmental difference in the stimulation of cardiac myofibrillar Mg2(+)-ATPase activity by calmidazolium. Pediat Res. 1990 ;28:46-49
29. Noland TA Jr, Raynor RL, Jideama NM, Guo X, Kazanietz MG, Blumberg PM, Solaro J, Kuo JF. Differential regulation of cardiac actomyosin S-1 MgATPase by protein kinase C isozyme-specific phosphorylation of specific sites in cardiac troponin I and its phosphorylation site mutants. Biochemistry. 1996 ;35(47):14923-14931
30. Leavis PC, Gergely J. Thin filament proteins and thin filament-linked regulation of vertebrate muscle contraction. CRC Crit Rev Biochem. 1984 ;16(3):235-305
31. Ishida A, Shigeri Y, Tatsu Y, Endo Y, Kameshita I, Okuno S, Kitani T, Takeuchi M, Yumoto N, Fujisawa HJ. Substrate specificity of Ca(2+)/calmodulin-dependent protein kinase phosphatase: kinetic studies using synthetic phosphopeptides as model substrates. J Biochem. 2001 ;129:745-752
Author contact
![]() Corresponding address:
Corresponding address:
Nathan Jideama, Department of Biological Sciences, Clark Atlanta University, P. O. Box 1733, Atlanta, Georgia 30314, USA. Tel.: 404-880-6873; Fax: (404) 880-8065; E-Mail: njideamaedu