Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2006; 2(1):10-16. doi:10.7150/ijbs.2.10 This issue Cite
Research Paper
NAD(H) recycling activity of an engineered bifunctional enzyme galactose dehydrogenase/lactate dehydrogenase
Virapong Prachayasittikul1, Sarah Ljung2, Chartchalerm Isarankura-Na-Ayudhya1, Leif Bülow2 ![]()
1. Department of Clinical Microbiology, Faculty of Medical Technology, Mahidol University, Bangkok, Thailand.
2. Department of Pure and Applied Biochemistry, Center for Chemistry and Chemical Engineering, Lund University, Lund, Sweden.
Received 2005-12-14; Accepted 2006-2-20; Published 2006-3-1
Abstract
A chimeric bifunctional enzyme composing of galactose dehydrogenase (galDH; from Pseudomonas fluorescens) and lactate dehydrogenase (LDH; from Bacillus stearothermophilus) was successfully constructed. The chimeric galDH/LDH possessed dual characteristics of both galactose dehydrogenase and lactate dehydrogenase activities while exhibiting hexameric rearrangement with a molecular weight of approximately 400 kDa. In vitro observations showed that the chimeric enzyme was able to recycle NAD with a continuous production of lactate without any externally added NADH. Two fold higher recycling rate (0.3 mM/h) than that of the native enzyme was observed at pH values above 8.5. Proximity effects became especially pronounced during the recycling assay when diffusion hindrance was induced by polyethylene glycol. All these findings open up a high feasibility to apply the NAD(H) recycling system for metabolic engineering purposes e.g. as a model to gain a better understanding on the molecular proximity process and as the routes for synthesizing of numerous high-value-added compounds.
Keywords: Chimeric bifunctional enzyme, NAD recycling, galactose dehydrogenase, lactate dehydrogenase, substrate channeling
1. Introduction
Vitalization and functioning achievements of cells are mainly based upon their metabolic activities which are frequently involved multiple enzymes in a compartmentalization of the subcellular structure arrangement. Channeling of substrate and proximity effects of enzymes in the metabolic compartment have been demonstrated and considered playing important role on metabolic regulation [1]. The channeling effect can be naturally occurred within a bifunctional/multifunctional enzyme or tightly associated multienzyme clusters or transient enzyme complexes. Tryptophan synthase from Neurospora crassa is an example of a single bifunctional enzyme channeling [2]. Meanwhile, in vitro, genetic engineering of novel bi- or multifunctional enzymes providing the putative channel in a close vicinity of two active sites has been reported [3]. These enzymes have further been applied as models for studying multienzyme reaction kinetics with particular focus on proximity and substrate channeling effects [4] both in vivo and in vitro [5]. However, proximity effect or channeling effect of coenzyme on the bifunctional enzyme still remains unexplored.
Coenzyme regeneration is essential in many enzymatic processes. Since the reduced forms of pyridine nucleotide cofactors are less stable and costly than the oxidized forms. Economically, to maximize the involving of cofactors, regeneration and recycling of the coenzyme within the enzyme reactor is often required. Engineering to generate the NAD(H) in a reaction has been performed by using both electrochemical [6] and photosensitized electron transfer [7]. However, the most common methods used are based on enzymatic regeneration [8, 9]. These include using a series of dehydrogenases e.g. alcohol dehydrogenase, formate dehydrogenase/hydrogenase and lipoamide dehydrogenase along with a low price sacrificial substrate [10, 11, 12]. In some circumstances, it is worth to note that encapsulation or immobilization of multienzyme cluster is needed to achieve the successful of sequential reactions [13, 14]. Therefore, in this study, the use of artificial bifunctional enzyme has been extended to a recycling reaction as compared to the previous systems. A chimeric enzyme composing of the two oligomeric enzymes, lactate dehydrogenase (LDH) from Bacillus stearothermophilus [15] is fused in-frame with the galactose dehydrogenase (galDH) from Pseudomonas fluorescens [16] and applied for the production of lactate under continuous recycling of NADH using pyruvate and galactose as substrates. Possibility of proximity effect of the coenzyme of the bifunctional engineered enzyme will also be explored.
2. Materials and methods
Chemicals and reagents
Enzymes used for DNA manipulation and galDH were purchased from Boehringer Mannheim. A plasmid-DNA purification kit was from Promega. High molecular weight standard proteins for gel filtration were from Pharmacia. Acrylamide and bisacrylamide were obtained from Bio-Rad Laboratories. Standard proteins for polyacrylamide gel electrophoresis were purchased from Sigma. Peroxidase-conjugated swine anti-rabbit IgG was purchased from DAKO. All other reagents were of analytical grade.
Bacterial strain and plasmids
E. coli strain F'11 recA [(lac,pro) Δthi, rifA, strA, recA/F'laclqZ- pro+] [17] was used as host. Plasmids pDZ10 [4] encoding galDH/β-gal and pLDH41 [18] were utilized as sources for the galDH gene and ldh gene, respectively.
Gel filtration of chimeric galDH/LDH
The chimeric galDH/LDH was isolated from E. coli carrying pDU712 grown to late exponential phase in LB broth supplemented with 50 mg/L ampicillin. Cells were harvested by centrifugation at 10,000 g for 10 minutes and then resuspended in 0.1 M sodium phosphate buffer, pH 6.0 containing 0.2 M NaCl and 1 mM DTT (buffer A). The following steps were all carried out at 4°C. After resuspension, cells were disrupted by sonic disintegration at output 5 with pulse on for 10×15 s (Sonifer B-30, Branson Sonic Power). Crude extract was clarified by centrifugation at 20,000 g for 30 minutes. Ammonium sulfate was added slowly to the supernatant until 45% saturation was reached. The precipitate was pelleted by centrifugation at 20,000 g for 30 minutes, redissolved and dialyzed in buffer A for overnight. Any precipitate was removed by centrifugation at 40,000 g for 10 minutes. The supernatant was further subjected to gel filtration using Sephacryl S-400 Superfine (Pharmacia) column (1.6 × 100 cm) equilibrated with buffer A. Fractions exhibiting both enzyme activities were collected. The molecular weight of the chimeric enzyme was determined using the standard protein markers: catalase (232 kDa); ferritin (440 kDa); aldolase (580 kDa); and thyroglobulin (669 kDa).
Enzyme assays
galDH activity was determined by using 16.6 mM galactose as substrate [19]. One unit of enzyme reduces 1 μmol of galactose per minute at room temperature in 0.09 M Tris-HCl, pH 8.5 containing 0.5 mM NAD. Assay of LDH activity was performed in 0.1 M MES buffer, pH 6.5 containing 0.2 mM NADH and 30 mM pyruvate. One unit of enzyme reduces 1 μmol of pyruvate per minute at room temperature.
The change in absorbance at 340 nm was followed spectrophotometrically in both assays. Protein concentrations were determined according to Bradford [20] using bovine serum albumin as standard. The pH profiles of LDH and galDH activities of the fusion and native enzymes were monitored in 0.1 M Tris-HCl in the pH range of 7.0 to 10.5 and in 0.1 M sodium phosphate buffer in the pH range of 5.0 to 7.0, respectively.
SDS-PAGE and Western blotting
SDS-PAGE was performed on 8% polyacrylamide slab gels using Tris-glycine, pH 8.3, discontinuous buffer system as described by Laemmli [21]. The molecular weights of the chimeric enzyme subunits were determined by comparing the relative mobilities with those of the standard proteins: myosin (205 kDa); β-galactosidase (116 kDa); phosphorylase b (97 kDa); bovine serum albumin (66 kDa); ovalbumin (45 kDa); and carbonic anhydrase (29 kDa).
For Western blotting, the proteins were transferred to a Millipore Immobilon TMPVDF transfer membrane using a Semi-Dry Electroblotter A (JKA-Biotech). To generate antibodies against galDH, rabbits were immunized with galDH. The antiserum was treated with ammonium sulfate to precipitate the antibodies and the precipitate was then resolved in 0.02 M Tris-HCl, pH 7.5 containing 0.5 M NaCl. Western blots were developed using the rabbit anti-bacterial galDH as first antibody and peroxidase-conjugated swine anti-rabbit IgG (DAKO, Denmark) as second antibody. Hydrogen peroxide and 4-chloro-1-naphtanol were used as substrates for peroxidase, according to instructions from supplier. Visualization of standard protein markers was performed using amido black staining.
Stability measurements
Each of the chimeric galDH/LDH obtained after gel filtration, native LDH and galDH was subjected to thermal denaturation at 50 and 60°C in 0.1 M sodium phosphate buffer, pH 6.0 containing 0.2 M NaCl and 1 mM DTT. Enzyme stability was also monitored in urea concentrations ranging from 1 to 7 M using the same buffer.
NADH recycling
The chimeric galDH/LDH obtained after gel filtration was added to 0.1 M Tris-HCl containing 0.5 mM NAD, 17 mM galactose and 30 mM pyruvate. The reactions were evaluated at pH 8.1, pH 8.9 and pH 9.6 by withdrawing samples of the reaction mixture at various time intervals. The samples were firstly deproteinized in an equal volume of 0.6 M perchloric acid and then neutralized with 0.3 M KOH. The levels of lactate produced in these samples were assayed enzymatically as described by Noll [22]. The kinetic properties of the chimeric enzyme were tested and compared with those obtained from the native enzyme. Production of lactate was also assayed in recycling reactions containing various concentrations of polyethylene glycol 20,000 ranging between 0 and 20 %.
3. Results
Construction of a chimeric gene encoding chimeric galDH/LDH
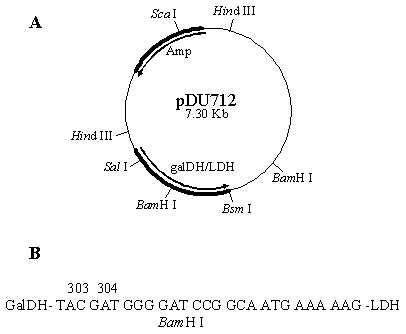
Experiment was initiated by partially digested the plasmid pUCLDH with Bsm I. A chemically synthesized DNA fragment coding for a BamH I restriction site along with 7 amino acids linker region was then inserted into the pUCLDH at the 5'- end of the ldh gene, generating the plasmid pUCLDHv. The pUCLDHv was subsequently digested with Sca I and partially with BamH I. The ldh fragment from pUCLDHv was then fused in-frame with the galdh gene originating from pDZ10 previously digested with BamH I and Sca I. The plasmid pDU712 (Figure 1) was transformed and expressed in E. coli strain F'11.
A. Schematic illustration of plasmid pDU7l2 encoding an in-frame fusion between galDH and LDH. B. Nucleotide sequence of the linker region between the fused galDH and LDH
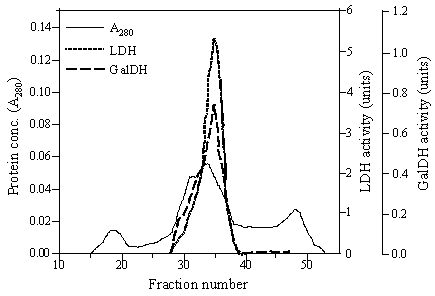
Molecular size estimation of chimeric galDH/LDH
The chimeric galDH/LDH was partially isolated by ammonium sulphate precipitation and gel filtration. The elution profiles from gel filtration showed that the chimeric enzyme possessed dual characteristics of both galDH and LDH activities as represented in Figure 2. The complete length of fusion protein eluted in one peak corresponded to a molecular weight of 400 kDa. Meanwhile, two additional peaks that could be recognized in the chromatogram demonstrated neither LDH nor galDH activity. The specific activities of the two enzyme moieties were in the range of 2-10 U/mg. Interestingly, the specific LDH activity was 5-6 time higher than the galDH activity (Table 1).
Isolation of galDH/LDH from E. coli F'11 carrying pDU712*
| Protein fraction | LDH | galDH | ||
|---|---|---|---|---|
| Activity (U) | Specific activity (U/mg) | Activity (U) | Specific activity (U/mg) | |
| Homogenate | 27.8 | 0.68 | 4.56 | 0.11 |
| Ammonium sulfate (0-45%) precipitation | 20.0 | 2.45 | 3.95 | 0.48 |
| Sephacryl S-400 | 20.0 | 6.54 | 3.25 | 1.04 |
*Results are obtained from a 250 ml bacterial culture grown to late exponential phase
Gel filtration of galDH/LDH. A Sephacryl S-400 Superfine column (1.6 × 100 cm) equilibrated with 0.1 M sodium phosphate buffer pH 6.0 containing 0.2 M NaCl and 1 mM DTT was used for the fractionation. Fraction volumes were 2.8 ml and the flow rate was 0.14 ml/min
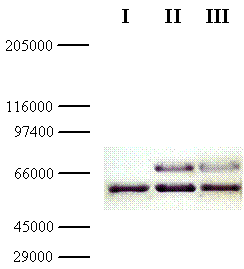
In parallel, the molecular weight of the chimeric galDH/LDH was determined via SDS-PAGE followed by Western blotting. From the Western blot, the molecular size of the chimeric galDH/LDH subunit was approximately 69 kDa (Figure 3; upper bands). Since the native galDH was a dimeric enzyme which has a molecular weight of 33 kDa per monomer [4] while the native LDH existed mainly as a tetrameric form with a monomeric molecular weight of 35 kDa. Our finding was in a good agreement with the theoretical calculation (68.5 kDa) of the complete length fusion protein. Taken together with the gel filtration experiments, these data supported the notion that the active form of galDH/LDH was present mainly as a hexameric form.
Western blot of galDH/LDH. Samples of the fusion protein from gel filtration diluted 10- (lane II) and 100- (lane III) fold were separated on SDS-PAGE with crude extract from the host cells as reference (lane I). The gel was blotted and detected using rabbit anti-galactose dehydrogenase IgG and peroxidase conjugated swine anti-rabbit IgG
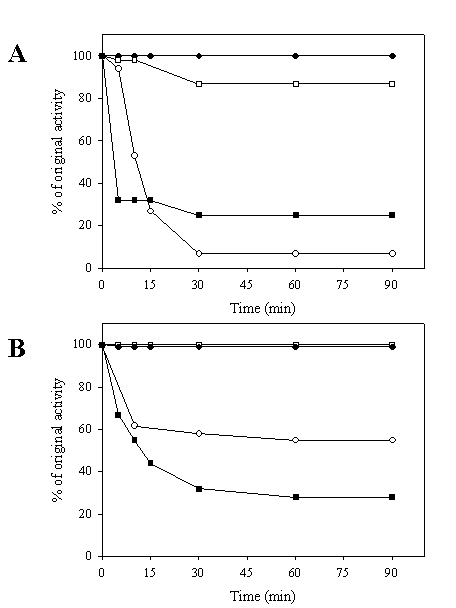
Thermal and urea stability of chimeric galDH/LDH
Stability against heat and urea denaturation of the chimeric galDH/LDH was tested. As shown in Figure 4A, it was obvious that the LDH portion of the chimeric galDH/LDH was more thermolabile than its native counterpart upon exposure to heat treatment at 60oC. A markedly decrease of LDH activity up to 90% was observed within 30 minutes. By contrarily, the galDH part was more stable than the native enzyme since the remaining activity up to 90% could be detected at 90 minutes incubation time. At 50oC, the denaturation profile of LDH was biphasic (Figure 4B). The LDH activity was rapidly reduced within 10 minutes and remained stable at approximately 60% of original activity. To evaluate whether the loss of activity was attributable to the decomposition into smaller oligomeric arrangements, the chimeric galDH/LDH was subjected to gel filtration after heat denaturation process (data not shown). From the chromatogram, it was concluded that the active chimeric enzyme still appeared in the hexameric form and no additional peaks was observed. Therefore, the loss of activity was most probably not due to decomposition of the hexameric structure but rather to internal polypeptide rearrangements.
Heat stability of galDH/LDH compared with native enzymes. The enzymes were heated at 60°C (A) and 50°C (B) over time intervals as indicated and residual activities were determined. LDH activity of native LDH (●), LDH activity of chimeric galDH/LDH (○), galDH activity of native galDH (■) and galDH activity of chimeric galDH/LDH (□)
Michaelis constants (Km) and pH profiles of chimeric galDH/LDH
Assay of the Michaelis constants (Km) of the chimeric galDH/LDH was performed. The Km of 0.10 mM for NADH (LDH activity) and 0.13 mM for NAD (galDH activity) were observed. These values were not much different from those obtained from native LDH (0.10 mM) and galDH (0.15 mM).
The maximal activity of chimeric galDH/LDH at various pHs was investigated. The highest activities of LDH and galDH were found at pH 6.5 and 10.5 as compared to 5.0 and 10.0 of the native LDH and native galDH, respectively. A similar shift in pH optima for galDH fused with β-galactosidase was previously observed by Ljungcrantz et al. [4].
NAD recycling capability of chimeric galDH/LDH
To test the NAD recycling capability of chimeric galDH/LDH, the optimum pH for NAD(H) was adjusted to give rise the enzyme activity ratio nearly close to one. The recycling reaction assays were performed at pH 8.1, 8.9 and 9.6 where the LDH and galDH activities were matched at pH 6.5 and 8.5, respectively. The recycling rate was determined as the amount of lactate produced per hour. Although, the galDH/LDH gave approximately 30 % lower recycling rates than those of the native at pH 8.1 and 8.9. However, it was shown that at pH 9.6 the rate was 2 fold greater than that of the native enzyme (Figure 5). Explanation on the disparity of cofactor regeneration between the native and chimeric enzyme could be drawn on the alteration of pH optima upon fusion. The native LDH retained only 5% of the maximum activity at pH 9.6 while approximately 20% LDH activity could be obtained in the case of galDH/LDH.
Recycling of NAD(H) at various pHs. The NAD(H) recycling reaction was initiated by adding 30 mM pyruvate, 17 mM galactose and 0.5 mM NAD at the pH values indicated. Samples were withdrawn and the amount of produced lactate was determined enzymatically
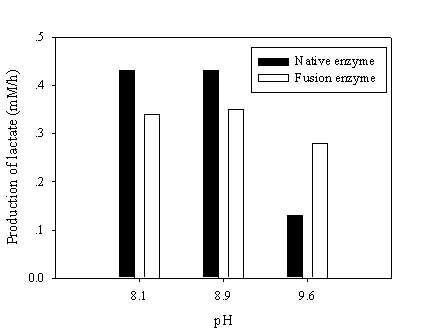
Proximity effects on NAD recycling of chimeric galDH/LDH
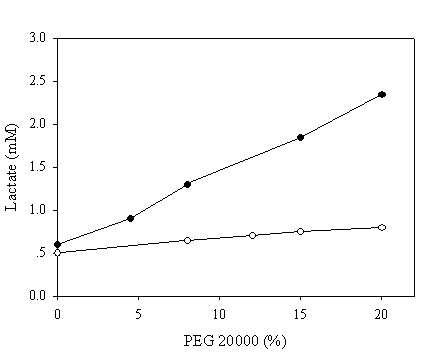
To mimic the intracellular milieu, proximity effect on the NAD recycling was further evaluated in reaction mixtures containing various concentrations of PEG 20,000. The pH chosen in this assay was 9.6 since all enzymatic activities were matched at this pH to ensure that the enzymatic activities of the fused and native systems were identical. Samples were withdrawn after 10 minutes and the production of lactate was assayed enzymatically. In the absence of PEG, a similar quantity of lactate was achieved in both systems (Figure 6). By contrarily, a markedly increase up to 5 fold in the production of lactate corresponding to the PEG concentration of 20% was observed in the case of chimeric galDH/LDH.
Recycling of NAD(H) at pH 9.6 in reaction medium containing polyethylene glycol 20,000. The amount of produced lactate was estimated after 10 minutes of reaction. The activities of the chimeric enzyme and native enzymes were matched at pH 9.6. Open and filled circles represent native and chimeric enzymes, respectively
4. Discussion
Simultaneous production of lactate through cofactor recycling of a bifunctional galDH/LDH
Herein, a chimeric redox enzyme (galDH/LDH) possessing the ability to produce lactate under continuous recycling of NAD(H) has successfully been constructed. Simultaneous generation of lactate at 0.3 mM/h can be achieved at a high basidic condition (pH 9.6). This rate is approximately 2 fold higher than that of the native enzyme (Figure 5). It is worth to state that the recycling of NAD(H) in this system does not show any immediate evidence for such physical channeling. Since the channeling may be dependent on physical transfer with the aid of ionic and hydrophobic interactions. Recent studies on the naturally occurring multienzyme dihydrofolate reductase/thymidylate synthase and on artificial bifunctional enzymes composed of enzymes in the citric acid cycle, e.g. malate dehydrogenase/citrate synthase, have shown evidences of electrostatic channeling, where the intermediate metabolite is transferred from one active site to the other via an electrostatic interaction on the surface of bifunctional enzyme [23, 24, 25]. In our case, when using a scavenger enzyme assay in vitro in which diaphorase is competing for the formed NADH, no difference between the fusion enzyme and the matched native enzymes can hence be detected (data not shown). Meanwhile, proximity effects are clearly observed when diffusion hindrance in the form of PEG addition has been applied to the reaction (Figure 6). Such effect may be particularly useful when utilizing chimeric NAD(H) recycling enzymes for metabolic engineering purposes. However, further work is required to understand whether the recycling occurs upon transiently gathering the two active sites into a close proximity to generate an enclosure that allows direct transmission of the intermediate and sterically prevents its leakage into the bulk phase. This has been believed to be involved in the channeling of NADH between dehydrogenases of opposite chiral specificity for the C-4 hydrogen of NADH [26, 27].
Physical characteristics and molecular arrangements of chimeric galDH/LDH
Based on our findings, no difference between the rate of reaction (Km) mediated by the chimeric galDH/LDH and native enzymes has been found. This clearly indicates that only minor structural changes may have occurred in the native coenzyme binding sites. Since the Michaelis constants provide a general indication of possible distortion around the active sites upon fusion to the other proteins. Fusion of the two enzymes in some circumstances affects the three-dimensional structure of the oligomeric protein in such a way that the subunit interactions are partly perturbed or the active site might be sterically hindered.
Molecular conformation and new sets of intra-polypeptide interactions in the chimeric protein are likely to drive the obtained quaternary structure into perhaps more than one arrangement. The most physical stability of these structures will emerge as the dominant one after purification and constitute the fusion enzyme studied in vitro. The fusion of the structural genes of two oligomeric proteins can be expected to give rise to the formation of protein polymers in vivo. The hexameric complex formed in this study, therefore, does not constitute the native subunit arrangement neither for galDH (dimeric) nor for LDH (tetrameric). These non-native interactions have strikingly opposite effects on the two enzyme moieties compared with their native counterparts. The stability of the galDH part of the fusion protein has hence been substantially increased. Similar stabilization effects of galDH fusions have previously been reported by Ljungcrantz et al. [4] between β-galactosidase and galDH and by Lindbladh et al. [5] between luciferase and galDH. The hexameric structure of the fusion protein apparently protects galDH from thermal and urea denaturation (Figure 4). However, the hexamer does not constitute an ideal arrangement for LDH, which precludes further purification of our hybrid. For instance, the LDH moiety rapidly lost its activity on ion exchange and affinity chromatography columns. To facilitate the handling of galDH/LDH it will be interesting to study if the folding pattern can be altered and given rise to other more stable three-dimensional arrangements. One approach can be co-expression with native subunits of one of the two fusion partners. Yet another approach will be to modify the design of the linker region in between the two enzyme moieties since it has been shown to have an impact on the stability and kinetics of fusion enzymes [28]. Moreover, previous studies on overexpression of oligomeric proteins together with chaperonins have resulted in elevated yields of correctly folded and assembled proteins [29]. Plasmid pDU712 has, therefore, been co-transformed with an expression plasmid encoding chaperonins GroES and GroEL [30], and the expressed proteins have been analyzed by gel exclusion chromatography. The oligomeric arrangement of enzymatic active protein complexes has been identical with expression of the fusion enzyme without chaperonins, a hexameric complex with the same characteristics as the original hexameric complex. However, the yield of correctly folded protein per litre cultivation has slightly been increased, approximately 10%. Such findings indicate that the gene fusion technique provides a simple way to express and purify artificial proteins carrying more than one enzymatic function within a single protein body in which the applications can be found both in metabolic engineering and biochemical analyses [3, 31].
Acknowledgements
This project was supported by the Swedish Research Council for Engineering Sciences (TFR). We also wish to acknowledge Dr. Anthony A. Gatenby for the kind gift of plasmid pGroESL.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Huang X, Holden HM, Raushel FM. Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu Rev Biochem. 2001 ;70:149-180
2. Matchett WH, DeMoss JA. The subunit structure of tryptophan synthase from Neurospora crassa. J Biol Chem. 1975 ;250(8):2941-2946
3. Bülow L. Preparation of artificial bifunctional enzymes by gene fusion. Biochem Soc Symp. 1990 ;57:123-133
4. Ljungcrantz P, Carlsson H, Mansson MO, Buckel P, Mosbach K, Bülow L. Construction of an artificial bifunctional enzyme, beta-galactosidase/galactose dehydrogenase, exhibiting efficient galactose channeling. Biochemistry. 1989 ;28(22):8786-8792
5. Lindbladh C, Persson M, Bülow L, Mosbach K. Characterization of a recombinant bifunctional enzyme, galactose dehydrogenase/bacterial luciferase, displaying an improved bioluminescence in a three-enzyme system. Eur J Biochem. 1992 ;204(1):241-247
6. Delecouls-Servat K, Bergel A, Basseguy R. Membrane electrochemical reactors (MER) for NADH regeneration in HLADH-catalysed synthesis: comparison of effectiveness. Bioprocess Biosyst Eng. 2004 ;26(4):205-215
7. Julliard M, Petit JL, Ritz P. Regeneration of NAD+ cofactor by photosensitized electron transfer in an immobilized alcohol dehydrogenase system. Biotech Bioeng. 1985 ;28:1774-1779
8. Mansson MO, Larsson PO, Mosbach K. Recycling by a second enzyme of NAD covalently bound to alcohol dehydrogenase. FEBS Lett. 1979 ;98(2):309-313
9. Ikemi M, Ishimatsu Y. The membrane bioreactor with coenzyme recycling system. J Biotechnol. 1990 ;14(2):211-220
10. Larsson KM, Adlercreutz P, Mattiasson B. Activity and stability of horse-liver alcohol dehydrogenase in sodium dioctylsulfosuccinate/cyclohexane reverse micelles. Eur J Biochem. 1987 ;166(1):157-161
11. Orlich B, Schomaecker R. Enzymatic reduction of a less water-soluble ketone in reverse micelles with NADH regeneration. Biotechnol Bioeng. 1999 ;65(3):357-362
12. Hilhost R, Laane C, Veeger C. Enzymatic conversion of apolar compounds in organic media using an NADH-regenerating system and dihydrogen as reductant. FEBS Lett. 1983 ;159:225-228
13. Ichinose H, Kamiya N, Goto M. Enzymatic redox cofactor regeneration in organic media: functionalization and application of glycerol dehydrogenase and soluble transhydrogenase in reverse micelles. Biotechnol Prog. 2005 ;21(4):1192-1197
14. Morikawa Y, Karube I, Suzuki S. NAD recycling in the collagen membrane. Biochim Biophys Acta. 1978 ;523(1):263-267
15. Barstow DA, Clarke AR, Chia WN, Wigley D, Sharman AF, Holbrook JJ, Atkinson T, Minton NP. Cloning, expression and complete nucleotide sequence of the Bacillus stearothermophilus L-lactate dehydrogenase gene. Gene. 1986 ;46(1):47-55
16. Blachnitzky EO, Wengenmayer F, Kurz G. D-Galactose dehydrogenase from Pseudomonas fluorescens. Purification, properties and structure. Eur J Biochem. 1974 ;47(2):235-250
17. Ruther U, Koenen M, Otto K, Muller-Hill B. pUR222, a vector for cloning and rapid chemical sequencing of DNA. Nucleic Acids Res. 1981 ;9(16):4087-4098
18. Carlsson H, Prachayasittikul V, Bülow L. Zinc ions bound to chimeric His4/lactate dehydrogenase facilitate decarboxylation of oxaloacetate. Protein Eng. 1993 ;6(8):907-911
19. Buckel P, Zehelein E. Expression of Pseudomonas fluorescens D-galactose dehydrogenase in E. coli. Gene. 1981 ;16(1-3):149-159
20. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976 ;72:248-254
21. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970 ;227(5259):680-685
22. Noll F. L-(+)-Lactate. In: (ed.) Bergmeyer HU. Methods of Enzymatic Analysis, 3rd ed. Deerfield Beach/Florida, Basel: Verlag Chemie Weinheim. 1984:582-588
23. Lindbladh C, Rault M, Hagglund C, Small WC, Mosbach K, Bülow L, Evans C, Srere PA. Preparation and kinetic characterization of a fusion protein of yeast mitochondrial citrate synthase and malate dehydrogenase. Biochemistry. 1994 ;33(39):11692-11698
24. Elcock AH, McCammon JA. Evidence for electrostatic channeling in a fusion protein of malate dehydrogenase and citrate synthase. Biochemistry. 1996 ;35(39):12652-12658
25. Elcock AH, Potter MJ, Matthews DA, Knighton DR, McCammon JA. Electrostatic channeling in the bifunctional enzyme dihydrofolate reductase-thymidylate synthase. J Mol Biol. 1996 ;262(3):370-374
26. Srivastava DK, Bernhard SA. Mechanism of transfer of reduced nicotinamide adenine dinucleotide among dehydrogenases. Biochemistry. 1985 ;24(3):623-628
27. Srivastava DK, Bernhard SA, Langridge R, McClarin JA. Molecular basis for the transfer of nicotinamide adenine dinucleotide among dehydrogenases. Biochemistry. 1985 ;24(3):629-635
28. Carlsson H, Ljung S, Bülow L. Physical and kinetic effects on induction of various linker regions in beta-galactosidase/galactose dehydrogenase fusion enzymes. Biochim Biophys Acta. 1996 ;1293(1):154-160
29. Teschke CM, King J. Folding and assembly of oligomeric proteins in Escherichia coli. Curr Opin Biotechnol. 1992 ;3(5):468-473
30. Van Dyk TK, Gatenby AA, LaRossa RA. Demonstration by genetic suppression of interaction of GroE products with many proteins. Nature. 1989 ;342(6248):451-453
31. Bülow L, Mosbach K. Multienzyme systems obtained by gene fusion. Trends Biotechnol. 1991 ;9(7):226-231
Author contact
![]() Corresponding address:
Corresponding address:
Assoc. Prof. Dr.Virapong Prachayasittikul, Department of Clinical Microbiology, Faculty of Medical Technology, Mahidol University, 2 Prannok Rd., Bangkok-Noi, Bangkok, Thailand 10700. Tel: 662-849-6318, Telefax: 662-849-6330, E-mail: mtvprmahidol.ac.th. Prof. Dr.Leif Bülow, Department of Pure and Applied Biochemistry, Center for Chemistry and Chemical Engineering, Lund University, P.O. Box 124 SE-221 00 Lund , Sweden. Tel: + 46 46 222 95 94, Telefax: + 46 46 222 46 11, E-mail: leif.bulowlth.se