Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2006; 2(4):179-185. doi:10.7150/ijbs.2.179 This issue Cite
Research Paper
The Inhibition and Treatment of Breast Cancer with Poly (ADP-ribose) Polymerase (PARP-1) Inhibitors
Joseph A. De Soto1, Xianyan Wang1, Yohei Tominaga1, Rui-Hong Wang1, Liu Cao1, Wenhui Qiao1, Cuiling Li1, Xiaoling Xu1, Amanda P. Skoumbourdis2, Sheila A. Prindiville3, Craig J. Thomas2, Chu-Xia Deng1 ![]()
1. Genetics of Development and Diseases Branch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health
2. Chemical Biology Core Facility, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health
3. Genetics Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, 10/9N105, 10 Center Drive, Bethesda, MD 20892, USA.
Received 2006-5-15; Accepted 2006-6-10; Published 2006-6-10
Abstract
BRCA1 and BRCA2 mutations are responsible for most familial breast carcinomas. Recent reports carried out in non-cancerous mouse BRCA1- or BRCA2-deficient embryonic stem (ES) cells, and hamster BRCA2-deficient cells have demonstrated that the targeted inhibition of poly(ADP-ribose) polymerase (PARP-1) kills BRCA mutant cells with high specificity. Although these studies bring hope for BRCA mutation carriers, the effectiveness of PARP-1 inhibitors for breast cancer remains elusive. Here we present the first in vivo demonstration of PARP-1 activity in BRCA1-deficient mammary tumors and describe the effects of PARP-1 inhibitors (AG14361, NU1025, and 3-aminobenzamide) on BRCA1-deficient ES cells, mouse and human breast cancer cells. AG14361 was highly selective for BRCA1-/- ES cells; however, NU1025 and 3-aminobenzamide were relatively non-selective. In allografts of naïve ES BRCA1-/- cells there was either partial or complete remission of tumors. However, in allografts of mouse, BRCA1-/- mammary tumors, there was no tumor regression or remission although a partial inhibition of tumor growth was observed in both the BRCA1-/- and BRCA1+/+ allografts. In human tumor cells, PARP-1 inhibitors showed no difference in vitro in limiting the growth of mammary tumors irrespective of their BRCA1 status. These results suggest that PARP-1 inhibitors may non-specifically inhibit the growth of mammary tumors.
Keywords: PARP inhibitors, BRCA1, breast cancer, therapeutic treatment, tamoxifen
1. Introduction
Germ line mutations of the breast cancer associated gene 1 (BRCA1) have been identified in about 50% of familial breast cancer cases and about 90% of familial breast and ovarian cancers. Individuals who harbor a BRCA1 mutation have a 50 to 80% lifetime risk of developing breast cancer by age 70 [1]. Current screening and chemoprevention strategies are suboptimal and consequently some women who are BRCA1 gene carriers elect to prophylactically have their breasts and/or ovaries removed to reduce their cancer risk [2-4]. Moreover, current chemo-preventative drugs such as tamoxifen often have adverse effects, primarily due to poor specificity of these compounds to mammary tumor tissues [5]. Thus, there is an urgent need to develop safe and effective drugs for the chemoprevention and therapeutic treatment of breast cancer. Recently, two studies in Nature and another in Cancer Research, demonstrated that PARP-1 inhibitors kill BRCA deficient cells with extremely high efficiency while the BRCA+/- and BRCA+/+ cells are relatively non-responsive to the treatment [6-8]. Thus, PARP-1 inhibitors might be the long-sought genetically specific drug that is both safe and effective for BRCA1/2-associated breast cancers. These results received worldwide attention with commentaries in the press and multiple Journals [9]. Yet, these data were obtained by using non-cancerous and genetically naïve BRCA1/2-/- ES cells, BRCA2 conditional mutant mouse intestine, BRCA2-/- V-C8 hamster cells, and two BRCA2-proficient breast cancer cell lines, MCF7 and MAD-MB-231 that carried acute suppression of BRCA2 by siRNA. Breast cancers originated from targeted disruption of BRCA1/2, or naturally occurring BRCA1/2 mutations were not tested. Our previous studies on BRCA1-associated mouse models indicate that BRCA1 mutant tumors arise after multiple genetic alterations that allow cells to survive and grow, while ES cells with a homozygous BRCA1 mutation lack viability dying in utero [10]. In addition, the non-cancerous hamster cells have also been shown to be exquisitely labile and sensitive to insult [11-13]. These observations cast significant doubts on the effectiveness and specificity obtained from BRCA1-/- ES cells in terms of application to BRCA1 associated breast cancer tumors. In order to have clinical relevance a more appropriate model should be used. To investigate a potentially more clinically relevant model, we studied twelve cell lines, including BRCA1-deficient and wild type ES cells, transgenic and BRCA1 knock out mouse mammary tumor cell lines, as well as human BRCA1+/+, BRCA1+/-, and BRCA1-/- breast cancer cell lines with three PARP-1 inhibitors: the very potent PARP-1 inhibitor AG14361 (KI=5nM), a moderately potent PARP-1 inhibitor NU1025 (KI =50nM) and a weak PARP inhibitor 3-aminobenzamide (3AB) (KI=500nM) [14].
2. Materials and Methods
Cells and Reagents
MCF-7, HCC1937, and MDA-MB-231 cells were obtained from ATTC (Mannassas VA). SUM1315M02 cells were obtained from Asterand (Detroit MI). The BRCA1 wild type and BRCA1 knockout mammary tumor cells were produced in this laboratory [10]. BRCA1+/- ES cells (24-28) were prepared as described previously, except that a hygmycin cassette was used instead of the neo gene [15]. The BRCA1-/- ES cells (24-27) were generated from the BRCA1+/- ES cells by deleting Brca1 exon 11 from the remaining Brca1 wild type allele using a conditional targeting vector described previously [[16] and data not shown]. 3-Aminobenzamide was obtained from Sigma (St. Louis, MO). NU1025 was obtained from EMB biosciences (La Jolla, Calif.), and AG14361 (>99.9% pure) was prepared as previously described (Skalitsky et al.). Cells were grown in DMEM media with 10% fetal bovine serum in a 5% CO2 incubator in 37oC. Cells at 80-85% confluence were trypsinized, washed with PBS and plated for each experiment.
Clonogenic assays
Three hundred ES cells, 500 mouse tumor cells or 5000 human tumor cells were plated in 12 well costar flasks. After 18 hours, the cells were exposed to PARP-1 inhibitor continuously, with changes in fresh media and PARP-1 inhibitor every four days. Colonies were grown for 10 days after which the colonies were fixed with methanol and stained with 1% Giemsa stain. Each experiment was performed in triplicate and each well counted three times
MTT proliferation assays
25,000 cells in the log phase of growth were plated in 12 well costar flasks. After 4 hours, PARP-1 inhibitor was added and the cells were incubated for 72 hours. The cells were then treated sequentially with 1% thiazolyl blue tetrazolium for 20 minutes and 2-propanol for 30 minutes. The absorption was read with the Perkin-Elmer 1420 multi-label counter. Each data point on each curve was replicated at least 8 times.
Tumor allograft studies
500,000 cells were implanted subcutaneously bilaterally in the flanks and on the back of 6-8 weeks old female nude mice. There were five mice in each of the control groups and five mice in each of the treatment groups. Treatments were initiated when measurable tumors were present in all mice for the treatment studies or on the day after injection with tumor cells. In the treatment studies animals received either AG14361 at 30 mg/kg in vehicle (35% ethanol/ 65% PBS) or vehicle alone intra-peritoneally for 5 days, or for three consecutive days every 5 days, all mice were monitored daily. For the tumor prevention studies, nude mice were injected intraperitoneally with either vehicle AG14361 30 mg/kg one day prior to implantation bilaterally with 500,000 cells. The mice were treated based on different protocols described in the text. Mice were followed up to 19-21 days when largest tumor reached about 1.6 cm in the diameter. Tumor volume (V) was calculated as V=2/3p__X)__Y)_rZ). "r" is radius, and X, Y and Z refer to each axis.
3. Results
Effects of PARP-1 inhibitors on mouse ES cells
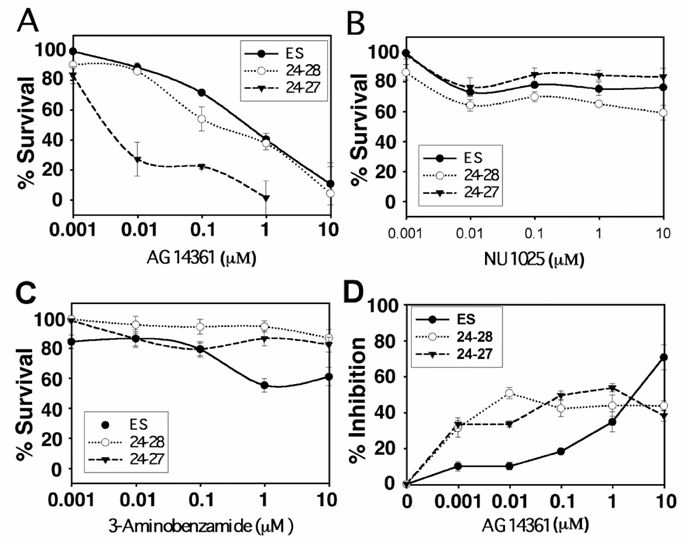
Our data on ES cells indicated that AG14361 is highly potent in killing BRCA1-/- cells in vitro and in vivo. The majority of BRCA1-/- ES cells were killed at a concentration of 0.01µM, and the surviving ES colonies had reduced size in the clonogenic assay (Fig. 1A and not shown). No surviving colonies were observed at a concentration of 1 µM (Fig. 1A). The survival of BRCA1+/+ and BRCA1+/- (24-28) colonies was however also inhibited by AG14361 at higher concentrations though to a lesser degree than that the BRCA1-/- (24-27) cells (Fig. 1A). The ability of either NU1025 (Fig. 1B) or 3AB (Fig. 1C) to inhibit the survival of ES cells was less than that of AG14361 as measured through the clonogenic assay. Hence, although the most potent PARP-1 inhibitor (AG14361, KI =5nM) is highly specific to BRCA1-/- ES cells, the clonogenic assays suggested that the weaker PARP-1 inhibitors (NU1025, KI =50nM; and 3AB, KI =500nM) are not. Next, the ability of AG14361 to inhibit the proliferation of ES cells as measured by MTT assay was evaluated (Fig. 1D). At levels of below 5 μM both the BRCA1-/- and BRCA1+/- ES cells were more sensitive to AG14361 growth inhibition than the wild type.
Response of ES cells to PARP-1 inhibitors. (A-C) Clonogenic survival assay of embryonic stem cells. 300 colonies of ES (BRCA1+/+), 24-28 (BRCA1+/-) and 24-27 (BRCA1-/-) ES cells were plated followed 18 hours later with treatment by PARP-1 inhibitors: AG14361 (A), NU1025 (B) and 3-Aminobenzamide (C) for 10 days. (D) Response of ES cells to AG 14361 revealed by MTT assay. Filled circles ES colonies, open circles 24-28 colonies and filled triangles 24-27 colonies.
We then evaluated the ability of AG14361 to prevent the development of ES tumors by treating the nude mice a day before implantation of ES cells with AG14361 and continuing for 9 days thereafter (Table 1a). There was a 90% reduction in the formation of BRCA1-/- (24-27) ES derived tumors relative to the control. The only tumor developed in the BRCA1-/- tumor was also very small. There was however, only a 22% reduction in the formation of BRCA1+/+ ES tumors.
Response of embryonic stem cells to AG14361 in xenographs. a) Prevention of embryonic stem cell tumor formation in xenographs. b) Treatment of embryonic stem cell tumor formation in xenographs.
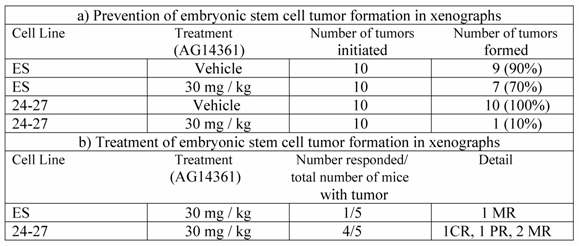
CR, complete response, total regression of tumor, no tumor at autopsy; PR, partial response 50% reduction in tumor ; MR, minor response 25% reduction in tumor
Next, we inoculated nude mice with ES cells and after the development of tumors treated the mice following an established procedure that efficiently killed BRCA2-/- V-C8 cells [7], ie. treatment of initiated tumors for five consecutive days with either vehicle or AG14361. The BRCA1-/- ES tumors underwent tumor regression in four out of the five mice treated with AG14361 (Table 1b). However, only one minor regression was seen out of the five mice with the BRCA1+/+ ES tumors. These results suggest that AG14361 is relatively specific in both preventing and treating BRCA1-/- ES tumors.
Effects of PARP-1 inhibitors on mouse mammary tumor cells
Thus our study, in principle, confirmed the previous finding that PARP-1 inhibitors could eliminate BRCA mutant cells both in vitro and in vivo. However, it remains unclear whether the PARP-1 inhibitors could prevent mammary cancer cell growth.
Next, we evaluated the growth of mouse mammary tumor cells in the presence of AG14361, NU1025 or 3AB. Five mouse mammary tumor cell lines were evaluated. This includes 3 cell lines derived from BRCA1Co/Co;MMTV-Cre or BRCA1Co/Co;WAP-Cre mice carrying a Cre-loxP mediated deletion of BRCA1 exon 11 (780 and 69) or complete deletion of BRCA1 (525), one cell line from MMTV-cNeu (NEU) and one line from MMTV-Ras (RAS) transgenic mice [10]. We have shown previously that many mammary tumors developed in BRCA1Co/Co;MMTV-Cre or BRCA1Co/Co;WAP-Cre mice had lost the entire BRCA1 coding region due to genetic instability in tumors (Figure No. 6 in Xu et al. 1999). The cell line 525 was derived from one of such tumors. Our data obtained from genomic PCR and RT-PCR confirmed the lack of BRCA1 coding region in this cell line (Fig. 2A,B). There was no significant difference in the sensitivity to growth inhibition as measured by growth inhibition (GI50) by MTT assay to AG14361 among all three BRCA1 deficient cancer cell lines (referred as BRCA1-/- for simplicity), while the BRCA1 wild type cell lines (NEU and RAS) exhibited 1.5 to 11 fold decrease in sensitivity to AG14361 inhibition of growth compared with BRCA1 mutant cells (Fig. 2C).
Treatment of mouse mammary tumors with PARP-1 inhibitors. (A, B), Genotyping of tumor cell lines 525 and 69 using PCR (A) and RT-PCR (B) analysis. Primers used for PCR are Brca1, 5' CTG GGT AGT TTG TAA GCA TCC 3' and 5' CTG CGA GCA GTC TTC AGA AAG 3' which amplify 621 bp from mutant allele after Cre-loxP mediated deletion; Smad4, 5'-GGG CAG CGT AGC ATA TAA GA -3' and 5'-GAC CCA AAC GTC ACC TTC AC -3' which amplify about 450 bp as a control for DNA. Primers used for RT-PCR are Brca1, 5' CTC AAG AAG CTG GAG ATG AAG G 3' and 5' GAA GAT GCC TAG AGC AGC CAT G 3' which amplify about 540 bp from Brca1 transcripts encoding BRCA1 short isoform; Gapdh, 5' ACA GCC GCA TCT TCT TGT GC 3' and 5' TTT GAT GTT AGT GGG GTC TCG C 3' which amplify about 450 bp as a control for RT-PCR. (C-E) MTT proliferation assay: BRCA1 wild type (NEU and RAS) and BRCA1-/- (780, 69, and 525) mouse mammary tumor cells were treated with AG14361 (C), NU1025 (D), and 3-Aminobenzamide (E) for 72 hours respectively. (F) Clonogenic survival assay. Five hundred 780, NEU, and 525 cells were exposed to AG14361 each for 10 consecutive days, 18 hours after plating.
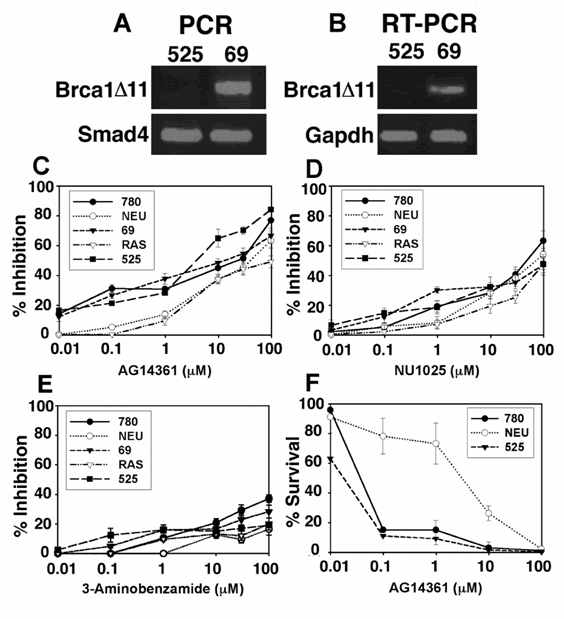
Next, the ability of NU1025 to inhibit these tumor lines was evaluated. NU1025 was only minimally more selective for the BRCA1-/- cell lines at levels below 1 µM of NU1025 Again the observed selectivity disappeared at about 10µM (Fig. 2D). The inhibitory ability of NU1025 was less than that observed for the stronger PARP-1 inhibitor AG14361 (Fig 2C). The weak PARP-1 inhibitor 3AB was less able to inhibit tumor cell proliferation in vitro than either AG14361 or NU1025 though 3AB also was more selective towards BRCA-/- cells at lower concentrations (Fig. 2E). Thus at low concentrations of PARP-1 inhibitors, their was some minimal to mild selectivity towards the inhibition of growth for BRCA1 knock out tumor cells, this difference however was not found at higher concentrations of PARP-1 inhibitors. We next performed clonogenic assays of the NEU, 780 and 525 cell lines, where we found that the BRCA1-/- tumor cells (780 and 525) were much more sensitive to AG14361 treatment than BRCA1+/+ tumor cells (NEU) (Fig. 2F). At concentrations of AG14361 of 0.1 and 1 μM, there was about 8-fold difference in survival rate. This difference, however, is much lower compared with an over 100 fold difference reported previously on pre-cancerous cells [6, 7].
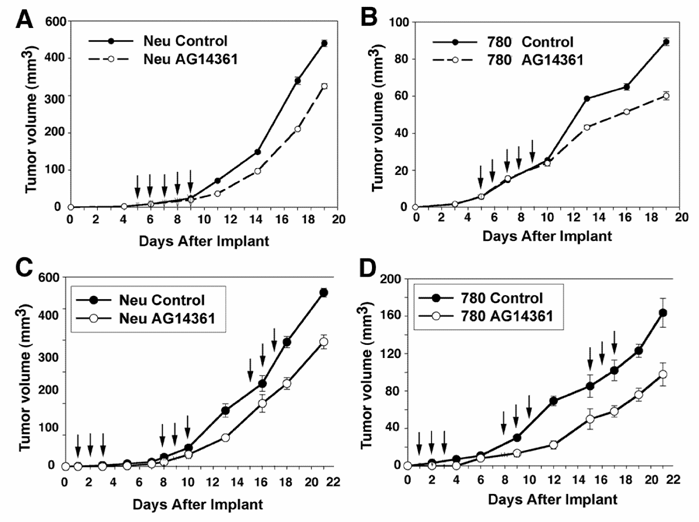
The in vitro experiments were followed by in vivo examination of BRCA1+/+ and BRCA1-/- mammary tumors exposed to 5 days of high dose AG14361 treatment. Inhibition of mammary tumor growth was observed two days after the fifth AG14361 injection relative to the control in both wild type and mutant cell lines (Fig. 3A,B). There were however no complete remissions, or partial remissions observed. Next, we followed a prevention protocol that was used to successfully eliminate sensitivity of BRCA-/- ES cell derived tumors to PARP-1 inhibitors [6]. However, our data indicated that the treatment failed to achieve a more than marginal specific killing of BRCA1 mutant cancer cells as both BRCA1-/- and BRCA1+/+ cells showed about a 30-40% reduction in growth (Fig. 3C,D). These observations revealed a significant difference between BRCA1 mutant ES cells and cancer cells in their response to PARP-1 inhibitors.
Response of in vivo allograft of BRCA1+/+ and BRCA1-/- mammary tumor cells to AG14361. Treatment intra-peritoneally with AG14361 (30 mg/kg) for five consecutive days as indicated by the arrows (days 5-9 after implantation) (A, B), or for three consecutive days starting on days 2, 9 and 16 (C, D). The mice were monitored daily. The average tumor size is indicated above for all five mice in both the control and treatment groups.
Effects of PARP-1 inhibitors on human breast cancer cells
Evaluation of human mammary tumor cells followed. Four cell lines derived from human breast cancers, MCF-7 (BRCA1+/+ and ERα+), MDA-MB-231 (BRCA1+/-, ERα-), HCC1937 (BRCA1-/-, ERα-) and SUM1315MO2 (BRCA1-/-, ERa-), were evaluated through their response to PARP-1 inhibitors. AG14361 showed little to no specificity in inhibiting the MDA-MB-231, HCC1937 or SUM1315MO2 cells as compared to its inhibition of MCF-7 cells (Fig. 4A). It is noted that the HCC1937 (5382insC) and the SUM1315MO2 (185delMO2) carry mutations of the BRCA1 genes that account for 85% of the human BRCA1 associated breast cancers [17]. The HCC1937 cells were slightly more sensitive to growth inhibition by NU1025 than MCF-7 cells (Fig. 4B). This was a bit surprising as NU1025 is less potent than AG14361. Next the ability of 3AB to inhibit breast cancer growth was evaluated. At concentrations below approximately 7-8 µM, 3-AB was slightly more effective in inhibiting the growth of HCC1937 and MDA-MB-436 cells as opposed to MCF-7 cells. This difference disappeared at higher doses (Fig. 4C). We next performed clonogenic assays and found that the survival fraction of both MDA-MB-231 and HCC1937 were slightly less that of MCF-7 above 1 µM (Fig. 4D).
Response of human mammary tumors to treatment with PARP-1 inhibitors. (A-C) MTT assay: 25000 MCF-7 (BRCA1+/+), MDA-MB-231 (BRCA1+/-), HCC1937 (BRCA1-/-), and SUM1315MO2 (BRCA1-/-) human breast cancer cells were treated for 48 hours with AG14361 (A), NU1025 (B) and/or 3-Aminobenzamide (C), respectively. (D) Clonogenic assay: MCF-7, MDA-MB-231 and HCC1937 cells were treated and exposed to AG14361 continuously for 10 days.
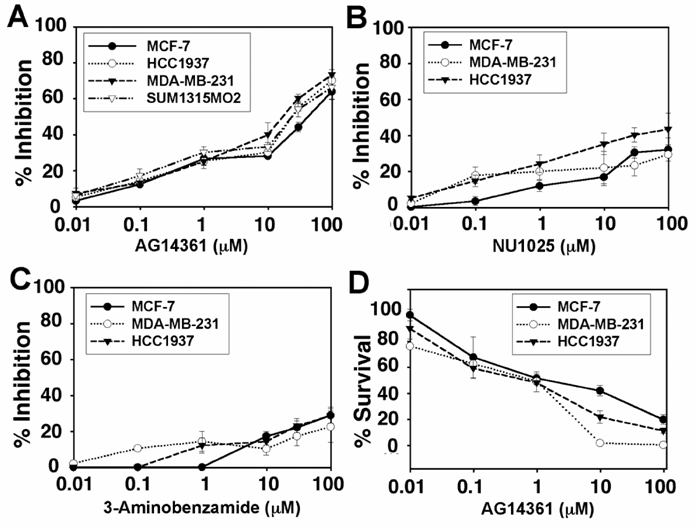
Thus, our studies indicate that the use of PARP-1 inhibitors in mouse BRCA1-/- mammary cancer cells seems to show only mild specificity in comparison with their controls, while PARP-1 inhibitors seem to uniformly inhibit growth of human breast cell lines regardless of the BRCA1 genotypes.
4. Discussion
We have shown that the PARP-1 inhibitor AG14361 selectively kill BRCA1-/- ES cells with high efficiency while it does not preferentially kill BRCA1-/- breast cancer cells compared with BRCA1+/+ controls. By definition a tumor cell represents a cell that has had multiple genetic mutations and that in tumor there may be numerous fundamentally different cancer cells when classified strictly by the type and frequency of individual genomic changes [10, 18, 19]. Thus, caution needs to be made in interpreting findings from BRCA1-/- ES cells, which have not gone through these genetic changes.
In addition, we found that the MTT assay is a more consistent model in predicting the sensitivity and specificity of tumor cells to PARP-1 inhibitors in comparison with profiles of tumor progression obtained from allograft models as opposed to the clonogenic assay. The higher sensitivity observed in clonogenic assay may be in part due to the extraordinary constant exposure to high drug concentrations, which would likely not be possible for such a length of time in any therapeutic treatment of human or in vivo animal systems. In addition, cancer cells are exquisitely dependant to other cancer cells for survival and growth in vivo and in vitro through paracrine cell/cell interactions. The low inoculating cell density in clonogenic assay may also be a factor for the increase sensitivity for the drug treatment.
Notably, we found that PARP-1 inhibitors can inhibit breast cancer cells irrespective of their BRCA1 and ERα status. This might potentially have significant clinical importance, as most BRCA1-/- breast cancers are ERα negative and thus do not exhibit a response to hormonal treatment such as tamoxifen [20, 21]. The use of PARP-1 inhibitors in combination with standard chemotherapy regiments may also prove to be a worthwhile endeavor as one can conceive of having to give only lower levels of PARP-1 inhibitors in combination with other drugs than alone in the treatment of breast cancers.
While this study tested effects of PARP-1 inhibitors on BRCA1 mutant cells, it is worthwhile to note that a recent investigation revealed no specific cell killing was found in pancreatic BRCA2-deficient human cancer, CAPAN-1, cells by NU1025 or 3AB [22], which is in a strong contrast to the results showing that NU1025 kills the non-cancerous hamster BRCA2-/- cells with high efficiency and specificity [7]. This was attributed to the weak activity of NU1025, as these cells can be efficiently killed by KU0058684 and KU0058948, which are more potent PARP-1 inhibitors [23]. Yet, clinically speaking as implied by Gallmeier and Kern [22], it is not the potency of a drug that is most important but the efficacy. That is the maximum biological effect, is perhaps the most important criteria by which clinicians treat patients. In addition, though we have shown that PARP-1 inhibitors are only minimally selective, if at all, to BRCA1-/- mammary tumors, our preliminary observation indicates that PARP-1 inhibitors may be useful in combination with standard chemotherapy in treating breast cancer (Our unpublished observation). We are now in the process of investigating combination therapy with AG14361, which is as at least as potent as KU0058684 and KU0058948, to test whether PARP-1 inhibitors can specifically/synergistically kill BRCA1/2-/- breast cancer cells with selective chemotherapeutic agents.
Acknowledgements
We thank Elise Kohn and members of Deng lab for critical discussion. This work was supported by the intramural Research Program of National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, USA.
Conflict of interests
The authors have declared that no conflict of interest exists.
References
1. Brody LC, Biesecker BB. Breast cancer susceptibility genes. BRCA1 and BRCA2. Medicine (Baltimore). 1998 ;77:208-26
2. Rebbeck TR, Lynch HT, Neuhausen SL. et al. Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med. 2002 ;346:1616-22
3. Kauff ND, Satagopan JM, Robson ME. et al. Risk-reducing salpingo-oophorectomy in women with a BRCA1 or BRCA2 mutation. N Engl J Med. 2002 ;346:1609-15
4. Metcalfe KA. Prophylactic bilateral mastectomy for breast cancer prevention. J Womens Health (Larchmt). 2004 ;13:822-9
5. Senkus-Konefka E, Konefka T, Jassem J. The effects of tamoxifen on the female genital tract. Cancer Treat Rev. 2004 ;30:291-301
6. Farmer H, McCabe N, Lord CJ. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005 ;434:917-21
7. Bryant HE, Schultz N, Thomas HD. et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005 ;434:913-7
8. Hay T, Jenkins H, Sansom OJ. et al. Efficient deletion of normal Brca2-deficient intestinal epithelium by poly(ADP-ribose) polymerase inhibition models potential prophylactic therapy. Cancer Res. 2005 ;65:10145-8
9. Brody LC. Treating cancer by targeting a weakness. N Engl J Med. 2005 ;353:949-50
10. Brodie SG, Xu X, Qiao W. et al. Multiple genetic changes are associated with mammary tumorigenesis in Brca1 conditional knockout mice. Oncogene. 2001 ;20:7514-23
11. Papouli E, Lafon C, Valette A. et al. Involvement of apoptosis in mitomycin C hypersensitivity of Chinese hamster cell mutants. Biochem Pharmacol. 2000 ;59:1101-7
12. Zdzienicka MZ. Mammalian X ray sensitive mutants: a tool for the elucidation of the cellular response to ionizing radiation. Cancer Surv. 1996 ;28:281-93
13. Overkamp WJ, Rooimans MA, Neuteboom I. et al. Genetic diversity of mitomycin C-hypersensitive Chinese hamster cell mutants: a new complementation group with chromosomal instability. Somat Cell Mol Genet. 1993 ;19:431-7
14. Calabrese CR, Almassy R, Barton S. et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst. 2004 ;96:56-67
15. Shen SX, Weaver Z, Xu X. et al. A targeted disruption of the murine Brca1 gene causes gamma-irradiation hypersensitivity and genetic instability. Oncogene. 1998 ;17:3115-24
16. Xu X, Wagner KU, Larson D. et al. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet. 1999 ;22:37-43
17. Elstrodt F, Hollestelle A, Nagel JH. et al. BRCA1 mutation analysis of 41 human breast cancer cell lines reveals three new deleterious mutants. Cancer Res. 2006 ;66:41-5
18. Weaver Z, Montagna C, Xu X. et al. Mammary tumors in mice conditionally mutant for Brca1 exhibit gross genomic instability and centrosome amplification yet display a recurring distribution of genomic imbalances that is similar to human breast cancer. Oncogene. 2002 ;21:5097-107
19. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000 ;100:57-70
20. Jones LP, Li M, Halama ED. et al. Promotion of mammary cancer development by tamoxifen in a mouse model of Brca1-mutation-related breast cancer. Oncogene. 2005 ;24:3554-62
21. Narod SA, Brunet JS, Ghadirian P. et al. Tamoxifen and risk of contralateral breast cancer in BRCA1 and BRCA2 mutation carriers: a case-control study. Hereditary Breast Cancer Clinical Study Group. Lancet. 2000 ;356:1876-81
22. Gallmeier E, Kern SE. Absence of Specific Cell Killing of the BRCA2-Deficient Human Cancer Cell Line CAPAN1 by Poly(ADP-ribose) Polymerase Inhibition. Cancer Biol Ther. 2005 ;4:703-6
23. McCabe N, Lord CJ, Tutt AN. et al. BRCA2-Deficient CAPAN-1 Cells are Extremely Sensitive to the Inhibition of Poly (ADP-Ribose) Polymerase: An Issue of Potency. Cancer Biol Ther. 2005 ;4:934-6
Author contact
![]() Correspondence to: Dr. Chu-Xia Deng, Phone: (301) 402-7225, Fax: (301) 480-1135. Email: chuxiadniddk.nih.gov
Correspondence to: Dr. Chu-Xia Deng, Phone: (301) 402-7225, Fax: (301) 480-1135. Email: chuxiadniddk.nih.gov