Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(1):40-46. doi:10.7150/ijbs.3.40 This issue Cite
Short Research Communication
Intrarenal Oxidative Stress and Augmented Angiotensinogen are Precedent to Renal Injury in Zucker Diabetic Fatty Rats
Yuki Suzaki, Yuri Ozawa, Hiroyuki Kobori ![]()
Department of Physiology, and Hypertension and Renal Center of Excellence, Tulane University Health Sciences Center, New Orleans, LA, USA
Received 2006-9-22; Accepted 2006-10-27; Published 2006-11-1
Abstract
The Zucker diabetic fatty (ZDF) rat is a model of type II diabetes and metabolic syndrome based on impaired glucose tolerance caused by the inherited insulin-resistance gene. The ZDF rat exhibits progressive nephropathy; however, the detailed mechanisms have remained unclear. This study was performed to examine the possible involvement of enhanced intrarenal angiotensinogen in the development of renal injury in ZDF rats. Genetic pairs of male ZDF rats and control lean rats (N=6 each) were maintained from 12 to 17 weeks of age. At 17 weeks of age, fasting blood glucose and urinary 8-isoprostane levels were significantly higher in ZDF rats compared with the controls. Systolic blood pressure progressively increased in ZDF rats from 120+/-1 to 137+/-1 mmHg during this period. In contrast, systolic blood pressure did not increase in the controls. Kidney angiotensinogen protein levels were significantly increased in ZDF rats compared with the controls (1.83+/-0.34 vs. 1.00+/-0.17, relative ratio). Expression of angiotensin II type 1a receptor mRNA was similar between these groups. The measured indices of renal damage in the present study (glomerular sclerosis, interstitial expansion, glomerular macrophage infiltration, and renal arterial proliferation) were not significantly increased at this stage in ZDF rats. However, we previously showed that the increased reactive oxygen species-related angiotensinogen enhancement plays an important role in the development of renal injury in a genetic salt-sensitive hypertension. Thus, the present data suggest that elevated reactive oxygen species and reactive oxygen species-associated augmentation of intrarenal angiotensinogen may initiate the development of renal injury in ZDF rats.
Keywords: renin-angiotensin system, diabetes mellitus, angiotensinogen, oxidative stress, renal injury
1. Introduction
Diabetic nephropathy is the most common cause of end-stage renal failure in patients starting dialysis in developed countries [1]. Clinical trials have demonstrated that high glucose levels are the principal cause of renal damage in both type I [2] and type II diabetes [3]. The mechanisms underlying the development of diabetic nephropathy are extremely complex; however, activation of the intrarenal renin-angiotensin (Ang) system (RAS) has been suggested [4-10].
Angiotensinogen is the only known substrate for the enzyme, renin, which cleaves angiotensinogen to release a biologically inactive decapeptide, AngI [11]. Angiotensin-converting enzyme (ACE) cleaves a dipeptide from AngI to produce a biologically active peptide, AngII [11]. AngII stimulates aldosterone secretion, cell proliferation and migration [12], thrombosis [13], and superoxide production [14-16]. All of these factors are involved in development and progression of nephropathy. Our previous reports suggest that AngII stimulates angiotensinogen synthesis and that enhancement of angiotensinogen may be a key component in the maintenance of elevated circulating and intrarenal AngII levels [17-21].
High glucose generates reactive oxygen species (ROS) as a result of glucose auto-oxidation, metabolism, and formation of advanced glycosylation end products [22]. AngII also stimulates ROS generation [23]. ROS is one of the important mediators of vascular complications in diabetes [4, 22]. In addition, we previously showed that angiotensinogen expression is augmented by ROS in salt-sensitive hypertensive rats [24].
The Zucker diabetic fatty (ZDF) rat is considered to be an excellent animal model of type II diabetes that presents a physiological and metabolic profile similar to that seen in humans [25, 26]. The ZDF rat is characterized by hyperglycemia, hyperinsulinemia, hyperlipidemia, moderate hypertension, moderate obesity, and progressive renal injury [27, 28]. Schafer et al. [29] showed that ZDF rats established diabetes at 17 weeks of age. Coimbra et al. [30] showed that ZDF rats developed focal and segmental sclerosis at 18 weeks of age, although severe hyperlipidemia, hyperglycemia, and obesity predated it. Hoshi et al. [25] reported that ZDF rats showed glomeruloscrerosis as early as 20 weeks of age. Chander et al. showed that ZDF rats developed focal and segmental sclerosis at 22 weeks of age [31]. Thus, there are clear data demonstrating that ZDF rats show renal injury starting at 18-20 weeks of age. Although ZDF rats steadily show renal injury around 20 weeks of age, early initiation factors involved in the development of renal injury has not been elucidated in ZDF rats. Mizuno et al. [26] demonstrated that a pharmacological blockade of the RAS with AngII type 1 (AT1) receptor blocker (ARB) reduced proteinuria and delayed the progression of renal disease independently from blood pressure control in ZDF rats. This result has suggested that the activated intrarenal RAS plays an important role in the development of renal injury in ZDF rats. Moreover, in salt sensitive hypertensive rats, we previously reported that the ROS-related activation of intrarenal angiotensinogen plays important roles in the development of renal injury [24]. These data prompted us to examine the possible involvement of enhanced ROS-associated augmentation of intrarenal angiotensinogen in the development of nephropathy at the early stage of diabetes in ZDF rats. Therefore, we have focused on intrarenal parameters of ZDF rats before the elicitation of renal injury.
2. Materials and Methods
Animals
The experimental protocol was approved by the Animal Care and Use Committee of Tulane University. Male ZDF rats (ZDF-fa/fa, 12 weeks of age, N=6) and age-matched control male lean rats (ZDF-+/?, 12 weeks of age, N=6) were purchased from Charles River Laboratories. These rats were maintained from 12 to 17 weeks of age. Rats were housed in metabolic cages and maintained in a temperature-controlled room regulated on a twelve-hour light/dark cycle with free access to water. Rats were fed with a commercially available rat chow containing normal salt and 16.7 kcal% fat (Formulab Diet 5008, LabDiet). Fasting blood glucose levels were measured using a glucose analyzer (TheraSense). Systolic blood pressures were measured by tail cuff plethysmography once a week as previously described [17-21, 32].
Urinary Measurements
Twenty-four hour urine samples were collected once a week in centrifuge tubes containing 10 µg/ml of indomethacin and 0.005% butylated hydroxytoluene. Urine samples were centrifuged and the supernatant was separated and stored at –80°C until assayed for total protein and 8-isoprostane. Urinary concentration of total protein was measured by colorimetric assays using a commercially available kit (Bio-Rad). 8-isoprostane is the major urinary metabolite of F2-isoprostanes and is formed non-enzymatically from the action of superoxide radicals on arachidonic acid [33]. The urinary 8-isoprostane excretions were measured by ELISA using a commercially available kit (Cayman).
Sample Collections
Blood and kidney samples were harvested at 17 weeks of age. After decapitation, trunk blood was collected into chilled tubes containing 5 mmol/l EDTA, 20 µmol/l enalaprilat, 10 µmol/l pepstatin A, and 1.25 mmol/l 1,10-phenanthroline. Plasma was separated and stored at –20°C until assayed for plasma AngII by radioimmunoassay as previously described [17-21, 32]. Immediately after removal, one kidney was homogenized in cold methanol and renal AngII was measured by radioimmunoassay as previously described [17-21, 32]. The contralateral kidneys were separated into three pieces and snap-frozen in liquid nitrogen for protein extraction, immersed in RNAlater (Ambion) for total RNA extraction and immersed in zinc-saturated formalin (Anatech) for tissue fixation.
Western Blot Analysis
Protein extraction of renal cortex and Western blot analysis for angiotensinogen were performed as previously described [17-21, 32]. Sheep polyclonal antibody against purified rat angiotensinogen was produced and characterized at University of Queensland, Australia [34]. Anti beta-actin antibody was purchased from Sigma. Data of Western blot analysis for angiotensinogen protein levels were normalized by beta-actin protein levels.
Quantitative Real-time RT-PCR
Total RNA extraction of renal cortex and quantitative real-time RT-PCR for AT1a receptor were performed as previously described [32]. The information of sequences are as follows: AT1a receptor; forward primer, 5'-CTT GTT CCC TTT CCT TAT CA-3', reverse primer, 5'-CGT TTC TTG GTT TGT TCT TT -3', probe, 5'- CAC CAG CTA TAC CCT TAT TTG GAA AGC -3'; GAPDH, forward primer, 5'- CAG AAC ATC ATC CCT GCA TC -3', reverse primer, 5'- CTG CTT CAC CAC CTT CTT GA -3', probe, 5'- CCT GGA GAA ACC TGC CAA GTA TGA TGA -3'. Data of quantitative RT-PCR for AT1a mRNA levels were normalized by GAPDH mRNA levels.
Histological Analysis
Histological samples were embedded in paraffin by an out-sourcing company (Mass Histology). The extent of glomerular sclerosis was evaluated with periodic acid Schiff-stained sections. The extent of interstitial expansion was evaluated with Picro-Sirius red-stained sections. Quantification was performed semi-automatically in a blind manner by using the Image-Pro plus software (Media Cybernetics) as previously described [32].
Immunohistochemistry
Paraffin-embedded sections from the rat kidneys were also used for immunohistochemical analysis with the ABC method (Vector Laboratories). The numbers of monocytes/macrophages in glomeruli were examined by immunohistochemistry using a commercially available antibody against CD68 (ED-1, Serotec) as previously described [32]. The magnitude of arterial proliferation of afferent arteriolar walls was evaluated by immunohistochemistry using a commercially available antibody against alpha-smooth muscle isoform of actin (Dako) with elastin stain (Sigma) as previously described [32].
Statistical Analysis
Statistical analysis was performed using paired or unpaired t-test. All data are presented as mean+/-SEM. p<0.05 was considered significant.
3. Results
General Animal Data
The rats' profiles of body weight, fasting blood glucose, urine volume and urine protein excretion at 17 weeks of age are shown in Table 1. The ZDF rats developed diabetes mellitus, as manifested by increases in water intake (ZDF rats: 66+/-17 ml/day vs. control lean rats: 29+/-6 ml/day at 17 weeks of age), urine volume, and blood glucose. The fasting glucose levels were significantly increased in ZDF rats compared to control lean rats. Urine volume and protein excretion also increased in ZDF rats compared to control lean rats.
Characteristics of ZDF rats and control lean rats at 17 weeks of age
| Lean | ZDF | |
|---|---|---|
| Body Weight, g | 288 ± 7 | 378 ± 18* |
| Fasting Blood Glucose, mg/dl | 47 ± 5 | 148 ± 36** |
| Urine Volume, ml/day | 9.8 ± 1.0 | 44.3 ± 16.6* |
| Urine Protein, mg/day | 6.8 ± 0.6 | 22.1 ± 2.7** |
**p<0.01, *p<0.05
Systolic Blood Pressure
Systolic blood pressure was similar between ZDF and control lean rats at 12 weeks of age. ZDF rats showed a moderate increase in systolic blood pressure, which reached a significant level by 15 weeks of age (137+/-1 mmHg) compared with 12 weeks of age (120+/-1 mmHg). The control lean rats did not have increased blood pressures.
Urinary 8-Isoprostane
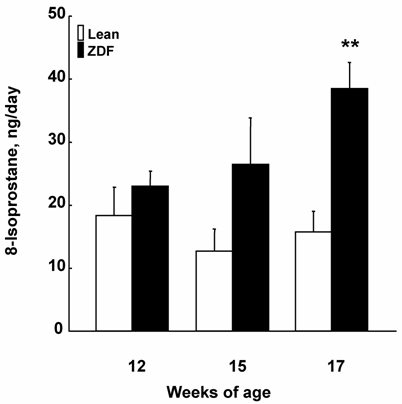
As shown in Figure 1, urinary 8-isoprostane excretion rates were not different between ZDF and control lean rats at 12 weeks of age. At 15 weeks of age, the urinary 8-isoprostane excretions showed a tendency to increase in ZDF rats and were significantly increased at 17 weeks of age.
The urinary 8-isoprostane excretions of ZDF and control lean rats. The urinary 8-isoprostane was significantly augmented in ZDF rats at 17 weeks of age. **p<0.01 vs. control lean rats at the same age.
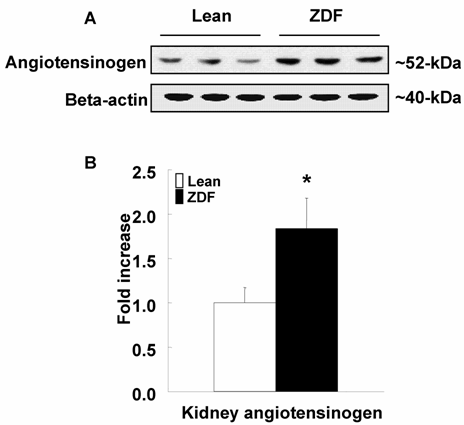
Kidney Angiotensinogen Protein
As shown in Figure 2, angiotensinogen protein expression was increased in ZDF rats at 17 weeks of age. No differences in the amount of beta-actin were observed among samples.
Kidney angiotensinogen levels in ZDF and control lean rats at 17 weeks of age. (A) A representative Western blot analysis of angiotensinogen and beta-actin. (B) The densitometric analysis shows that ZDF rats had significantly increased kidney angiotensinogen levels compared to control lean rats. *p<0.05 vs. control lean rats at the same age.
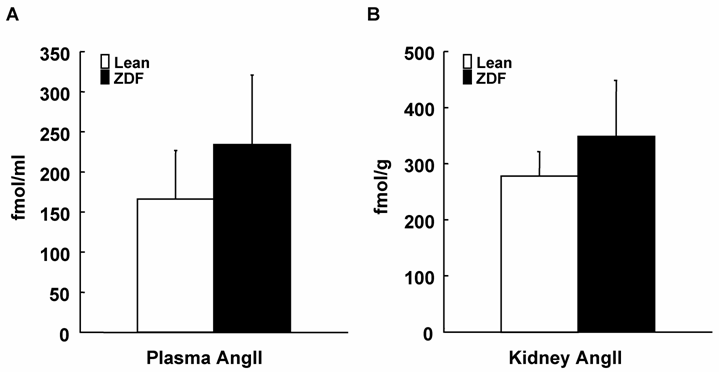
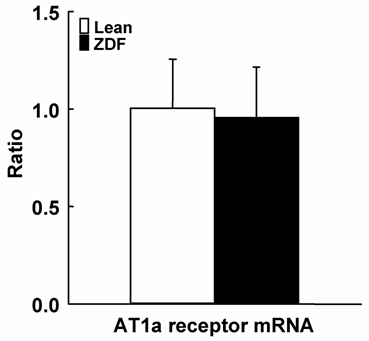
Plasma AngII, Kidney AngII and AT1a Receptor mRNA
As shown in Figure 3, ZDF rats showed slightly increased plasma AngII concentrations. Similar to plasma concentrations, intrarenal AngII contents were also slightly increased in ZDF rats. However, plasma AngII concentrations and kidney AngII contents were not significantly increased in ZDF rats. As shown in Figure 4, AT1a receptor mRNA was not different between ZDF and control lean rats.
Plasma (A) and kidney (B) AngII levels in ZDF and control lean rats at 17 weeks of age. Plasma AngII concentrations and kidney AngII contents were not significantly different between ZDF and control lean rats.
Kidney AT1a receptor mRNA in ZDF and control lean rats at 17 weeks of age. Kidney AT1a receptor mRNA in ZDF were not significantly different compared with control lean rats.
Renal Damage
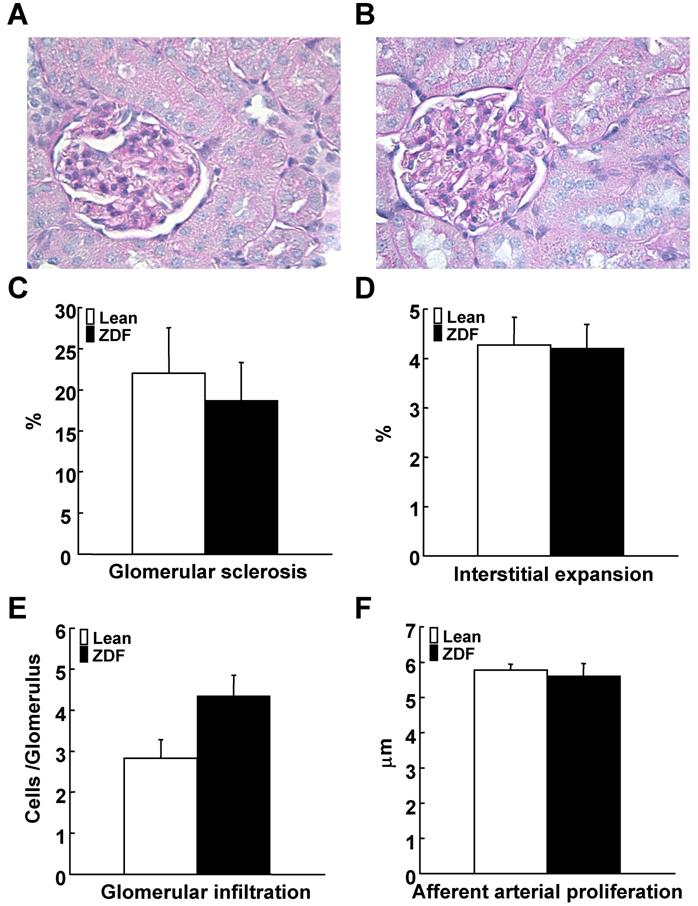
Glomerular sclerotic index were not different between ZDF and control lean rats (Figure 5A-5C). Interstitial expansive index showed similar levels between ZDF and control lean rats (Figure 5D). Glomerular macrophage infiltration was not significantly increased in ZDF rats (Figure 5E). Afferent arteriolar wall thickness was not altered in ZDF compared to control lean rats (Figure 5F).
Evaluation of renal damage in ZDF and control lean rats at 17 weeks of age. Renal damages were evaluated based on these four parameters: (A-C) glomerular sclerosis index using periodic acid-Schiff stained sections of ZDF (A) and control lean (B) rats, (D) interstitial expansion index using by Picro-Sirius red-stained sections, (E) glomerular macrophage
4. Discussion
In this study, emphasis was placed on evaluating early mediators of renal damage in ZDF rats at 17 weeks of age. Schafer et al. [29] showed that ZDF rats established diabetes at 17 weeks of age. Coimbra et al. [30] showed that ZDF rats developed focal and segmental sclerosis at 18 weeks of age, although severe hyperlipidemia, hyperglycemia, and obesity predated it. Hoshi et al. [25] reported that ZDF rats showed glomeruloscrerosis as early as 20 weeks of age. Chander et al. showed that ZDF rats developed focal and segmental sclerosis at 22 weeks of age [31]. Thus, there are clear data demonstrating that ZDF rats show renal injury starting at 18-20 weeks of age, and in the present study, ZDF rats at 17 weeks of age without nephropathy were examined whether augmented intrarenal angiotensinogen is involved in the development of renal injury. We observed that these ZDF rats show the angiotensinogen augmentation before the development of renal injury at 17 weeks of age in this study.
In the present study, we have investigated the possible involvement of the enhanced angiotensinogen in renal injury in ZDF rats. At 17 weeks of age, although ZDF rats were confirmed having high glucose concentration and urine protein, renal damage were not documented by glomerular sclerosis, interstitial expansion, glomerular macrophage infiltration or renal arterial proliferation. Importantly, the oxidative stress and intrarenal angiotensinogen were augmented at this time-point. Therefore, the augmentation of oxidative stress and intrarenal angiotensinogen are precedent to renal damage in ZDF rats.
Recent studies have provided evidence that AngII stimulates intracellular formation of ROS [35, 36]. AngII-induced ROS plays a pivotal role in several pathophysiological conditions such as hypertension, endothelial dysfunction, nitrate tolerance, atherosclerosis, and cellular remodeling [37]. Although plasma and kidney AngII levels were not significantly increased in ZDF rats, an oxidative stress marker, 8-isoprostane was significantly elevated in ZDF rats at 17 weeks of age. High glucose induces ROS generation via non-enzymatic glycation of proteins or glucose auto-oxidation [22]. Hyperglycemia, the main determinant of the initiation and progression of renal injury, not only generates more reactive oxygen metabolites, but also attenuates anti-oxidative mechanisms through non-enzymatic glycosylation of anti-oxidant enzymes [38]. It has been reported that 8-isoprostane is enhanced in the setting of diabetes in type I [39] and type II diabetic patients [40]. These studies support the concept that high glucose-induced ROS is involved in the setting of renal injury.
Changes in angiotensinogen levels can control the activity of the RAS, and its up-regulation may lead to elevated AngII levels and increases in blood pressure [41]. Our previous report suggested that stimulation of angiotensinogen synthesis by AngII may be a key component in the maintenance of elevated intrarenal AngII levels [17]. We also reported that angiotensinogen expression is elevated independently from blood pressure in AngII-dependent hypertension [20, 21]. Although plasma and kidney AngII levels were not significantly increased in the present study, kidney angiotensinogen protein was increased in ZDF rats. Systolic blood pressure in ZDF rats was also significantly higher than in control lean rats, but this elevation did not reach a hypertensive range. Interestingly, it is reported that angiotensinogen gene expression is induced by high glucose in proximal tubular cells [42]. In addition, Hsieh et al. [43] demonstrated that the stimulatory action of high glucose on angiotensinogen gene expression in immortalized renal proximal tubular cells is mediated at least in part via ROS generation. We also showed that augmentation of angiotensinogen is caused by ROS in hypertensive rats [24]. Our present data also demonstrate concomitant increases of ROS and angiotensinogen in ZDF rats compared with control lean rats before renal injury in ZDF rats was evident. Our present study thus suggests that ROS and ROS-mediated angiotensinogen activation may have an important role in early mediation of renal injury in ZDF rats.
We failed to demonstrate the enhanced levels of intrarenal AngII in ZDF rats even though the intrarenal AngII levels were maintained by the augmented levels of intrarenal angiotensinogen in ZDF rats. We did not measure plasma renin activity; however, it is well known that plasma renin activity is suppressed in both diabetic patients and animals [4-10]. The maintained levels of intrarenal AngII and the augmented levels of intrarenal angiotensinogen may partially account for the renoprotective effects of ARBs and ACE inhibitors in diabetic nephropathy even though the circulating RAS activity is suppressed in diabetes.
Our present study may imply that ZDF rats treated with antioxidant may demonstrate favorable effects on renal levels of angiotensinogen and AngII as well as urinary protein excretion; however, the emphasis of the present study was to report that intrarenal oxidative stress and augmented angiotensinogen are precedent to renal injury in ZDF rats. Practically, it is very difficult to use specific and effective antioxidants in vivo study. Further in vivo and/or in vitro study may be required to clarify these issues.
In summary, we have demonstrated in this study that the augmented oxidative stress and the enhanced intrarenal angiotensinogen precede the development of renal injury in ZDF rats. We previously reported that the ROS-related activation of intrarenal angiotensinogen plays important roles in the development of renal injury in salt sensitive hypertensive rats [24]. These data suggest that the augmented ROS and the ROS-associated activation of intrarenal angiotensinogen are involved in the development of renal injury. These mechanisms may partially account for the renoprotective effects of ARBs and ACE inhibitors in the diabetic nephropathy.
Acknowledgements
This study was supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (R01DK072408), the National Center for Research Resources (P20RR017659), the National Heart, Lung, and Blood Institute (R01HL026371), the Health Excellence Fund from Louisiana Board of Regents, and Sankyo Co. Ltd. (Tokyo, Japan). Y.S. is a recipient of a fellowship from the Kanae Foundation for Life and Socio-medical Science (Tokyo, Japan). The authors acknowledge critical reviews and valuable comments of L. Gabriel Navar, Ph.D. (Tulane University). The authors also acknowledge excellent technical assistances from My-Linh Rauv, Duy V. Tran, Dale M. Seth, and Mark A. Cabrera (Tulane University).
Conflict of interests
The authors have declared that no conflict of interest exists.
References
1. Joss N, Paterson KR, Deighan CJ, Simpson K, Boulton-Jones JM. Diabetic nephropathy: how effective is treatment in clinical practice? Qjm. 2002 ;95:41-9
2. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993 ;329:977-86
3. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998 ;352:837-53
4. Nagai Y, Yao L, Kobori H, Miyata K, Ozawa Y, Miyatake A, Yukimura T, Shokoji T, Kimura S, Kiyomoto H, Kohno M, Abe Y, Nishiyama A. Temporary angiotensin II blockade at the prediabetic stage attenuates the development of renal injury in type 2 diabetic rats. J Am Soc Nephrol. 2005 ;16:703-11
5. Carey RM, Siragy HM. The intrarenal renin-angiotensin system and diabetic nephropathy. Trends Endocrinol Metab. 2003 ;14:274-81
6. Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001 ;345:861-9
7. Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993 ;329:1456-62
8. Barnett AH, Bain SC, Bouter P, Karlberg B, Madsbad S, Jervell J, Mustonen J. Angiotensin-receptor blockade versus converting-enzyme inhibition in type 2 diabetes and nephropathy. N Engl J Med. 2004 ;351:1952-61
9. Luno J, Praga M, de Vinuesa SG. The reno-protective effect of the dual blockade of the renin angiotensin system (RAS). Curr Pharm Des. 2005 ;11:1291-300
10. Hollenberg NK. Renal implications of angiotensin receptor blockers. Am J Hypertens. 2001 ;14:237S-41S
11. Skeggs LT Jr, Kahn JR, Lentz K, Shumway NP. The preparation, purification, and amino acid sequence of a polypeptide renin substrate. J Exp Med. 1957 ;106:439-53
12. Dubey RK, Jackson EK, Luscher TF. Nitric oxide inhibits angiotensin II-induced migration of rat aortic smooth muscle cell. Role of cyclic-nucleotides and angiotensin1 receptors. J Clin Invest. 1995 ;96:141-9
13. Vaughan DE, Lazos SA, Tong K. Angiotensin II regulates the expression of plasminogen activator inhibitor-1 in cultured endothelial cells. A potential link between the renin-angiotensin system and thrombosis. J Clin Invest. 1995 ;95:995-1001
14. Hannken T, Schroeder R, Stahl RA, Wolf G. Angiotensin II-mediated expression of p27Kip1 and induction of cellular hypertrophy in renal tubular cells depend on the generation of oxygen radicals. Kidney Int. 1998 ;54:1923-33
15. Kawada N, Imai E, Karber A, Welch WJ, Wilcox CS. A mouse model of angiotensin II slow pressor response: role of oxidative stress. J Am Soc Nephrol. 2002 ;13:2860-8
16. Welch WJ, Mendonca M, Aslam S, Wilcox CS. Roles of oxidative stress and AT1 receptors in renal hemodynamics and oxygenation in the postclipped 2K,1C kidney. Hypertension. 2003 ;41:692-6
17. Kobori H, Harrison-Bernard LM, Navar LG. Expression of angiotensinogen mRNA and protein in angiotensin II-dependent hypertension. J Am Soc Nephrol. 2001 ;12:431-9
18. Kobori H, Harrison-Bernard LM, Navar LG. Enhancement of Angiotensinogen Expression in Angiotensin II-Dependent Hypertension. Hypertension. 2001 ;37:1329-1335
19. Kobori H, Harrison-Bernard LM, Navar LG. Urinary excretion of angiotensinogen reflects intrarenal angiotensinogen production. Kidney Int. 2002 ;61:579-85
20. Kobori H, Nishiyama A, Harrison-Bernard LM, Navar LG. Urinary angiotensinogen as an indicator of intrarenal Angiotensin status in hypertension. Hypertension. 2003 ;41:42-9
21. Kobori H, Prieto-Carrasquero MC, Ozawa Y, Navar LG. AT1 receptor mediated augmentation of intrarenal angiotensinogen in angiotensin II-dependent hypertension. Hypertension. 2004 ;43:1126-32
22. Ha H, Lee HB. Reactive oxygen species as glucose signaling molecules in mesangial cells cultured under high glucose. Kidney Int Suppl. 2000 ;77:S19-25
23. Liu J, Yang F, Yang XP, Jankowski M, Pagano PJ. NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy. Arterioscler Thromb Vasc Biol. 2003 ;23:776-82
24. Kobori H, Nishiyama A. Effects of tempol on renal angiotensinogen production in Dahl salt-sensitive rats. Biochem Biophys Res Commun. 2004 ;315:746-50
25. Hoshi S, Shu Y, Yoshida F, Inagaki T, Sonoda J, Watanabe T, Nomoto K, Nagata M. Podocyte injury promotes progressive nephropathy in zucker diabetic fatty rats. Lab Invest. 2002 ;82:25-35
26. Mizuno M, Sada T, Kato M, Koike H. Renoprotective effects of blockade of angiotensin II AT1 receptors in an animal model of type 2 diabetes. Hypertens Res. 2002 ;25:271-8
27. Vora JP, Zimsen SM, Houghton DC, Anderson S. Evolution of metabolic and renal changes in the ZDF/Drt-fa rat model of type II diabetes. J Am Soc Nephrol. 1996 ;7:113-7
28. Etgen GJ, Oldham BA. Profiling of Zucker diabetic fatty rats in their progression to the overt diabetic state. Metabolism. 2000 ;49:684-8
29. Schafer S, Linz W, Bube A, Gerl M, Huber J, Kurzel GU, Bleich M, Schmidts HL, Busch AE, Rutten H. Vasopeptidase inhibition prevents nephropathy in Zucker diabetic fatty rats. Cardiovasc Res. 2003 ;60:447-54
30. Coimbra TM, Janssen U, Grone HJ, Ostendorf T, Kunter U, Schmidt H, Brabant G, Floege J. Early events leading to renal injury in obese Zucker (fatty) rats with type II diabetes. Kidney Int. 2000 ;57:167-82
31. Chander PN, Gealekman O, Brodsky SV, Elitok S, Tojo A, Crabtree M, Gross SS, Goligorsky MS. Nephropathy in Zucker diabetic fat rat is associated with oxidative and nitrosative stress: prevention by chronic therapy with a peroxynitrite scavenger ebselen. J Am Soc Nephrol. 2004 ;15:2391-403
32. Kobori H, Ozawa Y, Suzaki Y, Nishiyama A. Enhanced intrarenal angiotensinogen contributes to early renal injury in spontaneously hypertensive rats. J Am Soc Nephrol. 2005 ;16:2073-80
33. Patrono C, FitzGerald GA. Isoprostanes: potential markers of oxidant stress in atherothrombotic disease. Arterioscler Thromb Vasc Biol. 1997 ;17:2309-15
34. Thomas WG, Sernia C. Immunocytochemical localization of angiotensinogen in the rat brain. Neuroscience. 1988 ;25:319-41
35. Onozato ML, Tojo A, Goto A, Fujita T, Wilcox CS. Oxidative stress and nitric oxide synthase in rat diabetic nephropathy: effects of ACEI and ARB. Kidney Int. 2002 ;61:186-94
36. Griendling KK, Ushio-Fukai M. Reactive oxygen species as mediators of angiotensin II signaling. Regul Pept. 2000 ;91:21-7
37. Wolf G. Free radical production and angiotensin. Curr Hypertens Rep. 2000 ;2:167-73
38. Wolff SP, Jiang ZY, Hunt JV. Protein glycation and oxidative stress in diabetes mellitus and ageing. Free Radic Biol Med. 1991 ;10:339-52
39. Davi G, Chiarelli F, Santilli F, Pomilio M, Vigneri S, Falco A, Basili S, Ciabattoni G, Patrono C. Enhanced lipid peroxidation and platelet activation in the early phase of type 1 diabetes mellitus: role of interleukin-6 and disease duration. Circulation. 2003 ;107:3199-203
40. Davi G, Ciabattoni G, Consoli A, Mezzetti A, Falco A, Santarone S, Pennese E, Vitacolonna E, Bucciarelli T, Costantini F, Capani F, Patrono C. In vivo formation of 8-iso-prostaglandin f2alpha and platelet activation in diabetes mellitus: effects of improved metabolic control and vitamin E supplementation. Circulation. 1999 ;99:224-9
41. Ichihara A, Kobori H, Nishiyama A, Navar LG. Renal renin-angiotensin system. Contrib Nephrol. 2004 ;143:117-30
42. Wang TT, Wu XH, Zhang SL, Chan JS. Effect of glucose on the expression of the angiotensinogen gene in opossum kidney cells. Kidney Int. 1998 ;53:312-9
43. Hsieh TJ, Fustier P, Wei CC, Zhang SL, Filep JG, Tang SS, Ingelfinger JR, Fantus IG, Hamet P, Chan JS. Reactive oxygen species blockade and action of insulin on expression of angiotensinogen gene in proximal tubular cells. J Endocrinol. 2004 ;183:535-50
Author contact
![]() Correspondence to: Hiroyuki Kobori, M.D., Ph.D., Director of the Molecular Core, Tulane Hypertension and Renal Center of Excellence, 1430 Tulane Avenue, #SL39, New Orleans, LA 70112-2699. Tel: +1-504-988-2591 Fax: +1-504-988-0911 E-mail: hkoboriedu
Correspondence to: Hiroyuki Kobori, M.D., Ph.D., Director of the Molecular Core, Tulane Hypertension and Renal Center of Excellence, 1430 Tulane Avenue, #SL39, New Orleans, LA 70112-2699. Tel: +1-504-988-2591 Fax: +1-504-988-0911 E-mail: hkoboriedu