Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(1):47-56. doi:10.7150/ijbs.3.47 This issue Cite
Research Paper
Distinct domain-dependent effect of syntaxin1A on amiloride-sensitive sodium channel (ENaC) currents in HT-29 colonic epithelial cells
Sunil K Saxena, Madhurima Singh, Simarna Kaur, Constantine George ![]()
Center for Cell and Molecular Biology, Department of Chemistry and Chemical Biology, Stevens Institute of Technology, Hoboken, NJ 07030, U.S.A.
Received 2006-8-16; Accepted 2006-10-30; Published 2006-11-6
Abstract
The amiloride-sensitive epithelial sodium channel (ENaC), a plasma membrane protein mediates sodium reabsorption in epithelial tissues, including the distal nephron and colon. Syntaxin1A, a trafficking protein of the t-SNARE family has been reported to inhibit ENaC in the Xenopus oocyte expression and artificial lipid bilayer systems. The present report describes the regulation of the epithelial sodium channel by syntaxin1A in a human cell line that is physiologically relevant as it expresses both components and also responds to aldosterone stimulation. In order to evaluate the physiological significance of syntaxin1A interaction with natively expressed ENaC, we over-expressed HT-29 with syntaxin1A constructs comprising various motifs. Unexpectedly, we observed the augmentation of amiloride-sensitive currents with wild-type syntaxin1A full-length construct (1-288) in this cell line. Both γENaC and neutralizing syntaxin1A antibodies blocked native expression as amiloride-sensitive sodium currents were inhibited while munc18-1 antibody reversed this effect. The coiled-coiled domain H3 (194-266) of syntaxin1A inhibited, however the inclusion of the transmembrane domain to this motif (194-288) augmented amiloride sensitive currents. More so, data suggest that ENaC interacts with multiple syntaxin1A domains, which differentially regulate channel function. This functional modulation is the consequence of the physical enhancement of ENaC at the cell surface in cells over-expressed with syntaxin(s). Our data further suggest that syntaxin1A up-regulates ENaC function by multiple mechanisms that include PKA, PLC, PI3 and MAP Kinase (p42/44) signaling systems. We propose that syntaxin1A possesses distinct inhibitory and stimulatory domains that interact with ENaC subunits, which critically determines the overall ENaC functionality/regulation under distinct physiological conditions.
Keywords: ENaC, Syntaxin1A, HT-29 colonic epithelial cells
1. Introduction
The regulation of sodium reabsorption and excretion in the kidney is central to the control of blood pressure and extra-cellular fluid volume. Regulation is chiefly mediated by the adrenal mineralocorticoid hormone, aldosterone, which effects the activity of the sodium channel. The molecular target for this regulation is the amiloride-sensitive epithelial sodium channel, a heteromultimer consisting of α, β and γ subunits 1. In aldosterone targeted epithelia, this amiloride-sensitive epithelial sodium channel (ENaC) represents the rate limiting step for sodium reabsorption 2, 3. Mutations in the human genes of β and γ subunits of ENaC cause a form of salt-sensitive hypertension described more effectively as Liddle's syndrome 4-7. These mutations either introduce frame-shifts, premature stop codons, or truncations 8, 9.
Several mechanisms are known to influence and tightly regulate Na+ entry through the apical membrane of epithelial cells. These mechanisms include changes in protein expression and the relative distribution of ENaC protein between intracellular vesicular pools and the plasma membrane 10, 11. Liddle's syndrome mutations lead to both: an increase in channel density at the cell surface and an increase in open channel probability 12 . Another mechanism described for ENaC regulation is through a group of proteins that could directly interact with ENaC and regulate its function. These are syntaxins, plasma membrane localized t-SNARES that are hypothesized to mediate vesicle trafficking. We 13 and others 14-16 have shown that SNAREs like syntaxin1A interact with and functionally regulate the amiloride-sensitive sodium channel (ENaC) in the Xenopus oocyte expression system and artificial lipid bilayer 17. Syntaxin1A also reportedly inhibits several other ion channels including Ca+2 channels 18, 19, CFTR chloride channels 20, 21, K+ channels 22 and GABA transporters 23 in the Xenopus oocyte expression system.
All these studies were performed in a system that lacks either ENaC or syntaxin de novo expression. In this communication, we are reporting that syntaxin1A over-expression up-regulates ENaC function in HT-29 colonic epithelial cells that natively express both ENaC 24, 25 and syntaxin1A. This up-regulation is the net result of the differential effect of multiple syntaxin motifs that involves a complex regulatory mechanism.
2. Materials and Methods
Materials and reagents
HT-29 cells were purchased from American type culture collection (ATCC), (Manassas, VA). Syntaxin1A constructs were a kind gift from Dr. Kevin Kirk, University of Alabama at Birmingham, (Birmingham, AL). ΔC syntaxin1A and Munc constructs were kindly donated by Dr. A. Naren, University of Tennessee at Memphis, (Memphis, TN). Munc18 antibodies were procured from Transduction Laboratories, (Lexington, KY). Horseradish peroxidase–conjugated (HRP-conjugated) secondary antibodies (anti-rabbit and anti-mouse) were from Pierce Chemical Co., (Rockford, IL). Goat anti-mouse IgG-Alexa 488 and Alexa 594 were purchased from Molecular Probes, (Eugene, OR). The anti-syntaxin1A monoclonal antibody (HPC-1), and other antibodies and reagents were obtained from Sigma Chemicals, (St. Louis, MO). In order to confirm our findings, ENaC antibodies available from various sources were used from time to time. The ENaC antibodies were raised by Research Genetics, (Huntsville, AL) and Genemed Synthesis, (San Francisco, CA). Alternatively, ENaC antibodies from other sources were also used to confirm the findings. αENaC antibody was a kind gift from Dr. Peter Smith, Department of Physiology and Biophysics, University of Alabama at Birmingham, (Birmingham, AL). The ENaC subunit specific antibodies were a generous gift by Dr. Bernard Rossier, University of Lausanne, (Lausanne, Switzerland). Lipofectamine was purchased from Invitrogen Life Technologies, (Carlsbad, CA). Chariot® protein delivery system was available from Active Motif, (Carlsbad, CA). RIPA buffer contained 50 mM Tris-Cl pH 7.4, 1% Triton X-100, 0.2% Sodium deoxycholate, and 0.2% Sodium dodecyl sulfate (SDS) with protease inhibitor.
Cell line
HT-29 cells were cultured in McCoy's 5a medium with 1.5 mM L-glutamine and 10% fetal bovine serum in 5% CO2 at 37°C. The cells were grown on Falcon 12 or 24 well inserts for all experiments and maintained to determine the amiloride-sensitive component of the Isc 26, 27.
Measurements of short circuit currents (Isc)
Amiloride-sensitive currents were recorded two-ways. The Isc were recorded with EVOMTM epithelial voltohmeter using STX2 electrode World Precision Instruments, (Sarasota, FL) as described before 24. Alternatively, some of the measurements were made in a modified Ussing chamber (Trans-24 mini perfusion chamber), Warner Instruments, (Hamden, CT). Apical and basolateral chambers were continuously bathed with medium and the Isc were measured with transepithelial voltage clamped at 0 mV with a DVC-1000 dual voltage clamp. Voltage pulses (10 mV) were applied every 3 min to monitor the transepithelial resistance. After the initial measurements, 10 µM amiloride were added to the apical side, and sodium currents expressed as the amiloride-sensitive component of the Isc.
Plasmids and transfection
Fusion proteins were made as described 28. Oligonucleotide-directed mutagenesis was used to produce truncations and deletions. All the constructs were confirmed by nucleotide sequencing. Abbreviations and the description of syntaxin1A constructs: syn1A-TMD, full length syntaxin, amino acid (aa) 1-288; syn1A∆c, syntaxin1A construct lacking the transmembrane domain, aa 1-266, syn1AH3-TMD, the H3 domain of syntaxin1A including TMD (aa 188-288); syn1AH3, the H3 domain of syntaxin1A (aa 188-266); syn1A∆H3-TMD, truncated syntaxin1A lacking aa 188-266 (aa 1-194); syntaxin1A∆C, syntaxin1A lacking TMD (aa 1-266). The cells were transfected with plasmid DNA constructs in lipofectamine according to the manufacturer's protocol. The expression of each protein was confirmed by SDS-PAGE, Western blot analysis, and detection of the protein with concomitant antibody.
Bacterially expressed fusion proteins
GST-fusion proteins were produced in Escherichia coli DH5a. RIPA cell lysates were centrifuged at 6000 rpm for 30 min, and the clarified supernatant was mixed with glutathione-sepharose beads and rocked overnight at 4 °C. The beads were then washed three times with wash buffer (150 mM NaCl, 25 mM Tris-HCl, pH 7.5, 1 mM dithiothreitol) supplemented with the protease inhibitors leupeptin (10 µg/mL), aprotinin (1% v/v final concentration), and phenylmethylsulfonyl fluoride (1 mM final concentration) before use. The protein bound to the beads was then used for coprecipitation experiments. For activity assays and immunoprecipitation, the fusion proteins were eluted with 25 mM glutathione solution in 20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, pH 8.0, and 0.5% Nonidet P-40 (NET-N) supplemented with these same protease inhibitors. For Chariot® experiments, the GST- was released by thrombin cleavage and dialysis against PBS wash buffer. Both bound and eluted proteins were quantitated by comparing their band intensities with those of known amounts of bovine serum albumin on silver-stained polyacrylamide gels.
Cell surface biotinylation, intracellular pool and ENaC detection
The biotinylation of cell surface proteins was performed using the kit and according to the protocol described by the manufacturer Pierce Biotechnology Inc., (Rockford, IL). In short, the proteins in the intact cells were surface-labeled with cell impermeant Sulpho-NHS-SS-biotin (0.5 mg/mL) at 4°C for 30 min. After washing three times with ice-cold quenching buffer, the cells were solubilized on ice in the presence of protease inhibitors. Surface, biotinylated proteins were adsorbed on 50% streptavidin–agarose bead slurry by rotating for 2 hr at 4°C. After brief centrifugation, supernatants representing the intracellular pool were collected and processed accordingly. Proteins were quantified by BCA method, separated by SDS–PAGE, and transferred to nitrocellulose for immunoblotting. The blots were analyzed for ENaC expression by Western blot analysis using subunit specific ENaC antibodies. The blots were raised using ECL and the films were developed using autoradiography.
Electrophoresis, immunoblotting and characterization of proteins
The proteins were solubilized at 70ºC for 15 min in Laemelli sample buffer and run through SDS-PAGE in 10% polyacrylamide gels. The proteins were transferred electrophoretically to the PVDF membrane in Towbin buffer. After blocking with 5% non-fat dry milk in TBS-Tween (Tris-buffered saline, 0.05% Tween-20, pH 7.4), the membranes were probed with polyclonal or mono-specific affinity purified anti-peptide antibodies. Blots were developed using enhanced chemiluminescence (ECL) and visualized by light-sensitive imaging film (Kodak). Quantification was carried out with densitometry.
Pull down assay
HT-29 cells were lysed in RIPA buffer and pooled lysates from 12 or 24 well plates were mixed at 4°C for 15 min on a rotary mixer. The lysates were centrifuged at 15,000xg for 10 min at 4°C. GST and GST-syntaxin1AΔC were added to clear supernatant and incubated for 30-60 min on a rotary shaker, after which glutathione sepharose beads (20 μL of 50% slurry in lysis buffer) were added and continue to mix at 4°C for 3 hr. At the end of 3 hr, beads were spun at 800xg for 2 min and the supernatant was discarded. The beads were washed 3 times with lysis buffer and the proteins were eluted with 20 μL of 5X sample buffer for 30 min at 37°C and subjected to Western blotting.
Delivery of antibodies
The Chariot® protein delivery system was utilized to target antibodies in HT-29 cells. Antibodies were complexed with the Chariot® reagent at a ratio of 1 ng IgG: 2 μL Chariot® in 100 μL PBS for 30 min. These IgG: Chariot® complexes were overlaid onto cultured cells in the presence of fresh serum free culture medium for 3 hr and amiloride-sensitive currents were recorded.
Inhibitor studies
The 60-70% confluent HT-29 cells were transiently expressed with syntaxin1A. Forty-eight hours later, the cells were exposed to different inhibitors. The following inhibitors (final conc. in parentheses) PI3 kinase inhibitor LY294002 (50 μM); MAP kinase inhibitor PD98059 (50 μM); protein kinase A inhibitor RpCPTcAMP (30 μM); phospholipase C (PLC) inhibitor U73122 (50 nM); and protein kinase C (PKC) activator phorbol myristate acetate PMA (200 nM) were diluted into protein-free medium just prior to use. Inhibition or activation in intact cells was measured by incubating HT-29 cells with the indicated concentration of reagents or dimethyl sulfoxide carrier (0.1%, v/v) for 45 min at 37°C. The amiloride-sensitive currents were recorded as described before.
Statistical analysis
A paired test or analysis of variance for multiple comparisons was used for statistical analysis. A p value less than 0.05 was considered significant.
3. Results
Effect of syntaxin1A on amiloride-sensitive currents
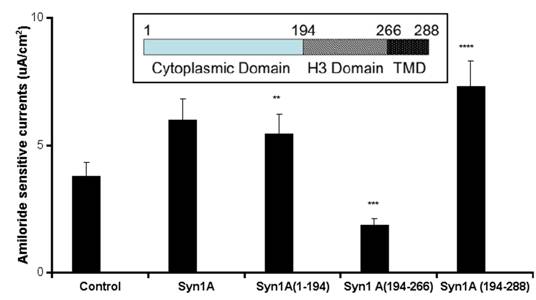
To test the hypothesis that syntaxin1A functionally modulates ENaC activity, amiloride-sensitive currents were recorded in HT-29 cells over-expressing syntaxin1A constructs comprising of specific domains (Fig 1). We observed that basal currents were augmented by the expression of full-length syntaxin1A in HT-29 cells, though the truncated constructs enumerate differential effect on amiloride-sensitive currents. For example, a modest stimulation was recorded with the syntaxin construct having the H1-H2 domains (1-194), while the expression of cytosolic H3 domain (194-266) inhibited amiloride-sensitive basal currents. Surprisingly, the inclusion of the transmembrane domain (TMD) to the H3 motif (194-288) augmented currents. These data suggest that expression of both H1-H2 domains (1-194) or H3-TMD (194-288) stimulate basal amiloride-sensitive currents and the enhanced currents observed with full length syntaxin1A expression might represent the cumulative effect of H1-H2 with H3-TMD combined. Interestingly, the H3 domain, which has dominant coiled-coiled domain and the TMD are reported to impart an inhibitory effect on ENaC function in Xenopus oocytes 16 and artificial-lipid bilayer system 17.
Syntaxin1A augments currents - HT-29 cells grown on cell inserts were transfected with wild-type syntaxin1A (Syn1A) and its truncated constructs. Inset- Structure of syntaxin1A. TMD represents the transmembrane domain. 48 hours later the amiloride-sensitive currents were recorded as described in the text. The data represents a mean of five individual experiments. Experimental conditions that resulted in a significant change (p < 0.05) from the relevant control values (Bar one) are denoted by multiple asterisks. Three asterisks denote higher statistical significance.
Dose-dependent stimulation of amiloride-sensitive currents
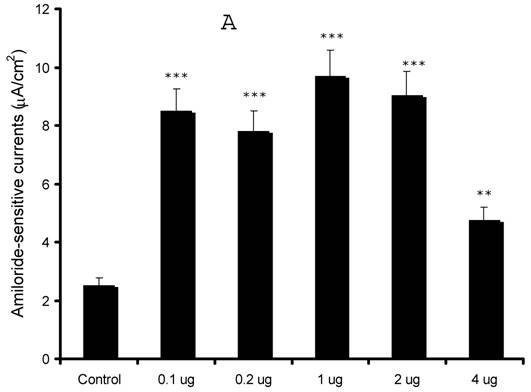
Our preliminary observations were contrary to the expected results as our initial results in the oocyte expression system indicated an inhibition of sodium currents 13. In order to confirm our findings, we over-expressed syntaxin1A at different concentrations (0.1 - 4.0 μg) and recorded amiloride-sensitive currents. As evident from (Fig 2), we observed an augmentation of amiloride-sensitive currents in all doses from 0.1 to 4.0 μg/well. Surprisingly, the stimulation was least with the highest dose (4.0 μg) used in the study, suggesting that higher expression might down-regulate channel function as a consequence of overdose.
Syntaxin1A up-regulates amiloride-sensitive currents in a dose-dependent manner - HT-29 cells grown on cell inserts were transfected with wild-type syntaxin1A (Syn1A) at different concentrations. 48 hours later amiloride-sensitive currents were recorded (A) as described in the text. (B) The protein extracts from the transfected cells were solubilized and analyzed by Western blot analysis. The blots were developed with syntaxin1A antibody. The data represents a mean of three individual experiments. Experimental conditions that resulted in a significant change (p < 0.05) from the relevant control values (Bar one) are denoted by multiple asterisks. Three asterisks denote higher statistical significance.

Effect of antibody delivery in the permeabilized cells
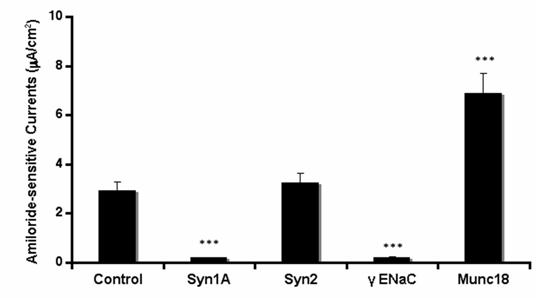
In order to further substantiate our observations, we introduced syntaxin1A monoclonal antibody by permeabilizing the cells with the Chariot® delivery system and recorded amiloride-sensitive currents. HT-29 cells natively express the proteins against which neutralizing antibodies were introduced in these cells (data not shown). As indicated in (Fig 3), the ENaC activity was completely abolished by the introduction of syntaxin1A and not with syntaxin2. Moreover, the complete elimination of sodium currents in the presence of γENaC antibody suggests that amiloride-sensitive currents are the consequence of ENaC function in these cells. Moreover, the syntaxin2 antibody failed to impart any effect on amiloride-sensitive currents while the antibody against syntaxin1A binding partner munc18-1 produced up-regulation of ENaC currents suggesting that the effect of syntaxin1A is specific and also stimulatory in HT-29 colonic epithelial cells. This observation is further supported by the introduction of munc18-1 antibody in the cells.
Syntaxin1A antibody inhibits amiloride-sensitive currents - The HT-29 cells were targeted with anti-syntaxin, or anti-ENaC antibodies or irrelevant IgG using the Chariot® protein delivery system. Antibodies were complexed with the Chariot® reagent at a ratio of 1 ng IgG: 2 μL Chariot® in 100 μL PBS for 30 min. Then the IgG: Chariot® complexes were overlaid onto cultured cells in the presence of fresh serum free culture medium for 3 hr and amiloride-sensitive currents were measured. Data represent a mean of three individual experiments. Experimental conditions that resulted in a significant change (p < 0.05) from the relevant control values (Bar one) are denoted by multiple asterisks.
Pull down of ENaC with syntaxin1A
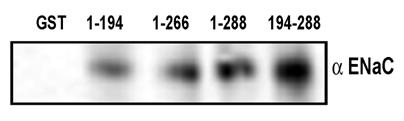
In order to understand if the functional result in the form of amiloride-sensitive currents is corollary to the physical association between ENaC and syntaxin under physiologically defined HT-29 cells, we performed co-immunoprecipitation studies in HT-29 cell lysates and also utilized pull down assay with GST-syntaxins as described in the experimental procedures (Fig 4). As expected, we observed that multiple syntaxin1A domains including H1-H3 interact with ENaC. As expected, the maximum binding was observed with the H3 domain of syntaxin1A. These observations lend support to the proposal that syntaxin1A, a protein with multiple coiled-coiled and binding domains, is capable of interacting with other proteins with variable affinity but with variable efficacy.
ENaC interacts with different syntaxin1A domains - Endogenous ENaC interacts with syntaxin in HT-29 cells - GST-syntaxin proteins were used to pull down proteins bound to natively expressed ENaC in HT-29 cell lysates. The immune complex was adsorbed on glutathione beads and then separated by SDS-Polyacrylamide gel electrophoresis and transferred to PVDF membrane. The blots were probed with affinity purified subunit specific γENaC antibody. The data shows interaction of multiple syntaxin1A domains with ENaC and points to its involvement and physiological significance in the regulation of the amiloride-sensitive epithelial sodium channel in native cells.
Biotinylation studies
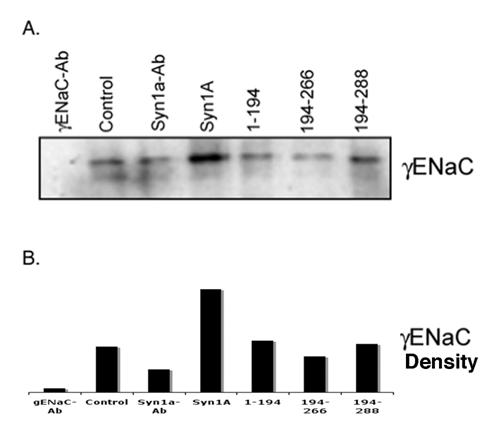
Our physical and functional observations implied that the modulation of amiloride-sensitive currents might be a consequence of net ENaC expression on the plasma membrane. In order to explore this possibility, we performed biotinylation studies on HT-29 cells transiently expressing syntaxin1A (Fig 5). We observed increased ENaC density at the plasma membrane in cells expressing full-length syntaxin1A. In addition to this, biotinylation experiments with multiple syntaxin constructs point to the moderate enhancement of αENaC expression in cells transfected with H3-TMD. However, ENaC density decreased considerably at the cell surface in cells expressed with H3 domain (Fig 5), otherwise it remained unaltered in other conditions.
Syntaxin1A domains modulate ENaC expression at the cell surface - HT-29 cells were transfected with wild-type syntaxin1A and its truncated constructs. Cell surface proteins were biotinylated with Sulpho-NHS-SS-biotin, pulled down with streptavidin-agarose separated by SDS-PAGE and transferred to PVDF membrane. The blots were probed with γENaC antibody (A). The protein was analyzed by densitometry (B). The data reflect increased expression of ENaC in HT-29 cells transfected with syntaxin1A, which is reflected in enhanced amiloride-sensitive currents reported in the text. Data represent three individual experiments each performed with different batches of HT-29 cells.
Intracellular (cytosolic) pool
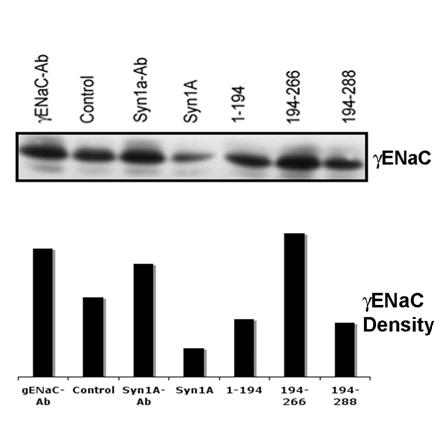
In order to further confirm that the changes in the apical expression of ENaC coincide with the decrease/increase of internal or cytosolic ENaC pools, we used the unbiotinylated pool as internal ENaC proteins since it could not get exposed on the cell surface29. The analysis of the internal pool by Western blotting using γENaC antibody (Fig 6) suggested that the cytosolic ENaC concentration(s) follows the pattern opposite to the ENaC expression at the plasma membrane. For example, we observed lower γENaC in the cytosolic pool as compared to the increased apical expression in the syntaxin1A transfected cells. This trend appears to continue in all the conditions tested (Fig 6) and when analyzed in conjunction with our data (Fig 5) suggests that the ENaC activity depends largely on the expressed channel at the plasma membrane.
Syntaxin1A domains modulate internal (cytosolic pool) of ENaC - HT-29 cells were transfected with wild-type syntaxin1A and its truncated constructs. Cell surface proteins biotinylated with Sulpho-NHS-SS-biotin were pulled out with streptavidin-agarose while the supernatant of the cell extract was separated by SDS-PAGE and transferred to PVDF membrane. The blots were probed with γENaC antibody (A). The protein was analyzed by densitometry (B). Data represent three individual experiments each performed with different batches of HT-29 cells.
Inhibitor studies
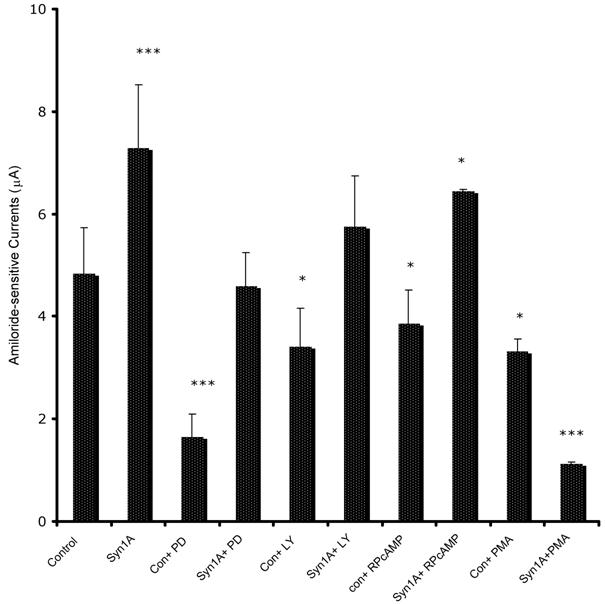
In order to define the molecular mechanism by which syntaxin1A stimulates amiloride-sensitive currents, we incubated HT-29 cells with different kinase inhibitors and recorded ENaC mediated currents (Fig 7). The inhibitors of PKA (RpCPTcAMP), PI3 kinase (LY294002) and MAP kinase (42/44 kD) (PD98059), all inhibited basal amiloride-sensitive currents, which could be reversed if the cells are transfected with syntaxin1A. These observations point to the complex regulatory mechanism by which syntaxin1A augments amiloride-sensitive ENaC function. We provide data suggesting an important role for cAMP, PI3 kinase and MAP kinase signaling in syntaxin1A dependent exocytosis of ENaC in HT-29 cells.
Syntaxin1A stimulates amiloride-sensitive channel (ENaC) activity by multiple signaling modules - HT-29 cells (60-70% confluent) on cell inserts were transfected with wild-type syntaxin1A (Syn1A) and incubated in the presence of indicated inhibitors; PD98059 (PD; 50 μM) or LY294002 (LY; 50 μM), RpCPTcAMP (RpcAMP; 30 μM) and PMA (200 nM) 48 hours later. The inhibition or activation in intact cells was measured with the indicated concentration of reagents or dimethyl sulfoxide carrier (0.1%, v/v) for 45 min at 37°C. The amiloride-sensitive currents were recorded as described in the text. The data represents a mean of three individual experiments. Experimental conditions that resulted in a significant change (p < 0.05) from the relevant control values (Bar one) are denoted by multiple asterisks. Three asterisks denote higher statistical significance.
4. Discussion
Syntaxin1A has been widely reported as an inhibitor of several ion channels, including ENaC. SNARE proteins are involved in the vesicle trafficking in both polarized and unpolarized systems 30, 31. Therefore, the inhibition of channel function by syntaxin, a t-SNARE, is puzzling. The reported inhibition of ENaC is based on observations in the cell systems which lack endogenous syntaxin1A expression and possibly other members of the SNARE family like VAMP and SNAP-23, which might be required to successfully assemble the SNARE complex with channel proteins into a proper assembly for delivery to the plasma membrane 32, 33. Additionally, all these experiments were performed with over-expressed syntaxin1A that might limit the availability of individual SNARE members. Thus, it is necessary to examine the role of syntaxin in a proper perspective. The information is currently unavailable in the cells, which express both ENaC and syntaxin1A. ENaC expression is well reported in both the proximal and distal colon 34-37 and HT-29 cells38. Our studies indicate that the colonic epithelial cell line HT-29 is an ideal cell system, which expresses the amiloride-sensitive epithelial sodium channel, responds to aldosterone stimulation 25 and natively expresses syntaxin1A (data not shown). Our lab has initially reported the inhibition of ENaC by syntaxin1A in the heterologous expression system of Xenopus oocytes 38 which lacks indigenous syntaxin1A. However, our experience with HT-29 colonic epithelial cells indicates that in a physiologically relevant cell system this trafficking protein acts otherwise, since in this cell line both ENaC and syntaxin1A are natively expressed. The major findings of our studies are (a) Syntaxin1A stimulates sodium channel activity; (b) ENaC interaction with syntaxin1A is not restricted to only the H3 domain; (c) Increased channel activity is a consequence of enhanced ENaC expression on the plasma membrane and (d) Syntaxin1A effect was dependent on multiple regulatory mechanisms comprising PKA, PI3 and MAP kinase. These data support the hypothesis that syntaxin1A is associated with ENaC trafficking under physiologically defined conditions.
In order to explore the effect of syntaxin1A on ENaC in HT-29 cells, we over-expressed syntaxin1A and its major cytosolic domains and recorded amiloride-sensitive currents (Fig 1). We recorded considerable currents (2-3 μA/cm2) that could be stimulated by aldosterone (>2.5 fold) (not shown) as reported in other cell systems like A6 39, 40, MDCK 41 and mpkCCDc14 42. Over-expression of syntaxin1A produced from 50% to 100% stimulation of amiloride-sensitive currents, while the expression of H3 domain showed the inhibition as reported in the Xenopus oocyte system 16. However, inclusion of the transmembrane domain reversed the inhibitory effect observed otherwise with the H3 domain. These observations support the notion that syntaxin1A stimulates ENaC activity in HT-29 cells, since the transmembrane domain is reportedly required for the inhibitory effect of syntaxin1A in ENaC 16 and other ion channels 43-45. Additionally, our data support that over-expression of syntaxin1A construct that lacks H3-TMD domains (1-194) had a slight stimulatory effect on amiloride-sensitive currents (30-40%). Our data further suggest (Fig 1) that H1-H2 (1-194) further also augments currents. Thus, our data establishes that syntaxin1A up-regulates basal ENaC function by a mechanism that requires the presence of transmembrane domain, since this motif is required in attaching the syntaxin to the plasma membrane 18, 46. We further confirmed that syntaxin1A over-expression augments ENaC function by performing the dose-dependent expression (Fig 2). Though these observations need to be examined carefully in the context of over-expressed proteins competing with endogenous syntaxin1A expression, these observations establish a definitive role for this t-SNARE in ENaC regulation in the epithelial cell system.
The fact that syntaxin1A stimulates ENaC in HT-29 cells was further confirmed by introducing neutralizing antibodies in the cells (Fig 3). We used the Chariot® delivery system 47 to introduce the antibodies in growing cells and recorded the complete inhibition of amiloride-sensitive currents using the syntaxin1A monoclonal antibody. A similar observation was made for the affinity purified γENaC subunit specific antibody. Anti-syntaxin2 as expected served as the control and showed no discernible effect on amiloride-sensitive currents. Since we neutralized natively expressed proteins, the data further support that syntaxin1A up-regulates ENaC in HT-29 colonic epithelial cells. Consistent with our observation, anti-munc18-1 augmented ENaC currents further supporting our data that syntaxin1A stimulates amiloride-sensitive currents in HT-29 cells. It should be noted that munc18-1, due to its high affinity binding 48, 49 for syntaxin1A reverses its regulatory effects 50 in a variety of cell systems.
In order to establish the physical protein-protein interaction between syntaxin1A and ENaC, we utilized in vitro pull down assay by incubating each purified ENaC subunit with GST-syntaxin1A. To test for a direct physical interaction between ENaC and syntaxin1A, we expressed major cytosolic domains of syntaxin1A (Fig 4). These fusion proteins were purified and tested for binding to the recombinant cytosolic domain of syntaxin1A (syntaxin1AΔC, where C refers to deletion of the C-terminal membrane anchor). We tested the functional relevance of the binding of syntaxin1A to ENaC in two ways: (i) by introducing reagents into cell by the Chariot® delivery system 51, 52 for performing neutralization experiments and (ii) introducing a peptide for performing blocking experiments. In the peptide blocking experiments, we observed that ENaC is acutely inhibited by syntaxin1A by introducing soluble GST-syntaxin fusion proteins (Data not shown). However, in HT-29 cells, introduction of γENaC acutely blocked the stimulatory effect of syntaxin1A over-expression suggesting that both proteins interact with each other confirming results published by other groups 16, 17.
In order to evaluate the domain-dependent modulation of ENaC at the protein level, we utilized various GST-syntaxin1A proteins to pull down ENaC from HT-29 RIPA lysates. The interaction between the H3 domain and ENaC has previously been documented 16, 17. GST-proteins on glutathione beads were solubilized and analyzed by Western blot analysis. The detection of blots with anti-αENaC revealed that besides H3 (194-266) or H3-TMD (194-288), the H1-H2 (1-194) domain of syntaxin1A also interacts with the epithelial sodium channel (Fig 4). More so, the binding affinity was low for 1-194 (H1-H2 domain) than 194-266 (H3) or 194-288 (H3-TMD) suggesting that the interaction between ENaC and syntaxin1A is complex and at multiple levels. We, however, at this time do not have any evidence to support the direct protein-protein interactions between ENaC and syntaxin1A.
These observations led us to believe that the augmentation of ENaC currents might be related to increased apical expression of the channel in the cells over-expressed with syntaxin1A. This is quite logical considering the confirmed role of syntaxin in exocytosis and fusion 53. In order to accomplish this objective, we initiated two sets of experiments, which could detect changes in apical expression. We first used cell impermeant Sulpho-NHS-SS-biotin to label cell surface proteins and then pull down biotinylated proteins with streptavidin-agarose (Pierce). The bound proteins were solubilized in SDS-buffer, separated by electrophoresis and transferred to PVDF membrane. The blots were probed with α or γENaC subunit specific antibody (Fig 5). In continuity with our previous observations, we detected increased ENaC density or enhanced apical expression of channel protein in transfected cells compared to control or untransfected cells. At the same time we detected lower internal or cytosolic ENaC pools from the similar set of conditions outlined above (Fig 6). Likewise, the immunofluorescence and confocal microscopic observations (data not shown) supported the increased expression of ENaC in syntaxin1A transfected HT-29 cells. However, it is possible that the molecular mechanism of the syntaxin1A effect is more complex than anticipated. Our efforts to delineate a definite mechanism supported the participation of several key signaling molecules (Fig 7) and thus suggest a complicated scenario. It is beyond the scope of this study to describe a detailed molecular mechanism of the syntaxin1A effect. Yet, the mechanism is diverse and may also include changes in open channel probability or gating 54-56. Previous studies have supported aldosterone induced translocation of αENaC in rat kidney, 57, 58 and other cell lines 59.
We have provided evidence for an interaction between the multiple domains of syntaxin1A, a possibility proposed for the CFTR-syntaxin model 43. The biotinylation observations indicate the enhanced expression of ENaC in HT-29 cells over-expressed with syntaxin1A constructs that augment amiloride-sensitive currents. The differential effect of syntaxin1A in the heterologous expression system and in the cells endogenously expressing syntaxin1A on ENaC channel regulation is not mutually exclusive. Our data further point to a possibility that the syntaxin1A expression leads to the redistribution of the channel protein. In T84 colonic epithelial cells, which has a considerable syntaxin1A expression, the inclusion of syntaxin1AΔC stimulates CFTR currents 50. Our data suggest that the role of syntaxin in the epithelial cell system is in line with its expected function, which is essentially trafficking. Since, this cell line expresses all the components of SNARE machinery, syntaxin1A effect on ENaC function is stimulatory and supports its primary function in exocytosis 30, 60. Our results also raise the possibility that the over-expression of syntaxin might result in its binding with other molecules (e.g. CFTR and other ion channels) 18, 19, 22, 61, 62 and may limit the availability of other SNARE molecules for participation in membrane fusion reactions in certain cell types and intracellular compartments 63, 64. SNAP-25 and syntaxin1A independently inhibits the L-type Ca+2 channel. However, when both are present, they actually exhibit stimulatory effects on this calcium channel 65. This observation is indicative of the positive regulation conferred on the ion channel by a multi-SNARE protein complex. We believe that syntaxin1A possess distinct inhibitory and stimulatory domains that interact with and regulate ENaC function. The inclusion of the membrane binding or transmembrane domain (TMD) reverses the effect of the H3 domain since TMD is involved in hooking up the channel proteins to the plasma membrane which appears to effect (increase) the channel expression at the cell surface. This increase is reflected in the enhanced channel function. A recent report with H+ATPase suggests that the H3 portion itself has three distinct motifs which characteristically binds to H (+)-ATPase (aa 235-264), and SNAP-23 and VAMP (aa 190-234) to an equivalent degree as full-length syntaxin 66. Additionally, we propose syntaxin1A as an important constituent of ENaC function associated with aldosterone stimulation. It would be interesting to know if syntaxin1A physically and functionally interacts with disease-associated mutants of ENaC. Whether or not syntaxin1A also limits the function of wild- type in renal cortical collecting ducts remains to be determined. If so, maneuvers that could potentially neutralize the modulatory effect of syntaxin1A on ENaC will be of great therapeutic interest.
Acknowledgements
Authors are thankful to Dr. Kevin Kirk, University of Alabama at Birmingham, Birmingham, AL and Dr. A. Naren, University of Tennessee at Memphis, Memphis, TN for the generous gift of syntaxin1A constructs. Authors wish to acknowledge the technical assistantship of Rina Shah, Danielle DeLuca and Natalia Barszcz during the course of study. Authors also appreciate National Institutes of Health for the Grant DK-57717 awarded to SKS.
Conflict of interest
The author has declared that no conflict of interest exists.
References
1. Canessa CM, Horisberger JD, Rossier BC. Epithelial sodium channel related to proteins involved in neurodegeneration. Nature. 1993 ;361:467-70
2. Rossier BC, Canessa CM, Schild L, Horisberger JD. Epithelial sodium channels. Curr Opin Nephrol Hypertens. 1994 ;3:487-96
3. Rossier BC. The epithelial sodium channel (ENaC): new insights into ENaC gating. Pflugers Arch. 2003 ;446:314-6
4. Auberson M, Hoffmann-Pochon N, Vandewalle A, Kellenberger S, Schild L. Epithelial Na+ channel mutants causing Liddle's syndrome retain ability to respond to aldosterone and vasopressin. Am J Physiol Renal Physiol. 2003 ;285:F459-71
5. Kamide K, Tanaka C, Takiuchi S, Miwa Y, Yoshii M, Horio T, Kawano Y, Miyata T. Six missense mutations of the epithelial sodium channel beta and gamma subunits in Japanese hypertensives. Hypertens Res. 2004 ;27:333-8
6. Nakano Y, Ishida T, Ozono R, Matsuura H, Yamamoto Y, Kambe M, Chayama K, Oshima T. A frameshift mutation of beta subunit of epithelial sodium channel in a case of isolated Liddle syndrome. J Hypertens. 2002 ;20:2379-82
7. Kyuma M, Ura N, Torii T, Takeuchi H, Takizawa H, Kitamura K, Tomita K, Sasaki S, Shimamoto K. A family with liddle's syndrome caused by a mutation in the beta subunit of the epithelial sodium channel. Clin Exp Hypertens. 2001 ;23:471-8
8. Hiltunen TP, Hannila-Handelberg T, Petajaniemi N, Kantola I, Tikkanen I, Virtamo J, Gautschi I, Schild L, Kontula K. Liddle's syndrome associated with a point mutation in the extracellular domain of the epithelial sodium channel gamma subunit. J Hypertens. 2002 ;20:2383-90
9. Hannila-Handelberg T, Kontula K, Tikkanen I, Tikkanen T, Fyhrquist F, Helin K, Fodstad H, Piippo K, Miettinen HE, Virtamo J, Krusius T, Sarna S, Gautschi I, Schild L, Hiltunen TP. Common variants of the beta and gamma subunits of the epithelial sodium channel and their relation to plasma renin and aldosterone levels in essential hypertension. BMC Med Genet. 2005 ;6:4
10. Snyder PM. The epithelial Na+ channel: cell surface insertion and retrieval in Na+ homeostasis and hypertension. Endocr Rev. 2002 ;23:258-75
11. Shimkets RA, Lifton RP, Canessa CM. The activity of the epithelial sodium channel is regulated by clathrin-mediated endocytosis. J Biol Chem. 1997 ;272:25537-41
12. Firsov D, Schild L, Gautschi I, Merillat AM, Schneeberger E, Rossier BC. Cell surface expression of the epithelial Na channel and a mutant causing Liddle syndrome: a quantitative approach. Proc Natl Acad Sci U S A. 1996 ;93:15370-5
13. Saxena S, Quick MW, Tousson A, Oh Y, Warnock DG. Interaction of syntaxins with the amiloride-sensitive epithelial sodium channel. J Biol Chem. 1999 ;274:20812-7
14. Qi J, Peters KW, Liu C, Wang JM, Edinger RS, Johnson JP, Watkins SC, Frizzell RA. Regulation of the amiloride-sensitive epithelial sodium channel by syntaxin 1A. J Biol Chem. 1999 ;274:30345-8
15. Condliffe SB, Zhang H, Frizzell RA. Syntaxin 1A regulates ENaC channel activity. J Biol Chem. 2004 ;279:10085-92
16. Condliffe SB, Carattino MD, Frizzell RA, Zhang H. Syntaxin 1A regulates ENaC via domain-specific interactions. J Biol Chem. 2003 ;278:12796-804
17. Berdiev BK, Jovov B, Tucker WC, Naren AP, Fuller CM, Chapman ER, Benos DJ. ENaC subunit-subunit interactions and inhibition by syntaxin 1A. Am J Physiol Renal Physiol. 2004 ;286:F1100-6
18. Arien H, Wiser O, Arkin IT, Leonov H, Atlas D. Syntaxin 1A modulates the voltage-gated L-type calcium channel (Ca(v)1.2) in a cooperative manner. J Biol Chem. 2003 ;278:29231-9
19. Kang Y, Huang X, Pasyk EA, Ji J, Holz GG, Wheeler MB, Tsushima RG, Gaisano HY. Syntaxin-3 and syntaxin-1A inhibit L-type calcium channel activity, insulin biosynthesis and exocytosis in beta-cell lines. Diabetologia. 2002 ;45:231-41
20. Naren AP, Di A, Cormet-Boyaka E, Boyaka PN, McGhee JR, Zhou W, Akagawa K, Fujiwara T, Thome U, Engelhardt JF, Nelson DJ, Kirk KL. Syntaxin 1A is expressed in airway epithelial cells, where it modulates CFTR Cl(-) currents. J Clin Invest. 2000 ;105:377-86
21. Cormet-Boyaka E, Di A, Chang SY, Naren AP, Tousson A, Nelson DJ, Kirk KL. CFTR chloride channels are regulated by a SNAP-23/syntaxin 1A complex. Proc Natl Acad Sci U S A. 2002 ;99:12477-82
22. Ji J, Salapatek AM, Lau H, Wang G, Gaisano HY, Diamant NE. SNAP-25, a SNARE protein, inhibits two types of K channels in esophageal smooth muscle. Gastroenterology. 2002 ;122:994-1006
23. Wang D, Deken SL, Whitworth TL, Quick MW. Syntaxin 1A inhibits GABA flux, efflux, and exchange mediated by the rat brain GABA transporter GAT1. Mol Pharmacol. 2003 ;64:905-13
24. Saxena S, Singh M, Engisch K, Fukuda M, Kaur S. Rab proteins regulate epithelial sodium channel activity in colonic epithelial HT-29 cells. Biochem Biophys Res Commun. 2005 ;337:1219-23
25. Saxena SK, Singh M, Shibata H, Kaur S, George C. Rab4 GTP/GDP modulates amiloride-sensitive sodium channel (ENaC) function in colonic epithelia. Biochem Biophys Res Commun. 2006 ;340:726-33
26. Huber SM, Braun GS, Horster MF. Expression of the epithelial sodium channel (ENaC) during ontogenic differentiation of the renal cortical collecting duct epithelium. Pflugers Arch. 1999 ;437:491-7
27. Chang CT, Bens M, Hummler E, Boulkroun S, Schild L, Teulon J, Rossier BC, Vandewalle A. Vasopressin-stimulated CFTR Cl- currents are increased in the renal collecting duct cells of a mouse model of Liddle's syndrome. J Physiol. 2005 ;562:271-84
28. Naren AP, Cormet-Boyaka E, Fu J, Villain M, Blalock JE, Quick MW, Kirk KL. CFTR chloride channel regulation by an interdomain interaction. Science. 1999 ;286:544-8
29. Saxena SK, Kaur S. Rab27a negatively regulates CFTR chloride channel function in colonic epithelia: involvement of the effector proteins in the regulatory mechanism. Biochem Biophys Res Commun. 2006 ;346:259-67
30. Salaun C, James DJ, Greaves J, Chamberlain LH. Plasma membrane targeting of exocytic SNARE proteins. Biochim Biophys Acta. 2004 ;1693:81-9
31. Wenthold RJ, Safieddine S, Ly CD, Wang YX, Lee HK, Wang CY, Kachar B, Petralia RS. Vesicle targeting in hair cells. Audiol Neurootol. 2002 ;7:45-8
32. Kweon DH, Kim CS, Shin YK. Regulation of neuronal SNARE assembly by the membrane. Nat Struct Biol. 2003 ;10:440-7
33. Zhang F, Chen Y, Kweon DH, Kim CS, Shin YK. The four-helix bundle of the neuronal target membrane SNARE complex is neither disordered in the middle nor uncoiled at the C-terminal region. J Biol Chem. 2002 ;277:24294-8
34. Fuller PJ, Brennan FE, Burgess JS. Acute differential regulation by corticosteroids of epithelial sodium channel subunit and Nedd4 mRNA levels in the distal colon. Pflugers Arch. 2000 ;441:94-101
35. Greig ER, Baker EH, Mathialahan T, Boot-Handford RP, Sandle GI. Segmental variability of ENaC subunit expression in rat colon during dietary sodium depletion. Pflugers Arch. 2002 ;444:476-83
36. Greig ER, Boot-Handford RP, Mani V, Sandle GI. Decreased expression of apical Na+ channels and basolateral Na+, K+-ATPase in ulcerative colitis. J Pathol. 2004 ;204:84-92
37. Staub O, Yeger H, Plant PJ, Kim H, Ernst SA, Rotin D. Immunolocalization of the ubiquitin-protein ligase Nedd4 in tissues expressing the epithelial Na+ channel (ENaC). Am J Physiol. 1997 ;272:C1871-80
38. Mick VE, Itani OA, Loftus RW, Husted RF, Schmidt TJ, Thomas CP. The alpha-subunit of the epithelial sodium channel is an aldosterone-induced transcript in mammalian collecting ducts, and this transcriptional response is mediated via distinct cis-elements in the 5'-flanking region of the gene. Mol Endocrinol. 2001 ;15:575-88
39. De La Rosa DA, Paunescu TG, Els WJ, Helman SI, Canessa CM. Mechanisms of Regulation of Epithelial Sodium Channel by SGK1 in A6 Cells. J Gen Physiol. 2004 ;124:395-407
40. Spindler B, Mastroberardino L, Custer M, Verrey F. Characterization of early aldosterone-induced RNAs identified in A6 kidney epithelia. Pflugers Arch. 1997 ;434:323-31
41. Blazer-Yost BL, West TA, Stack J, Peck K, Lahr TF, Gekle M. Effect of the mycotoxin, ochratoxin A, on hormone-stimulated ion transport in a cultured cell model of the renal principal cell. Pflugers Arch. 2005 ;450(1):53-60
42. Bens M, Vallet V, Cluzeaud F, Pascual-Letallec L, Kahn A, Rafestin-Oblin ME, Rossier BC, Vandewalle A. Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J Am Soc Nephrol. 1999 ;10:923-34
43. Naren AP, Quick MW, Collawn JF, Nelson DJ, Kirk KL. Syntaxin 1A inhibits CFTR chloride channels by means of domain-specific protein-protein interactions. Proc Natl Acad Sci U S A. 1998 ;95:10972-7
44. Cui N, Kang Y, He Y, Leung YM, Xie H, Pasyk EA, Gao X, Sheu L, Hansen JB, Wahl P, Tsushima RG, Gaisano HY. H3 domain of syntaxin 1A inhibits KATP channels by its actions on the sulfonylurea receptor 1 nucleotide-binding folds-1 and -2. J Biol Chem. 2004 ;279(51):53259-65
45. Bezprozvanny I, Zhong P, Scheller RH, Tsien RW. Molecular determinants of the functional interaction between syntaxin and N-type Ca2+ channel gating. Proc Natl Acad Sci U S A. 2000 ;97:13943-8
46. Lewis JL, Dong M, Earles CA, Chapman ER. The transmembrane domain of syntaxin 1A is critical for cytoplasmic domain protein-protein interactions. J Biol Chem. 2001 ;276:15458-65
47. Zwermann O, Beuschlein F, Klink A, Stahl M, Reincke M. The role of the ACTH receptor in adrenal tumors: identification of a novel microsatellite marker. Horm Metab Res. 2004 ;36:406-10
48. Yang B, Steegmaier M, Gonzalez LC Jr, Scheller RH. nSec1 binds a closed conformation of syntaxin1A. J Cell Biol. 2000 ;148:247-52
49. Gladycheva SE, Ho CS, Lee YY, Stuenkel EL. Regulation of syntaxin1A-munc18 complex for SNARE pairing in HEK293 cells. J Physiol. 2004 ;558:857-71
50. Naren AP, Nelson DJ, Xie W, Jovov B, Pevsner J, Bennett MK, Benos DJ, Quick MW, Kirk KL. Regulation of CFTR chloride channels by syntaxin and Munc18 isoforms. Nature. 1997 ;390:302-5
51. Selim AA, Abdelmagid SM, Kanaan RA, Smock SL, Owen TA, Popoff SN, Safadi FF. Anti-osteoactivin antibody inhibits osteoblast differentiation and function in vitro. Crit Rev Eukaryot Gene Expr. 2003 ;13:265-75
52. Bardag-Gorce F, Riley N, Nguyen V, Montgomery RO, French BA, Li J, van Leeuwen FW, Lungo W, McPhaul LW, French SW. The mechanism of cytokeratin aggresome formation: the role of mutant ubiquitin (UBB+1). Exp Mol Pathol. 2003 ;74:160-7
53. Rowe J, Corradi N, Malosio ML, Taverna E, Halban P, Meldolesi J, Rosa P. Blockade of membrane transport and disassembly of the Golgi complex by expression of syntaxin 1A in neurosecretion-incompetent cells: prevention by rbSEC1. J Cell Sci. 1999 ;112(Pt 12):1865-77
54. Berdiev BK, Shlyonsky VG, Karlson KH, Stanton BA, Ismailov II. Gating of amiloride-sensitive Na(+) channels: subunit-subunit interactions and inhibition by the cystic fibrosis transmembrane conductance regulator. Biophys J. 2000 ;78:1881-94
55. Fyfe GK, Canessa CM. Subunit composition determines the single channel kinetics of the epithelial sodium channel. J Gen Physiol. 1998 ;112:423-32
56. Ji HL, Fuller CM, Benos DJ. Intrinsic gating mechanisms of epithelial sodium channels. Am J Physiol Cell Physiol. 2002 ;283:C646-50
57. Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest. 1999 ;104:R19-23
58. Dijkink L, Hartog A, Deen PM, van Os CH, Bindels RJ. Time-dependent regulation by aldosterone of the amiloride-sensitive Na+ channel in rabbit kidney. Pflugers Arch. 1999 ;438:354-60
59. Alvarez de la Rosa D, Li H, Canessa CM. Effects of aldosterone on biosynthesis, traffic, and functional expression of epithelial sodium channels in A6 cells. J Gen Physiol. 2002 ;119:427-42
60. Reales E, Mora-Lopez F, Rivas V, Garcia-Poley A, Brieva JA, Campos-Caro A. Identification of soluble N-ethylmaleimide-sensitive factor attachment protein receptor exocytotic machinery in human plasma cells: SNAP-23 is essential for antibody secretion. J Immunol. 2005 ;175:6686-93
61. Li C, Roy K, Dandridge K, Naren AP. Molecular assembly of cystic fibrosis transmembrane conductance regulator in plasma membrane. J Biol Chem. 2004 ;279:24673-84
62. Ling S, Sheng JZ, Braun JE, Braun AP. Syntaxin 1A co-associates with native rat brain and cloned large conductance, calcium-activated potassium channels in situ. J Physiol. 2003 ;553:65-81
63. Hohne-Zell B, Galler A, Schepp W, Gratzl M, Prinz C. Functional importance of synaptobrevin and SNAP-25 during exocytosis of histamine by rat gastric enterochromaffin-like cells. Endocrinology. 1997 ;138:5518-26
64. Linial M. SNARE proteins--why so many, why so few? J Neurochem. 1997 ;69:1781-92
65. Ji J, Yang SN, Huang X, Li X, Sheu L, Diamant N, Berggren PO, Gaisano HY. Modulation of L-type Ca(2+) channels by distinct domains within SNAP-25. Diabetes. 2002 ;51:1425-36
66. Li G, Yang Q, Alexander EA, Schwartz JH. Syntaxin 1A has a specific binding site in the H3 domain that is critical for targeting of H+-ATPase to apical membrane of renal epithelial cells. Am J Physiol Cell Physiol. 2005 ;289:C665-72
Author contact
![]() Correspondence to: Sunil K Saxena, Center for Cell and Molecular Biology, Department of Chemistry and Chemical Biology, Stevens Institute of Technology, 314 McLean Hall, 5th and River Street, Hoboken, NJ 07030, Telephone: 201-216-8956; Fax: 201-216-8240; E-Mail: ssaxenaedu
Correspondence to: Sunil K Saxena, Center for Cell and Molecular Biology, Department of Chemistry and Chemical Biology, Stevens Institute of Technology, 314 McLean Hall, 5th and River Street, Hoboken, NJ 07030, Telephone: 201-216-8956; Fax: 201-216-8240; E-Mail: ssaxenaedu