Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(3):129-131. doi:10.7150/ijbs.3.129 This issue Cite
Editorial
Swine Genome Science Comes of Age
Zhihua Jiang1, Max F. Rothschild2 ![]()
1. Department of Animal Sciences, Washington State University, Pullman, WA 99164-6351, USA
2. Department of Animal Science and the Center for Integrated Animal Genomics, Iowa State University, Ames, IA 50011, USA
Received 2007-1-25; Accepted 2007-2-6; Published 2007-2-9
Abstract
Pigs were among the first animals to be domesticated and pork is one of the most widely eaten meats in the world today. The pig has also been an excellent biomedical model for understanding a variety of human health issues such as obesity, diabetes, cancer, female reproductive health, cardiovascular disease, and infectious diseases. Genome sequencing, mapping, expression and functional analyses have significantly advanced our ability to unravel the secrets of the pig. Therefore, this edition, with six reviews from leading scientists, offers the opportunity for all interested researchers and readers to see the big picture of porcine genomics.
Keywords: swine genome, sequencing, functional analyses.
Introduction
16th century English Proverb: “You can't make a silk purse from a sow's ear”.
During the past decade, the swine genome community has generated a tremendous amount of useful information and tools for facilitating genome mapping, quantitative trait loci (QTL) discovery and transcriptome analyses in pigs. While these new developments and achievements still won't allow silk purses to be made from sow's ears, they have created real opportunities for scientific discovery and practical achievements. In particular, approximately 1.3 million sequences have already been submitted to the GenBank database and a 4x genome sequence will be likely released to the public in early 2008. These unique resources allow researchers to investigate the pig genome structure, function and evolution, understand the genetic variation underlying economically important traits for pig improvement and develop pig models for unraveling the genetic complexity of human diseases.
It has been widely believed that pigs were first domesticated from the wild boars (Sus scrofa) approximately 9,000 years ago. Domestication, selective breeding and adaptation to diverse environments have led to development of more than 250 breeds worldwide. Pigs mature faster than other domesticated animals, have large litters and can feed on a variety of feed sources including human garbage, making them efficient and valuable parts of many agricultural systems. Pigs are mostly used for food. However, people have also used their hide for shields, their bones for tools and weapons, their bristles for brushes, their skins for clothes and shoes and even their acute sense of smell for a variety of tasks. Wild pigs have been also hunted for sport (Figure 1). Pigs are known to be intelligent animals and, in particular, some miniature pigs have become popular as house pets.
A sassanid rock relief of boar hunting (http://en.wikipedia.org).

The pig genome is of similar size, complexity and genetic information as the human genome. In 1993, the public databases had only gathered approximately 600 pig sequences, while at the end of 2006, the total number of pig sequences has reached nearly 1.3 million entries in the public domain. Among these pig sequences, over half of them are ESTs (expressed sequence tags) derived from more than 250 cDNA libraries [1]. The Wellcome Trust Sanger Institute has been the biggest player in the GSS (genome survey sequence) sequencing, contributing ~75% of the GSS entries. Through funding provided by the CSREES-USDA, the Sanger Institute is currently engaged in whole genome sequencing of the pig with the goal of finishing sequencing of a 4X genome coverage by January 2008 [2]. To date, there are 1,850 working draft sequences from the pig genome that have been released to the public domain. All of these provide the pig genome community with a solid foundation and a unique resource for advancing human and animal health and animal agriculture in the 21st century.
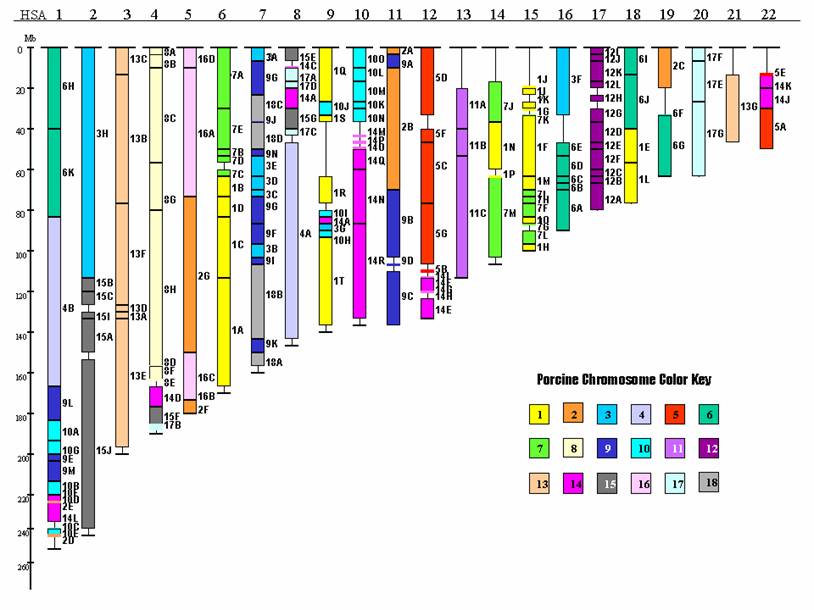
The early versions of whole genome linkage maps in pigs were mostly with microsatellite markers and a limited number of genes. However, the pig genome community has shifted the focus from the whole genome linkage maps to whole genome radiation hybrid (RH) maps since then and this has allowed for many more genes to be mapped and anchored using microsatellites. Although some discrepancies exist among these maps, they have contributed to an identification of over 170 conserved segments between the human and pig genomes, which have helped to further determine the evolutionary relationship between these two species (Figure 2). As the numbers of common markers have grown, integration of both genetic maps and RH maps are now a reality. Certainly, these well-integrated maps will assist in the genome sequence assembly of the pig.
Conserved segments between human and pig autosome genomes.
Most economically important phenotypes in pig production are quantitative in nature, and often result from complex interactions between numerous environmental factors and alleles of many genes. Since the initial major locus for fat deposition on chromosome 4 was discovered in 1994, quantitative trait loci (QTL) results from 110 papers have identified 1,675 QTL [3]. Among them, 1,285 were QTL related to meat quality. The identification of genes affecting complex traits has been considered to be one of the most difficult and challenging tasks of genetics today. The major challenge to identifying the genes underlying QTL is not the detection and localization of a QTL, but rather the endless process to narrow a QTL region to a few candidate genes that can be rigorously tested. The draft sequence will of course aid in this process. As an example, the Animal Genome Group at Monsanto has genotyped more than 4,000 autosome single nucleotide polymorphisms (SNPs) on a total of approximately 6,000 pigs for investigating the extent and range of linkage disequilibrium (LD) in the pig genome [4]. Hopefully the LD structures within/across chromosomes will provide useful information for whole genome association studies and characterization of candidate genes for these complex traits. On the other hand, to date most of the useful genes and markers for economically important traits have been either patented or kept as trade secrets and in part these approaches have limited the use of these gene tests to only larger genetics companies. Changes in the patent approval process and patent expense will likely limit the future numbers of patents for gene tests.
During the past five years there has been a tremendous rise in porcine transcriptome analyses. A variety of techniques have been used, such as cDNA libraries and EST production, differential display PCR, serial analyses of gene expression, suppression subtractive hybridization, real-time quantitative PCR and microarray-based techniques [1]. It has been proposed that transcriptome analysis would allow an efficient, objective, quantitative evaluation of genes contributing to QTL and that transcriptome analysis has the potential to reduce the overall effort needed in identifying genes causally associated with quantitative traits of interest.
Pigs are increasingly appreciated as excellent models for medical research, as pigs and humans are so similar in many aspects of both infant and adult anatomy, physiology, biochemistry, pathology and pharmacology. Because there is no placental transfer of antibodies to the developing pig fetus, newborn piglets lack maternal antibodies, making them good for studies of the mechanisms of immunity. As early as 1775, pigs were shown to develop atherosclerosis as a natural part of aging. Normal, healthy swine are known to develop spontaneous atherosclerotic plaques similar to that seen in humans. Pigs are also useful to medical researchers studying other cardiovascular problems and gastric ulcers. As mammals, both the human and the pig have a highly conserved genome size and presumably share most of their orthologous genes. In addition, there is also a high degree of sequence conservation between the orthologous genes as well as a high degree of map synteny between these two species. These features make the pig a unique model to probe pathways regulating normal development as well as disease processes [5].
The pig genome project is entering a stage where the level of genomic data is rapidly accelerating. We anticipate that the genome sequence for the pig should be completed over the next two years. During that time period, the swine genome community will annotate the sequences and determine the features of biological relevance and interest. These results will provide an important foundation for investigation of the structure and function of the pig genome as well as provide a powerful toolbox for understanding the genetic variation underlying economically important and complex phenotypes and be of value to further understand human health. Overall, advancing swine genomics promises to significantly benefit the pig industry and consumers by providing knowledge and technologies that can help optimize production, quality, nutritional value and resistance to diseases [6]. This edition, with six reviews from leading scientists, offers the opportunity to all interested researchers to see the big picture of porcine genomics. Finally, let us remind the readers that February 18, 2007, is the start of the Chinese New Year, which is the “Year of the Pig.” This series of papers is devoted to the advances from the past and opportunities the bright future will make possible in this “Year of the Pig” and beyond.
Acknowledgements
The authors thank the following colleagues: P. J. Burfening, Kefei Chen, A. C. Clutter, O. Couture, Fengxing Du, R. D. Green, D.L. Hamernik, Zhi-Liang Hu, M. M. Lohuis, J. A. Long, J. L. Lunney, M. A. Qureshi, L. B. Schook, C. K. Tuggle and Yanfang Wang for their contributions to this special issue.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Tuggle CK, Wang Y-F, Couture O. Advances in Swine Transcriptomics. Int J. Biol Sci. 2007;3:132-152
2. Chen K, Baxter T, Muir W, Groenen M, Schook L. Genetic resources, genome mapping and evolutionary genomics of the pig (Sus scrofa). Int J. Biol Sci. 2007;3:153-165
3. Rothschild MF, Hu Z-L, Jiang Z. Advances in QTL Mapping in Pigs. Int J. Biol Sci. 2007;3:192-197
4. Du F-X, Clutter AC, Lohuis MM. Characterizing Linkage Disequilibrium in Pig Populations. Int J. Biol Sci. 2007;3:166-178
5. Lunney JK. Advances in Swine Biomedical Model Genomics. Int J. Biol Sci. 2007;3:179-184
6. Green RD, Qureshi MA, Long JA, Burfening PJ, Hamernik DL. Identifying the Future Needs for Long-Term USDA Efforts in Agricultural Animal Genomics. Int J. Biol Sci. 2007;3:185-191
Author contact
![]() Correspondence to: Dr. Zhihua Jiang, Tel: +509 335 8761; Fax: +509 335 4246; E-mail: jiangzedu
Correspondence to: Dr. Zhihua Jiang, Tel: +509 335 8761; Fax: +509 335 4246; E-mail: jiangzedu