Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2009; 5(5):411-420. doi:10.7150/ijbs.5.411 This issue Cite
Research Paper
In vivo measurement of protein functional changes
Aili Wang, Zhicheng Zhang, Qinyi Zhao ![]()
ICU, Navy General Hospital, Beijing 100037, PR China
Received 2009-2-24; Accepted 2009-6-1; Published 2009-6-5
Abstract
Conformational changes in proteins are fundamental to all biological functions. In protein science, the concept of protein flexibility is widely used to describe protein dynamics and thermodynamic properties that control protein conformational changes. In this study, we show that urea, which has strong sedative potency, can be administered to fish at high concentrations, and that protein functional changes related to anesthesia induction can be measured in vivo. Ctenopharyngodon idellus (the grass carp) has two different types of N-methyl d-aspartate (NMDA) receptors, urea-insensitive and urea-sensitive, which are responsible for the heat endurance of fish. The urea-sensitive NMDA receptor showed high protein flexibility, the gamma aminobutyric acid (GABA) receptor showed less flexibility, and the protein that is responsible for ethanol anesthesia showed the lowest flexibility. The results suggest that an increase in protein flexibility underlies the fundamental biophysical mechanisms of volatile general anesthetics.
Keywords: Urea, volatile anesthetics, protein flexibility, receptor, grass carp
Introduction
The mechanism of action of volatile general anesthetics still remains an enigma in medical science and biology (1-4). By the turn of the 20th century, Meyer-Overton rules had proposed that volatile anesthetics would increase in potency as a function of their solubility in olive oil (1). This discovery has dominated our understanding of action of volatile anesthetics for a century, indicating an underlying premise that the lipid bilayer of the cell membrane is a primary target of volatile general anesthetics. Thus, changes in membrane density (critical volume hypothesis) and membrane fluidity were invoked as the mechanism of action of volatile anesthetics (1).
However, the work of Frank and Lieb fundamentally changed this original attitude toward anesthetic function (5, 6). Their work showed that the target of volatile general anesthetics was protein, not the cell membrane. The Meyer-Overton rule could be also interpreted by nonspecific interactions between volatile general anesthetics and hydrophobic areas of a protein (5, 6). In recent years, a number of gene mutants have been isolated that code for proteins related to anesthetics (7, 8).
Volatile general anesthetics can act on all proteins in vivo (1-3, 9). Consequently, it is necessary to differentiate the diversified effects of anesthesia induction. Theoretically speaking, the protein target of an anesthetic can be classified into four groups: 1) full target protein of anesthetics, in which the change of function (protein conformation) induces a full anesthesia state; 2) contributing protein targets of anesthetics, in which the change of conformation alone does not induce full anesthesia state, but contributes to the depth of the anesthesia depth and its effect can be seen in combined anesthesia; 3) complementary protein target of anesthetics, which do not influence the depth of anesthesia, but can satisfy other requirement of operation, such as muscle relaxation; or 4) the protein target of the anesthetic has nothing to do with the anesthesia effect.
Currently, there are two different approaches for investigating the action mechanism of anesthetics. The first approach is based on the hypothesis that different volatile general anesthetics act on different in vivo protein targets (1-4, 9). The second approach views volatile general anesthetics as all acting on the same protein target and all other effects are independent of anesthesia depth. The underlying conclusion of first hypothesis is that the Meyer-Overton rule is nonsense and that the interaction between a protein target and a particular anesthetic is specific. In contrast, the second hypothesis views the interaction of protein and volatile general anesthetics as unspecific.
There is evidence in support of both of these hypotheses. For the first hypothesis, different volatile general anesthetics have been shown to have different profiles on the activity of ion channels, the NMDA receptor, the GABA receptor, and a number of other proteins (1-8). No single protein that has been studied can account for all properties of action of volatile anesthetics (1, 4). This hypothesis recognizes that all volatile general anesthetics do not have to act in the same way.
Evidence in support of the second hypothesis includes: 1) the required concentration of volatile general is quite high; for example, it is 0.5 mol/L for ethanol. Chemically speaking, the specific interaction between a protein and ligand normally occurs below a ligand concentration of 100 mmol/L. This high concentration of anesthetic indicates that the interaction between it and the target protein is unspecific. 2) this unspecific interaction between protein and volatile general anesthetics can account for Meyer-Overton rules; 3) changes in pressure can reverse the anesthetic effect (1); and 4) it is unlikely that the number of volatile general anesthetics can act on so many different proteins.
If a volatile general anesthetic acts on proteins in an unspecific manner, it must be influencing the nature of protein dynamics (protein flexibility). A conformational change (or alteration of function) of specific proteins in the brain (or the peripheral nervous system) is the molecular mechanism underlying anesthesia (10, 11). Based upon the above considerations and reviewing protein thermodynamic structural theory, Zhao has predicted that an increase in protein flexibility induced by volatile general anesthetics is the fundamental biophysical mechanism of anesthesia (12). If this is the case, we have postulated that urea and guanidine hydrochloride, two protein denaturants that are most commonly used in biochemistry (13), should show anesthetic potency to some degree and thus should show cooperativity with volatile general anesthetics. Our tests have confirmed this prediction to be true. Because guanidine hydrochloride shows high toxicity even at low concentrations (0.2 M) (data not shown), in this study, we have examined urea in more detail.
Urea increases protein flexibility and is widely used in vitro to disturb protein conformation in order to measure this flexibility (13). However, there are very few reports about urea endurance of cells and intact fish (e.g. 14). In the current study, we show that fish can tolerate high concentrations of urea and that the flexibility of proteins related to the induction of anesthesia can be measured in vivo.
Small organic solvents, such as ethanol, chloroform, phenol, are also known in biochemistry to be protein conformation disturbers or denaturants (15-18). Among the consideration that most volatile anesthetics are organic solvent with small molecular weight, theoretically speaking they are protein denaturant or protein flexibility enhancer. When protein flexibility is disturbed by a denaturant, the equilibrium between different conformational states of that protein can be disrupted, which may result in the alteration of the functional conformation of the protein (19, 20). Although urea and organic substances influence protein conformation through different chemical mechanisms, their net effects on protein conformational changes overlap. Therefore, they can act cooperatively in the induction of anesthesia.
Based on the discovery that high in vivo concentrations of urea can be tolerated by fish, we have developed a new technique to measure in vivo protein functional changes. Unlike in vitro tests, in which the protein activity can be determined directly, we cannot determine protein activity (or enzyme activity) directly through in vivo methods. We can, however, determine the behavioral changes in a fish that has had a protein conformational change induced. By analyzing the dose-response curve of a ligand for a protein target in the presence of urea, we can indirectly measure the functional change of that protein. The protein flexibility, or more specifically, its sensibility to urea, can then be judged.
By applying this technique, we have been able to successfully measure the functional changes of several proteins involved in anesthesia induction. These include two types of N-methyl d-aspartate (NMDA) receptors (urea-sensitive and urea-insensitive) that are present in grass carp. None of the GABA or NMDA receptors appear to be full protein targets of ethanol anesthesia.
The grass carp (21) was selected for use in our experiments because it can be obtained in vast quantities in Beijing. In addition, urea can be readily removed from the body of the fish, and the full recovery of the fish from the anesthetized state has been well established.
Experimental Procedures
Materials
Larval fish of the species Ctenopharyngodon idellus (grass carp, or white amur) with body weights between 2 and 5 g were purchased in the Beijing market (China). Beijing winter grass carp are fish caught in the winter in Beijing; these fish cannot endure temperatures above 30℃. Beijing summer fish are those caught in the summer, and these fish can tolerate temperatures up to at least 37℃. Beijing winter grass carp can transform into summer grass carp when they are grown at 22℃ for more than 10 days.
Methods
Chemicals were added to the tap water in which the fish were kept, and the behavior of the fish was observed. Levels of chemicals were added at rates that kept their concentrations constant throughout the experimental period (data not shown).
In studying the anesthetic effect of urea, several criteria were used to determine the level of anesthesia in the fish. These criteria included the loss of response to stimuli (mechanical and sound), loss of equilibrium (floating belly up), loss of the sense of direction, loss of muscle tone, and cessation of voluntary swimming (22). Fish were fasted for 12-16 hours prior to receiving anesthesia and oxygen was supplied during all experiments. In general, 6-10 fish were utilized in each experiment. For the measurement of protein flexibility, the endpoint of anesthesia was taken to be the point at which equilibrium was lost and the fish floated upside down; this point could be easily judged. During induction of anesthesia, fish typically had difficulty remaining upright, and finally swam on their sides or upside down (the fully anesthetized state).
For the purposes of the experiment, the fully anesthetized state was required to persist for at least 5 minutes, during which the fish could not swim upright. The anesthetic concentration of the drug in question was defined as the concentration at which all fish were anesthetized within 10 minutes, but could not be anesthetized within 6 minutes. If all fish were anesthetized within 6 minutes, the drug concentration was deemed to be too high and the test was repeated. The accuracy of the determination of anesthetic concentration was approximately 5%, and the precision (or repeatability) was approximately 2% for tests performed simultaneously. Because precise results could be consistently obtained, statistical analysis of data was deemed unnecessary.
Principle of Protein Functional Change Measurements
At concentrations below 1.2 M, urea does not overcome physiological homeostasis, but it can influence protein function by increasing protein flexibility. The urea treatment caused subtle conformational changes in numerous receptors (proteins) that were not detectable at the behavioral level, so we could not directly measure the functional change of a specific protein. In order to measure the functional change of a protein of interest, a specific ligand of this protein was used to strengthen its functional change, and thus, a change in its biological function could be observed at behavioral level.
By analyzing the change of the dose-response of a ligand for a target protein in the presence of urea (or the ligand-urea titration for anesthesia in this case), we can indirectly measure the functional change of the protein. Protein flexibility can be judged from the slope of the ligand-urea titration during anesthesia. As a protein with higher flexibility will usually show a greater functional change at low concentrations of urea, the amount of ligand (drug) required for anesthesia induction should be less. The onset of the anesthetized state (the equivalence point for the ligand-urea titration) can be easily and precisely determined, and therefore the flexibility of proteins related to anesthesia induction can be judged.
Theoretically, a ligand such as alcohol may produce its measured effects through many receptors, rather than only through a single one. However, in practice, only the interaction between ethanol and a full protein target of ethanol anesthesia can be measured. If there is no full target for ethanol, the result may be very complex and the figure will not be easily analyzed. However, we did not encounter this type of case in our study. Another consideration is that many proteins (and receptors) of a ligand may contribute to the induction of anesthesia. For the same reasons, only a full target of a ligand involved in anesthesia induction can be measured. We were therefore able to judge the flexibility of a single protein.
Results
The toxicity and anesthetic potency of urea
Because there have been no previous reports regarding the toxicity and anesthetic potency of urea, we first tested fish for sensitivity to this compound. The results are shown in Table 1.
The toxicity and anesthetic potency of urea. (T) Under this urea condition, the anesthesia test can be performed within 10 minutes. (a) The safe concentration refers to the urea concentration at which no death of fish occurs within 20 minutes and at which fish continue to survive after removal of urea. The conditional death concentration refers to the urea concentration at which no death occurs within 20 minutes but death may be seen after removal of urea, or fish are unable to eat. The acute death concentration refers to the urea concentration at which the death of fish may be seen within 20 minutes. (b) The convulsion potency of urea can be fully suppressed by ethanol, urethane, and midazolam, but not by ketamine. This finding rules out the possibility that excitability of fish is induced by the osmotic effect of urea solution. The test temperature was 14℃.
| Urea | Classification | Phase and biological trait |
|---|---|---|
| 0-0.3 M | (T) | Sedative phase, safety concentration (a) (The sensitivity to stimuli is decreased. The fish are less active.) |
| 0.4-1.0 M | (T) | Agitated phase, safety concentration (a) (The sensitivity to stimuli is greatly decreased. The fish move purposelessly. Convulsions can occasionally be seen (b]. The fish lose sense of direction.) |
| 1.1-1.4 M | (T) | Conditional death concentration (The fish will die over a long period, or more than 30 minutes.) |
| 1.5-3 M | Acute death concentration (All fish die within a short period. At 3 M, all fish die within 3 minutes. At 2.4 M, all fish die within 15 minutes. At 1.8 M, fish die within more than 10 minutes.) |
Two phases, sedation and agitation, were clearly observed as responses of grass carp to urea. The first response was the loss of sensitivity to sound and to vibrating stimuli; this was followed by the loss of directional sense (fish would frequently swim into the walls), and finally, sensitivity to mechanical stimuli would be lost. The ability to maintain equilibrium was not fully destroyed by urea even at very high concentrations (e.g. 2.0 M). No effect on respiration was observed in our experiments. These results clearly suggest that urea has a weak anesthetic potency and it alone cannot induce a full anesthesia state of a fish. However, urea cannot be classified as a traditional anesthetic because of its convulsive effects.
At high concentrations, urea showed a potential to cause convulsions, with the convulsions and the sedative effects occurring simultaneously. However, it was possible to study the two processes separately (see the following discussion and figure 1).
The activity of fish. A) The activity of fish was measured by recording the frequency of movement; 100% represents fish activity in the absence of drugs (40 turns/minute, waving tail). The solution contained 60 mg/L of ketamine and 0.6 M urea. The fish were Beijing summer grass carp. The movements of fish were counted and the frequencies of tail waving were calculated. Ketamine had no impact on movement of fish; therefore, urea was seen to have a strong sedation potency. The fish become very excited after 5 minutes and counting became impossible. The curve was drawn based upon experimental data as well as behavioral observation (simulation). 10 fish were used in this test.
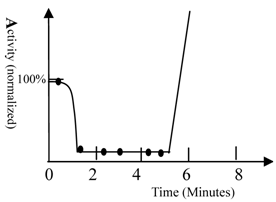
High concentrations of urea could also be lethal to the fish. Although the mechanism leading to death was not studied, it is obvious that physiological homeostasis can be destroyed by high concentrations of urea, as many biochemical processes would be expected to be disturbed.
The osmotic effect of urea was not a factor in our experiments
The osmotic effect of high concentrations of urea can induce specific signal transduction systems (14). Consequently, it was necessary to rule out the possibility that the anesthetic potency of urea was induced by osmotic effects of the high molarity urea solution. It was impossible to experimentally distinguish the osmotic effects of urea solution from other effects of urea. However, the following evidence indicates that, although the osmotic effect occurred in vivo, it did not interfere with induction of anesthesia. First, the osmotic effect took place predominantly in the region of the gills, as the concentration difference for urea was high at this site. However, no abnormalities in respiration were observed, even at urea concentrations as high as 2.4 M. Since the urea reached the brain gradually, which is judged based upon the onset of anesthesia, the osmotic effect of urea on the nerve cell membranes of fish was assumed to have been very small and could not be maintained. Second, the co-addition of urea and ethanol at high concentrations did not induce convulsions, suggesting that the potential of urea to cause convulsions does not result from its osmotic pressure. Third, the osmotic effect should disappear when the difference in urea concentration between the body of the fish and the medium is reduced. However, the anesthesia state of the fish induced by urea was stable, suggesting that the osmotic effect of urea was unrelated to anesthesia production. This hypothesis was further confirmed by the results of the measurement of protein flexibility.
The time scale of biological phenomena
The time scale of biological phenomena occurring during the study was measured. An example of such a biological phenomenon (waving the tail of fish) is shown in figure 1.
When ketamine and urea were added simultaneously to the water, the fish became less active within 40 seconds. The fish then became very excited after 5 minutes. This indicates that approximately 5 minutes is required for an unknown pathway of signal transduction to exert its effects. Ketamine had little impact on fish movement; thus, the sedative effect observed was induced by urea. This result confirmed that different pathways of signal transduction mediated the sedative and convulsive activities of urea. The time required for transport of the anesthetic from the medium to the brain of the fish varied between approximately 40 seconds and 3 minutes. The drug reached its equilibrium concentration in the body within 10 minutes. This conclusion was supported by fact that prolonging the time did not increase the number of fish anesthetized.
The NMDA receptor and the full protein target of ethanol anesthesia
The above experiments cannot be used to identify the main targets of urea or those of other general anesthetics. One way to investigate their identity is to compare the flexibility of protein targets. Therefore, we measured the flexibility of the NMDA receptor (23) and the full protein target of ethanol anesthesia.
As shown in Figure 2, urea at low concentrations (0.2M) could greatly amplify the effect of ketamine. In contrast, cooperativity between urea and ethanol only occurred at very high concentrations (> 1.2 M urea). Use of urethane gave similar results to those obtained with ethanol (data not shown). These results suggest that the NMDA receptor for ketamine shows a high protein flexibility, while the full protein target of ethanol anesthesia is more rigid.
The anesthesia titration curve of ketamine and ethanol vs. urea. The normalized concentration of ketamine (○) or ethanol (▲) required for anesthesia production. The concentration of ketamine (125 mg/L) or ethanol (2% by volume) required for anesthesia production in the absence of urea was 100% (in order to analyze and compare results between different tests, we explored normalized concentration, rather than absolute concentration). The temperature was 22℃ and Beijing summer grass carp were used in this test. The titration tests were done at variable urea concentrations and the ketamine (ethanol) concentration, at which the fish can be fully anesthetized, was measured. The urea could cooperate with ethanol in anesthesia induction at high concentrations; (see at 1.2 M). In contrast, urea inhibited NMDA receptor activity at rather low concentrations, e.g. 0.2 M. Each of data points was from one test (10 fish), and it was further ascertained once or more at same concentration.
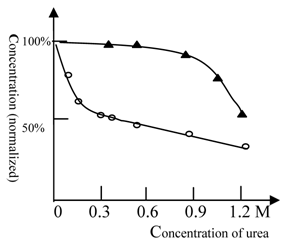
Urea-sensitive and urea-insensitive NMDA receptors
Distinct sensitivity differences were seen between winter grass carp and summer grass carp in response to urea.
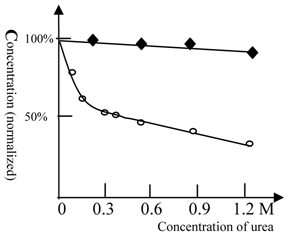
As shown in figure 3, ketamine anesthesia of Beijing winter carp showed a reduced sensitivity to urea. For Beijing summer grass carp, the concentration of ketamine required for anesthesia at 0.3 M urea was 50% lower than that for control test (with no urea). However, urea at even higher concentrations did not affect the concentration of ketamine required for anesthesia in Beijing winter grass carp (Figure 3). This result indicates that Beijing winter grass carp do not have urea-sensitive NMDA receptors, but only possess the urea-insensitive NMDA receptor. The anesthesia curves for winter and summer grass carp are shown in Figure 4.
The anesthesia titration of ketamine vs. urea for summer and winter grass carp. The concentration of ketamine (125 mg/L for summer and 63 mg/L for winter grass carp) required for anesthesia production in the absence of urea was taken as 100%. (◆) represents Beijing winter grass carp and (○) represents Beijing summer grass carp. The temperature was 22℃. The titration tests were done at variable urea concentrations and the ketamine concentration, at which the fish can be fully anesthetized, was measured. In contrast with summer grass carp, the ketamine anesthesia of winter grass carp was insensitive to urea. This indicated that the NMDA receptor of winter grass carp differed from that of summer grass carp. Each of data points was from one test (10 fish), and it was further ascertained once or more at same concentration.
The anesthesia curve of ketamine for winter and summer grass carp. (●) represents Beijing winter grass carp and (○) represents Beijing summer grass carp. The summer fish were transformed from winter fish after 10 days culture at 22 ℃. The temperature was 22℃. The ED50 of ketamine for winter grass carp is 65mg/L, whereas the ED50 of ketamine for summer grass carp is 115mg/L. The transformation from winter to summer fish was accompanied by substantial physiological changes and an increase in the concentration of ketamine required for anesthesia. Each of data points was from one test (10 fish), and it was further ascertained once or more at same concentration.
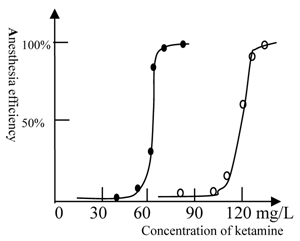
However, our result does not indicate the precise reasons for this difference in NMDA receptors; thus, further study using in vitro experiments are still required. Because a prolonged period of at least 10 days is required to transform a winter fish to a summer fish, it is evident that a substantial physiological change occurs in the fish during this transformation.
NMDA receptors are temperature sensitive
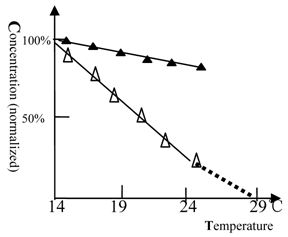
The discovery that Beijing winter grass carp do not possess urea-sensitive NMDA receptors implies that NMDA receptors play an important role in the heat endurance of fish. We then tested the temperature dependence of the response of Beijing grass carp to ketamine and ethanol anesthesia.
As shown in figure 5, when the temperature was elevated, the anesthetic concentration of ketamine dropped rapidly and tended to zero at 29 ℃. The Beijing summer grass carp gave a similar profile but at higher temperature (data not shown). This result shows that NMDA receptors are temperature sensitive. In contrast, the full protein target of ethanol anesthesia was less sensitive to temperature.
The anesthetic potency of ketamine or ethanol for winter grass carp vs. temperature. (△) represents ketamine and (▲) represents ethanol. The concentration of ketamine (150 mg/L) or ethanol (2.6% by volume) required for anesthesia production at 14℃ was 100%. The titration tests were done at variable temperature and the ketamine (ethanol) concentration, at which the fish can be fully anesthetized, was measured. At 27 or 29 °C, the fish become very excited and measurement of anesthesia became impossible. The ketamine concentration dropped to zero near 29 ℃, at this temperature heat coma state of fish could be found. The NMDA receptor was temperature sensitive. Each of the data points was from one test (10 fish), and it was further ascertained once or more at same concentration.
Titration of ketamine vs. urea at a constant concentration of midazolam and promethazine
Because there are no satisfactory drugs to use for the determination of protein flexibility related to the GABA receptor (24-27) or the histamine H1 and H2 receptors (28, 29), the protein flexibility of these receptors cannot be measured directly. Accordingly, we explored the technique of balanced anesthesia. Promethazine (a ligand of the H1 and H2 receptors) and midazolam (a powerful agonist of the GABAA receptor) can cooperate with ketamine in anesthesia production. Titration of ketamine against urea was performed at a constant concentration of midazolam or promethazine. These results are shown in figure 6.
Balanced anesthesia titration (ketamine vs. urea). The 100% concentration represents 38 mg/L of ketamine. (◆) represents promethazine (6.3 mg/L in all tests) and (▲) represents midazolam (5 mg/L in all tests). The titration tests were done at variable urea concentration and the ketamine concentration, at which the fish can be fully anesthetized, was measured. Beijing winter grass carp were selected because they lack the urea-sensitive NMDA receptor. Therefore, the alteration of the anesthetic concentration of ketamine reflects the alteration of GABA (or H1 and H2) receptor activity (or its flexibility) induced by urea, but not NMDA receptor. The temperature was 22℃. In this figure, the GABA receptor activity is biphasic. At urea concentrations below 0.6 mol/L, the protein flexibility was increased and GABA receptor activity rose. At high concentrations of urea, protein denaturation occurred and the GABA receptor gradually lost its activity. A similar phenomenon can be seen with many enzymes (e.g., 30). Each of data points was from one test (10 fish), and it was further ascertained once or more at same concentration.
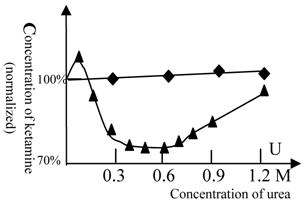
As NMDA receptors of winter fish are insensitive to urea, the change in the concentration of ketamine required for anesthesia production reflects the alteration in the activity of the GABA receptor (or the H1 and H2 receptors) induced by urea. The results show that urea slightly antagonizes the effect of promethazine. At a concentration of 0.1 M, urea slightly lessened the effect of midazolam, but at 0.3-0.6 M, urea greatly strengthened the effect of midazolam. This indicates that GABA receptor activity was greatly influenced by concentrations of 0.3-0.6 M urea.
Conclusion and discussion
The observed effect of urea is induced by increasing protein flexibility of a target receptor
According to well-established knowledge of biochemistry, the urea-protein interaction can be analyzed by two different mechanisms. The first is specific interaction between protein and urea which occurs at urea concentration below 0.1 M, within this mechanism the urea acts a ligand of a protein. The second is unspecific interaction between urea and protein, which occurs at urea concentration above 0.1 M, within this mechanism the urea acts molecules of solvent. The protein functional changes we measured in our tests were at urea concentrations of 0.1 M to 1.2 M, therefore the effect of urea is to increase protein flexibility (15-18). We use a broad sense of the concept of protein conformational change here, which would include protein denaturation, alteration of ligand-receptor (or drug) binding constants (30, 31), and dissociation or rearrangement of protein complexes (32).
It can be argued that there are undoubtedly a huge number of biological consequences following the addition of urea to a fish environment. To affirm our contention that anesthesia state is induced by increasing protein flexibility of desired receptor, we point out that:
- The time scale of anesthesia induction (1 minute), which is the time required for drug conveyance from solution to the fish brain (see figure 1), indicates that anesthesia is induced by direct alteration of protein functional conformation, otherwise, the time required for anesthesia induction would be longer, perhaps as long as 4 minutes.
- Only the co-effect (desired effect) of a urea-receptor-ligand can induce the full state of anesthesia and the observed behavioral change.
- If an undesired effect of other receptors were substantially taking place, this would have disturbed the experimental measurements. However, an undesired effect will show similar profiles in different tests. The figures we obtained differed greatly from each other, so we concluded that the undesired effect must be very small and it did not influence our judgment.
- The different effects of urea on different receptors (or proteins) could be separately measured (Figure 2-6).
These facts rule out many possible explanations for our test, such as disturbing the physiological state of fish (disturbing the equilibrium of biochemical networks, distribution of soluble biomaterials, etc.), altering cell permeability to ions and soluble compounds, or reducing binding constants due to changes in the ionic environment.
Ethanol at low concentration, such as 100 mmol/L, has a great impact on the in vivo function of many proteins, such as the NMDA receptor (33, 34), the GABA receptor (35, 36), kinases (37), and signal transduction activities (38-41). However, these effects are not detectable at the behavioral level (see figure 2), as the anesthesia concentration of ethanol is about 0.5 mol/L. This result indicates that the protein conformational change of the full protein target (as yet unknown) for ethanol anesthesia occurs at 0.5 M of ethanol. Therefore, the NMDA receptor and GABA receptor are not the full protein target of ethanol anesthesia. The NMDA receptor, as well as GABA receptor, are also not coupled to the full protein target of ethanol in anesthesia induction.
Ketamine is a ligand of many ion channels, including the NMDA receptor, nicotinic acetylcholine receptors (42), 5HT3 receptors (43), voltage-gated sodium channels (44) and the GABA receptor (45). It should be only cautiously asserted that the behavioral changes in fish are induced by the NMDA receptor and not other proteins. It is well-established that the NMDA receptor is a major excitatory receptor and it is the full protein target of ketamine anesthesia (1). Therefore, although ketamine can influence the functions of many proteins, it is reasonable to conclude that behavioral changes in fish are mainly induced by functional changes of the NMDA receptor.
Generally speaking, unless we know the essential background of ligand-receptor interaction, the explanation of behavioral change is very tenuous and we should be very cautious in explaining these data.
An increase in protein flexibility contributes to the mechanism of action of volatile general anesthetics
The discovery of the anesthetic potency of urea is significant for our understanding of the mechanism of action of volatile general anesthetics. It provides powerful evidence for the hypothesis that protein flexibility underlies the fundamental mechanism of anesthesia.
An increase in protein flexibility could result in many types of changes within a protein, such as a protein conformational change (46, 47), a decrease of protein conformational coupling between sensor and effector of a receptor (48), and dissociation or rearrangement of protein complexes. These changes can trigger signal transduction activity and influence many biological functions (38, 49). Urea and volatile general anesthetics are all enhancers of protein flexibility. Urea is weakly anesthetic and amphiphilic; its denaturing effect does not target hydrophobic sites only, but acts on all parts of a protein molecule. Volatile anesthetics are strong anesthetics and are targeted to only the hydrophobic sites of a protein. Therefore, we conclude that an increase of flexibility in the hydrophobic sites of proteins contributes to anesthesia production.
The GABA and NMDA receptors are not the full protein targets of ethanol anesthesia
There are many types of NMDA and GABA receptors (23-27). The particular type of receptor that is responsible for anesthesia was not investigated in this study. The full protein target of ethanol anesthesia is rigid, while the urea-sensitive NMDA and GABA receptors show high flexibility. Therefore, the GABA and NMDA receptors are not the full protein targets of ethanol anesthesia, although volatile general anesthetics have a major impact on their activities (1). Both GABA and NMDA receptors are flexible. Urea enhances the activity of GABA receptors (at a concentration of 0.2-0.5 M) and suppresses activity of the NMDA receptor. This is in agreement with the physiological functions of these receptors. As proteins show high flexibility at high temperatures, when most nerve cell receptors are easily excited, the requirement for NMDA receptor activity decreases and the requirement for GABA increases. The loss of NMDA receptor activity that is induced by high temperature results in heat coma, not anesthesia.
The opposite effect of NMDA and GBAB receptor activity on stimulus could be seen in many volatile anesthetics (1), such as ethanol (33-37), nitrous oxide (50, 51), and Xenon (50, 51). The protein flexibility of receptors can account for these phenomena.
The NMDA receptors play an important role in heat endurance of fish
Our studies demonstrate that NMDA receptors have the lowest heat stability. Heat coma in fish was accompanied by the loss of NMDA receptor function (Figure 5). If fish heat coma is not fully induced by the NMDA receptor, the receptor must at least play an important role in the phenomenon. The other evidence is that different types of NMDA receptors have been observed to act during adaptation to high temperatures. Heat coma in humans may be induced by the same mechanism, but further study of this syndrome is required.
Our results clearly show that in vivo administration of urea can have a major impact on biological function, and that this can be explored experimentally. The technique described in this paper is applicable to many research fields.
Acknowledgements
We would like to thank Professor HA Nash (Laboratory of Molecular Biology, National Institute of Mental Health, Building 36/Room 1B08, 9000 Rockville Pike, Bethesda, MD 20892-4034, USA), Dr. Zhenbao Yu (Pharmaceutical Sector, Biotechnology Research Institute, National Research Council of Canada, Montreal, Quebec, Canada H4P 2R2) for their valuable comments and suggestion during the writing of this manuscript.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Evers AS & Crowder CM. Cellular and molecular mechanisms of anesthesia. In: (ed.) Barash PG, Cullen BF, Stoelting RK. Clinical Anesthesia, 5th ed. Philadelphia: Lippincott Williams & Wilkins. 2006:111-132
2. Franks NP. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci. 2008;9:370-86
3. Campagna JA, Miller KW, Forman SA. Mechanisms of Actions of Inhaled Anesthetics. N Engl J Med. 2003;348:2110-2124
4. Eger EI, Raines DE, Shafer SL, Hemmings HC & Sonner JM. Is a New Paradigm Needed to Explain How Inhaled Anesthetics Produce Immobility? Anesth Analg. 2008;107:832-848
5. Franks NP, Lieb WR. Do general anaesthetics act by competitive binding to specific receptors? Nature. 1984;310:599-601
6. Franks NP, Lieb WR. Mapping of general anesthetic target sites provides a molecular basis for cutoff effects. Nature. 1985;316:349-351
7. Nash HA. In vivo genetics of anaesthetic action. Br J Anaesth. 2002;89:143-55
8. Sedensky MM, Pujazon MA & Morgan PG. Tail clamp responses in stomatin knockout mice compared with mobility assays in Caenorhabditis elegans during exposure to diethyl ether, halothane, and isoflurane. Anesthesiology. 2006;105:498-502
9. Ma D, Williamson P, Januszewski A, Nogaro MC, Hossain M, Ong LP, Shu Y, Franks NP & Maze M. Xenon mitigates isoflurane-induced neuronal apoptosis in the developing rodent brain. Anesthesiology. 2007;106:746-53
10. Yonkunas MJ, Xu Y & Tang P. Anesthetic interaction with ketosteroid isomerase: insights from molecular dynamics simulations. Biophys J. 2005;89:2350-2356
11. Ueda I. Effects of diethyl ether and halothane on firefly luciferin bioluminescence. Anesthesiology. 1965;26:603-606
12. Zhao Q. Irreversible thermodynamics theory of protein folding and protein thermodynamics structure. Progr Biophys biochem. 2001;28:429-435
13. Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Meth Enzymol. 1986;131:266-280
14. Cohen DM, Chin WW, Gullans SR. Hyperosmotic urea increases transcription and synthesis of Egr-1 in murine inner medullary collecting duct (mIMCD3) cells. J Biol Chem. 1994;269:25865-25870
15. Lopez MM & Kosk-Kosicka D. Spectroscopic Analysis of Halothane Binding to the Plasma Membrane Ca2+-ATPase. Biophys J. 1998;74:974-980
16. Griebenow K & Klibanov AM. On protein denaturation in aqueous-organic mixtures but not in pure organic solvents. J Am Chem Soc. 1996;118:11695-11700
17. Solt K, Johansson JS & Raines DE. Kinetics of anesthetic-induced conformational transitions in a four-alpha-helix bundle protein. Biochemistry. 2006;45:1435-41
18. Hartsough DS & Merz KM. Protein flexibility in aqueous and nonaqueous solutions. J. Am. Chem. Soc. 1992;114:10113-10116
19. Frauenfelder H, Parak F & Young RD. Conformational substrates in proteins. Annu Rev Biophys Chem. 1988;17:451-479
20. Tetreau C & Lavalette D. Dominant features of protein reaction dynamics: conformational relaxation and ligand migration. Biochem Biophys Acta. 2005;1724:411-424
21. Horvath L, Tamas G & Seagrave C. Carp and pond fish culture. London: Fishing News Books. 2002
22. Schaeffer DO. Nontraditional Laboratory Animals. In: (ed.) Kohn DF, Wixson SK, White WJ, Benson GJ. Anesthesia and Analgesia in Laboratory Animals. London: Academic Press. 1997:362-364
23. Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7-61
24. Korpi ER, Grunder G & Luddens H. Drug interactions at GABA(A) receptors. Prog Neurobio. 2002;67:113-59
25. Erwin S. The Benzodiazepine Recognition Site on GABAA Receptors. Med Chem Rev. 2005;2:251-256
26. Evans MS & Viola-McCabe KE. Midazolam inhibits long-term potentiation through modulation of GABAA receptors. Neuropharmacology. 1996;35:347-57
27. Rudolph U, Mohler H. Analysis of GABAA receptor function and dissection of the pharmacology of ben- zodiazepines and general anesthetics through mouse genetics. Annu Rev Pharmacol Toxicol. 2004;44:475-498
28. Hill SJ, Ganellin CR, Timmerman H, Schwartz JC, Shankley NP, Young JM, Schunack W, Levi R & Haas HL. International Union of Pharmacology. XIII. Classification of Histamine Receptors. Pharmacol Rev. 1997;49:253-278
29. Jafri MS, Moore KA, Taylor GE, Weinreich D. Histamine H1 receptor activation blocks two classes of potassium current, IK(rest) and IAHP, to excite ferret vagal afferents. J Physiol. 1997;503:533-546
30. Maurizi G, Pitari G, Duprè S. Conformational changes in pantetheine hydrolase as a function of guanidinium chloride concentration. J Protein Chem. 1995;14:373-9
31. Sigman JA, Patwa TH, Tablante AV, Joseph CD, Glucksman MJ & Wolfson A J. Flexibility in substrate recognition by thimet oligopeptidase as revealed by denaturation studies. Biochem J. 2005;388:255-261
32. Slater SJ, Cook AC, Seiz JL, Malinowski SA, Stagliano BA & Stubbs CD. Effects of Ethanol on Protein Kinase C α Activity Induced by Association with Rho GTPases. Biochemistry. 2003;42:12105-12114
33. Lovinger DM, White G, Weight FF. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science. 1989;243:1721-1724
34. Kumari M & Ticku MK. Regulation of NMDA receptors by ethanol. Prog Drug Res. 2000;54:152-189
35. Mameli M, Botta P, Zamudio PA, Zucca S & Valenzuela CF. Ethanol Decreases Purkinje Neuron Excitability by Increasing GABA Release in Rat Cerebellar Slices. J Pharmacol Exp Ther. 2008;327:910- 917
36. Roberto M, Madamba SG, Moore SD, Tallent MK & Siggins GR. Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons. Proc Natl Acad Sci USA. 2003;100:2053-2058
37. Lai CC, Kuo TI & Lin HH. The Role of Protein Kinase A in Acute Ethanol-Induced Neurobehavioral Actions in Rats. Anesth Analg. 2007;105:89-96
38. Ron D. Signaling cascades regulating NMDA receptor sensitivity to ethanol. Neuroscientist. 2004;10:325-336
39. Chen S & Charness ME. Ethanol inhibits neuronal differentiation by disrupting activity-dependent neuroprotective protein signaling. Proc Natl Acad Sci USA. 2008;105:19962-19967
40. Harvey-Girard E, Dunn RJ. Excitatory amino acid receptors of the electrosensory system: the NR1/NR2B N-methyl-D-aspartate receptor. J Neurophysiol. 2003;89:822-32
41. Mitchell DC, Lawrence JTR & Litman BJ. Primary alcohols modulate the activation of the G protein-coupled receptor rhodopsin by a lipid-mediated mechanism. J Biol Chem. 1996;271:19033-19036
42. Furuya R, Oka K, Watanabe I, Kamiya Y, Itoh H & Andoh T. The effects of ketamine and propofol on neuronal nicotinic acetylcholine receptors and P2x purinoceptors in PC12 cells. Anesth Analg. 1999;88:174-80
43. Peters JA, Malone HM & Lambert JJ. Ketamine potentiates 5-HT3 receptor-mediated currents in rabbit nodose ganglion neurones. Br J Pharmacol. 1991;103:1623-1625
44. Reckziegel G, Friederich P & Urban BW. Ketamine effects on human neuronal Na+ channels. Eur J Anaesthesiol. 2002;19:634-640
45. Irifune M, Sato T, Kamata Y, Nishikawa T, Dohi T & Kawahara M. Evidence for GABAA Receptor Agonistic Properties of Ketamine: Convulsive and Anesthetic Behavioral Models in Mice. Anesth Analg. 2000;91:230-236
46. Kenakin T. Inverse, protean, and ligand-selective agonism: matters of receptor conformation. Faseb J. 2001;15:598-611
47. Kaech S, Brinkhaus H & Matus A. Volatile anesthetics block actin-based motility in dendritic spines. Proc. Natl. Acad. Sci. USA. 1999;96:10433-10437
48. Zhao Q. Protein thermodynamic structure. IUBMB life. 2009;61:600-606
49. Dickinson R, Peterson BK, Banks P, Simillis C, Martin JC, Valenzuela CA, Maze M & Franks NP. Competitive inhibition at the glycine site of the N-methyl-D-aspartate receptor by the anesthetics xenon and isoflurane: evidence from molecular modeling and electrophysiology. Anesthesiology. 2007;107:756-67
50. Hapfelmeier G, Zieglgänsberger W, Haseneder R, Schneck H & Kochs E. Nitrous Oxide and Xenon Increase the Efficacy of GABA at Recombinant Mammalian GABAA Receptors. Anesth Analg. 2000;91:1542-1549
51. Dinse A, Fohr KJ, Georgieff M, Beyer C, Bulling A & Weigt HU. Xenon reduces glutamate-, AMPA-, and kainate-induced membrane currents in cortical neurons. Br J Anaesth. 2005;94:479-485
Author contact
![]() Correspondence to: Qinyi Zhao, P.O. Box 2619, Beijing, 100068 PR China. Email: qyzhcom
Correspondence to: Qinyi Zhao, P.O. Box 2619, Beijing, 100068 PR China. Email: qyzhcom