Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2009; 5(5):428-437. doi:10.7150/ijbs.5.428 This issue Cite
Research Paper
Nuclear Localization of p38 MAPK in Response to DNA Damage
C. David Wood1, Tina M. Thornton1, Guadalupe Sabio2, Roger A. Davis2, Mercedes Rincon1 ![]()
1. Department of Medicine/Immunobiology Program, University of Vermont, Burlington, VT 05405. USA.
2. Program in Molecular Medicine, University of Massachusetts, Worcester, MA 01605. USA.
Received 2009-4-30; Accepted 2009-6-10; Published 2009-6-16
Abstract
p38 MAP kinase (MAPK) is activated in response to environmental stress, cytokines and DNA damage, and mediates death, cell differentiation and cell cycle checkpoints. The intracellular localization of p38 MAPK upon activation remains unclear, and may depend on the stimulus. We show here that activation of p38 MAPK by stimuli that induce DNA double strand breaks (DSBs), but not other stimuli, leads to its nuclear translocation. In addition, naturally occurring DSBs generated through V(D)J recombination in immature thymocytes also promote nuclear accumulation of p38 MAPK. Nuclear translocation of p38 MAPK does not require its catalytic activity, but is induced by a conformational change of p38 MAPK triggered by phosphorylation within the active site. The selective nuclear accumulation of p38 MAPK in response to DNA damage could be a mechanism to facilitate the phosphorylation of p38 MAPK nuclear targets for the induction of a G2/M cell cycle checkpoint and DNA repair.
Keywords: p38 MAP kinase, DNA damage, cell cycle checkpoint, DNA repair
Introduction
The p38 MAPK family are a group of kinases belonging to the MAPK family, together with the JNK and ERK groups. There are four different isoforms of p38 MAPK (p38α, p38β, p38γ, and p38δ). p38α is the most abundant and widely expressed, but it is also the only isoform with a non-redundant function in vivo [1-5]. p38 MAPK is activated following phosphorylation at Thr180/Tyr182 within the active site. This phosphorylation is mediated primarily by upstream MKK3 and MKK6, although in vitro, MKK4 may also contribute to p38 MAPK activation in the absence of MKK3/MKK6 [6-8]. MKK3 and MKK6 in turn are regulated by phosphorylation through upstream MAPK kinase kinases (MAPKKK).
The p38 MAPK pathway is activated in response to environmental stress (UV, ionizing radiation, oxidative stress, and FAS ligand) and cytokines (e.g. TNFα) [9]. Activation of the p38 MAPK pathway by cytokines and receptor ligands, normally leads to cell differentiation. Activation of p38 MAPK through environmental stress can mediate cell death. In response to DNA damage stimuli that induce DSBs (ionizing radiation, UV, chemotherapeutic drugs) activation of p38 MAPK can also lead to the induction of a G2/M cell cycle checkpoint through p53-dependent and independent mechanisms [10-15]. p38 MAPK can also be activated by naturally generated DSBs in immature thymocytes at the CD4-CD8- double negative (DN)3 stage, whilst they undergo V(D)J recombination of the T cell receptor (TCR)β gene.
Despite the number of studies showing activation of p38 MAPK in response to DNA damage inducing stimuli and its role in the G2/M checkpoint, the mechanism by which p38 MAPK is activated, and its intracellular distribution is unclear. Unlike other MAPK, p38 MAPK has no nuclear localization signal, and has been shown to be distributed throughout the cytosol and nucleus [16]. However, in response to specific stimuli p38 MAPK has been shown to preferentially accumulate in the cytosol [17]. Depending on the stimuli, p38 MAPK can have a variety of substrates, including transcription factors (ATF2, MEF2), protein kinases (MnK, MAPKAPK2), death/survival molecules (Bcl2, caspases), and cell cycle control factors (cyclin D1) [18-22]. Thus, it is possible that the intracellular distribution of p38 MAPK is associated with its substrate specificity and determined by the nature of the stimuli.
We show here that p38 MAPK translocates to the nucleus specifically upon activation by stimuli that induce DSBs, and that p38 MAPK nuclear translocation is triggered by a conformational change induced by phosphorylation within the active site. This specific nuclear translocation could be relevant for this pathway to regulate the initiation of a G2/M cell cycle checkpoint and promote DNA repair.
Results
DNA Damage Induces the Nuclear Translocation of p38 MAPK
Despite its role in the induction of cell cycle checkpoints in response to DSBs inducing stimuli, little is known about the intracellular distribution of p38 MAPK following its activation by DSBs. Unlike other MAPKs, p38 MAPK lacks a specific nuclear localization sequence. We therefore investigated the intracellular localization of p38 MAPK in response to UV radiation, which induces DNA single strand damage that can lead to the formation of DNA DSBs [23]. 293T cells were transfected with p38 MAPK and exposed to UV irradiation. Cells were stained for p38 MAPK and examined by confocal microscopy. In unexposed cells, p38 MAPK was distributed throughout the cell, but predominantly outside the nucleus (Fig 1A). Interestingly, following exposure to UV, p38 MAPK accumulated in the nucleus (Fig 1A). The generation of DSBs in response to UV exposure was monitored by staining cells for the presence of phosphorylated H2AX at Ser139 (termed γH2AX), a known indicator of DNA DSBs (Fig 1A) [24, 25]. To corroborate that this nuclear translocation was due to the presence of DSBs, cells were exposed to X-radiation, a known source of ionizing radiation that induces DNA DSBs. Similar to UV exposure, X-radiation also caused a translocation of p38 MAPK to the nucleus (Fig 1A). To visualize phospho-p38 in this study, an antibody that recognizes p38 MAPK which has been dually phosphorylated on both Thr180/Tyr182 was used. Staining for phospho-p38 MAPK showed no phosphorylation of p38 MAPK in unexposed cells (Fig 1B), but high levels of phosphorylated p38 MAPK specifically in the nuclei of cells exposed to either UV or X-radiation (Fig 1B). Thus, nuclear localization of p38 MAPK in response to DNA damage inducing stimuli is associated with its phosphorylation.
Nuclear Translocation of p38 MAPK in Response to DNA Damage Stimuli. (A) 293T cells were transiently transfected with wild type p38 MAPK and left unstimulated (unstim), or treated with UV irradiation (UV), or X-radiation (X-Ray). Cells were stained for p38 MAPK (green) and γH2AX (red). TOPRO-3 (blue) was used as a nuclear marker. (B) 293T cells were transfected and treated as in (A). Cells were stained for p38 MAPK (green), and phospho-p38 MAPK (red). 'Merge' indicates the merged images of TOPRO-3 and p38 MAPK or TOPRO-3, p38 MAPK, and phospho-p38 MAPK. (C) and (D) 293T transfected cells as in (A) were treated with TNFα (10 ng/ml), Ig-FASL (200 ng/ml) and cross linker (4 μg/ml), or PMA (10 ng/ml) for 30 mins. Staining and confocal analysis were performed as described in (A). Data representative of at least three independent experiments.
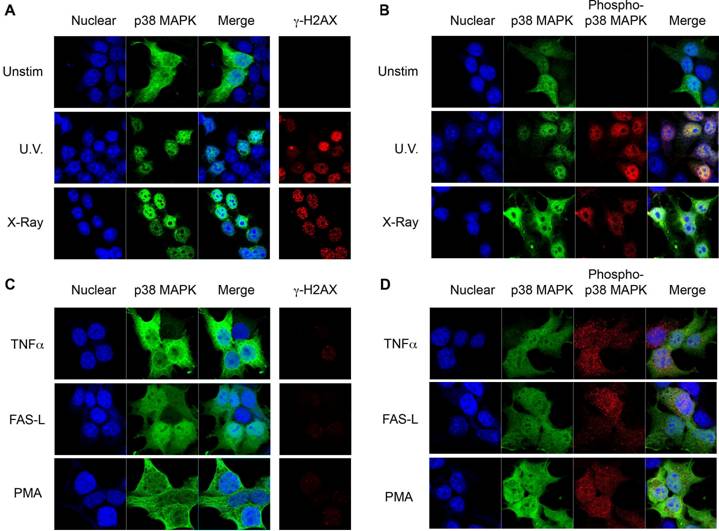
Since a nuclear translocation of p38 MAPK upon cell stimulation has not been previously reported, we examined the distribution of p38 MAPK in response to other known activators which do not induce DNA damage. Treatment with TNFα, one of the first identified activators of p38 MAPK, did not cause p38 MAPK nuclear accumulation (Fig 1C), although p38 MAPK phosphorylation was induced (Fig 1D). Similarly, Fas ligation or PMA activated p38 MAPK (Fig 1D), but did not promote its nuclear accumulation (Fig 1C). Thus, p38 MAPK is selectively translocated to the nucleus in response to stimuli that generate DSBs.
Phosphorylation of p38 MAPK is Essential for its Nuclear Translocation in Response to DNA Damage
p38 MAPK is activated following phosphorylation at Thr180/Tyr182 by upstream MAPKKs [16]. In order to assess whether phosphorylation of p38 MAPK was required for its nuclear translocation following DNA damage, cells were transfected with either wild type p38 MAPK or a dominant negative p38 MAPK (dnp38) mutant where Thr180/Tyr182 have been substituted with non-phosphorylatable Ala/Phe residues respectively [16]. Whilst wild type p38 MAPK was distributed throughout the cells, dnp38 was totally excluded from the nucleus in untreated cells (Fig 2A). In contrast to wild type p38 MAPK which accumulated preferentially in the nucleus, dnp38 remained in the cytosol and was excluded from the nucleus following exposure to X-radiation (Fig 2A). The presence of γH2AX showed that cells transfected with dnp38 have DSBs, indicating the activation of the DNA damage response (Fig 2A). Similar to X-radiation, UV exposure failed to induce nuclear translocation of dnp38 (Fig 2B). Thus, phosphorylation of p38 MAPK at Thr180/Tyr182 is required for the nuclear translocation of p38 MAPK following DNA damage.
p38 MAPK Phosphorylation is Required for its Nuclear Translocation. (A) 293T cells were transiently transfected with either wild type or dnp38 MAPK and left unstimulated (unstim), or treated with X-Ray. Cells were stained for p38 MAPK (green) and γH2AX (red). TOPRO-3 (blue) was used as a nuclear marker. (B) 293T cells were transfected with dnp38 MAPK and left unstimulated (unstim), or treated with UV irradiation (UV). Staining and confocal analysis were performed as described in (A). (C) and (D) 293T cells transiently transfected with wild type p38 MAPK, were treated with SB203580 (5 μM) 40 mins, and left unstimulated or exposed to X-Ray (C) or UV (D). Staining and confocal analysis were performed as described in (A). 'Merge' indicates the merged images of TOPRO-3 and p38 MAPK. Data representative of at least three independent experiments.
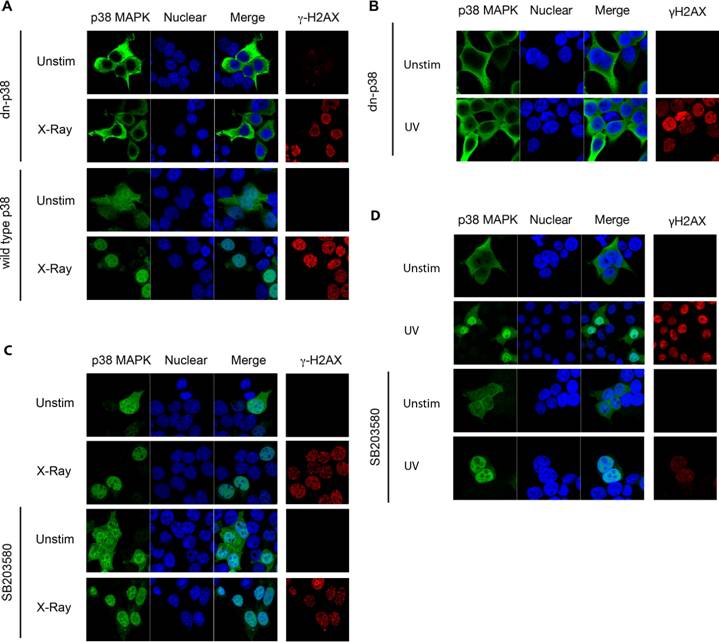
Phosphorylation of p38 MAPK leads to its activation by inducing a conformational change. To dissociate whether p38 MAPK activity, or solely the conformational change caused by the phosphorylation was required for nuclear translocation we examined the effect of a pharmacological p38 MAPK inhibitor, SB203580 [26], which binds at the catalytic site and blocks its activity. Cells were transfected with wild type p38 MAPK and pretreated with SB203580 prior to X-radiation exposure. Interestingly, SB203580 did not prevent nuclear translocation of p38 MAPK in response to X-radiation (Fig 2C). Similarly, no effect of SB203580 on UV-induced nuclear translocation of p38 MAPK could be detected (Fig 2D). Thus, catalytic activity of p38 MAPK is not required for its nuclear translocation in response to DNA damage, suggesting that a conformational change caused by upstream phosphorylation promotes its nuclear transport.
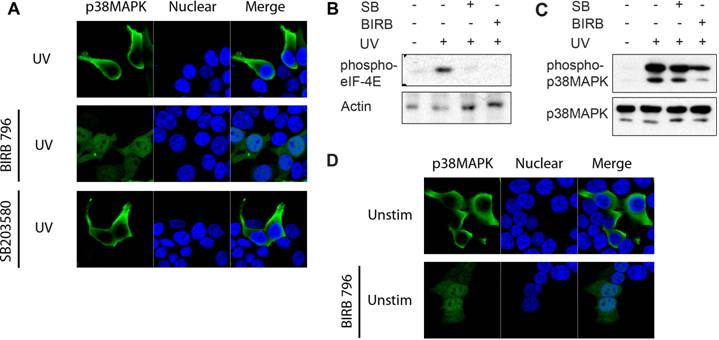
Unlike SB203580, BIRB 796 belongs to a new class of p38 MAPK inhibitors, inhibiting p38α MAPK via a novel mechanism which indirectly competes with ATP binding by inducing a conformational change [27]. Binding of BIRB 796 to p38 MAPK causes a large structural reorganization of the activation loop (residues 171-184) and the resulting structure is non-compatible with ATP-binding. This conformational change can resemble that induced by phosphorylation at Thr180/Tyr182. Since SB203580 did not affect p38 MAPK translocation, despite inhibiting p38 MAPK catalytic activity, we investigated whether a conformational change induced by BIRB 796 could affect the localization of the dnp38 mutant by mimicking the effect of phosphorylation. Cells were transfected with dnp38 and pretreated with BIRB 796 prior to UV exposure. Although dnp38 remained in the cytosol, the presence of BIRB 796 restored its nuclear translocation (Fig 3A). In contrast, dnp38 did not translocate to the nucleus in the presence of SB203580 (Fig 3A). To show that both compounds were able to inhibit the activity of p38 MAPK in these cells, we examined downstream targets of p38 MAPK. We analyzed the activation of Mnk kinase since it is a primary target for p38 MAPK but not for JNK [19]. Mnk1/2 mediate phosphorylation and activation of the translation initiator factor 4E (eIF-4E), and phosphorylation of eIF-4E is usually used as a read out system for activity of Mnk [19]. eIF-4E phosphorylation was induced upon UV exposure, but the presence of either SB203580 or BIRB 796 blocked its phosphorylation (Fig 3B). Thus, both pharmacological compounds are able to interfere with the catalytic activity of p38 MAPK. We also examined the effect of both inhibitors on the phosphorylation of p38 MAPK at the Thr180/Tyr182 motif by upstream MAPKK. According to its mechanism of action, SB203580 did not interfere with the phosphorylation of p38 MAPK (Fig 3C). In contrast, BIRB 796 reduced the phosphorylation of p38 MAPK at this domain, supporting the conformational change effect that this compound has (Fig 3C). Analysis of p38 MAPK cellular localization in non-UV exposed cells treated with BIRB 796, indicated that BIRB 796 by itself was also able to promote accumulation of dnp38 to the nucleus in the absence of other stimuli (Fig 3D). This further supports that p38 MAPK activation loop phosphorylation is critical to its nuclear translocation by inducing a conformational change.
p38 MAPK Phosphorylation Induces a Conformational Change that is Required for Nuclear Translocation. (A) 293T cells transfected with dnp38 MAPK were treated with BIRB 796 (5 μM) 40 mins, and exposed to UV irradiation (UV). Cells were stained for p38 MAPK (green) and TOPRO-3 (blue). 'Merge' indicates the merged images of TOPRO-3 and p38 MAPK. (B) 293T cells transiently transfected with wild type p38 MAPK, were left unstimulated or exposed to UV in the presence or absence of SB203580 (5 μM) or BIRB 796 (5 μM). Whole cell extracts were then examined for phosphorylated eIF-4E by Western blot analysis. Actin levels were examined as a loading control. (C) Cells were exposed and treated as described in (B) and whole cell extracts were used to examine phospho-p38 MAPK and total p38 MAPK by Western blot analysis. (D) 293T cells transfected with dnp38 MAPK were treated with vehicle or BIRB 796 for 40 mins. Cells were stained as described in (A). Data representative of at least three independent experiments.
Nuclear Translocation of Endogenous p38 MAPK in Thymocytes Following Ionizing Radiation
To show that nuclear accumulation of endogenous p38 MAPK in response to ionizing radiation also occurs in primary cells, we examined endogenous p38 MAPK distribution in thymocytes. Total thymocytes from wild type mice were exposed to X-radiation, and stained for endogenous p38 MAPK. The intracellular distribution of p38 MAPK was examined by confocal microscopy. Unlike 293T cells, thymocytes have a very limited amount of cytoplasm (immediately surrounding the nucleus). Nonetheless, p38 MAPK could be detected in the cytosol of thymocytes prior to exposure (Fig 4A). However, exposure to ionizing radiation resulted in a nuclear accumulation of p38 MAPK (Fig 4A). These results further demonstrate that p38 MAPK specifically translocates to the nucleus in response to DNA damage.
Nuclear Translocation of Endogenous p38 MAPK in Thymocytes in Response to DNA Damage. (A) Total thymocytes from wild type mice were left unstimulted (unstim), or exposed to X-Ray, and stained for p38 MAPK (red) and YoYo (green). (B) Whole cell extracts from total and purified DN thymocytes from wild type and MKK3-/-/MKK6+/- mice were examined for phospho-p38 MAPK and p38 MAPK by western blot analysis (C) Total thymocytes from both wild type and MKK3-/-/MKK6+/- mice were left unstimulted (unstim), or treated with X-Ray, and stained for p38 MAPK (red), γH2AX (green), and TOPRO-3 (blue). 'Merge' indicates the merged images of TOPRO-3 and p38 MAPK. A higher magnification view of the insets (marked with white boxes) in the 'merge' panels is shown on the right side. (D) Graph displaying the percentage of cells demonstrating p38 MAPK nuclear accumulation. Determined in total thymocytes from WT and MKK3-/-/MKK6+/- mice left unsitmulated or exposed to X-Ray (total count >150 cells per population, counted from 3 independent areas of visualization). Error bars represent SEM. Data representative of at least three independent experiments.
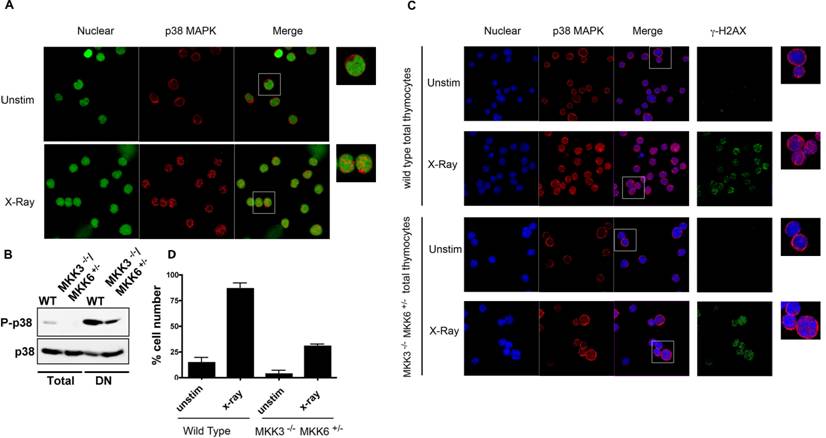
MKK3 and MKK6 are the main MAPKKs that phosphorylate and activate p38 MAPK [28]. Double deficiency for both MKK3 and MKK6 (MKK3-/-/MKK6-/-) in mice causes embryonic lethality, similar to p38α MAPK deficiency [8]. In contrast, MKK3-/-/MKK6+/- mice are viable, but p38 MAPK activation is severely compromised (Sabio, G. et al unpublished data). To confirm that activation of p38 MAPK was impaired in thymocytes from these mice we examined the levels of phospho-p38 MAPK by Western blot analysis. The levels of phospho-p38 MAPK were reduced in total thymocytes from MKK3-/-/MKK6+/- mice compared with the levels in wild type thymocytes (Fig 4B). We also examined the levels of activated p38 MAPK in the double negative (DN) thymocyte population where p38 MAPK is constitutively activated [29]. The levels of phospho-p38 MAPK were also substantially reduced in DN thymocytes from MKK3-/-/MKK6+/- mice (Fig 4B). Thus, activation of p38 MAPK is impaired in MKK3-/-/MKK6+/- thymocytes. Wild type and MKK3-/-/MKK6+/- thymocytes were then exposed to X-radiation, and p38 MAPK intracellular distribution was assessed. As described above, X-radiation induced the nuclear accumulation of p38 MAPK in wild type total thymocytes (Fig 4C and 4D). In contrast, nuclear translocation of p38 MAPK was severely compromised in MKK3-/-/MKK6+/- thymocytes (Fig 4C). The presence of DSBs, as determined by γH2AX staining, was comparable between exposed wild type and MKK3-/-/MKK6+/- thymocytes (Fig 4C), indicating that MKK3-/-/MKK6+/- thymocytes responded to X-radiation. These results demonstrate that in primary cells, endogenous p38 MAPK translocates to the nucleus in response to DNA damage, and that nuclear translocation requires its phosphorylation.
V(D)J recombination mediated DSBs Induce the Nuclear Translocation of p38 MAPK
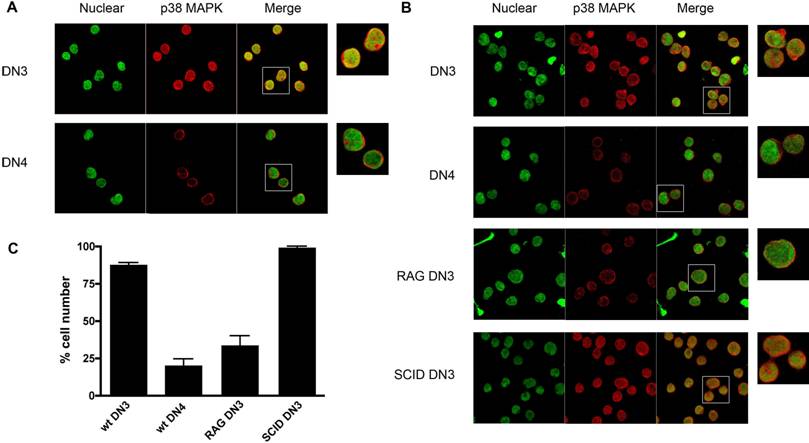
p38 MAPK is activated by V(D)J-mediated DSBs during recombination of the TCRβ gene in DN3 (CD25+ CD44-) thymocytes and it is inactivated once DN3 thymocytes differentiate into DN4 (CD25- CD44-) thymocytes, and DNA repair has taken place [30]. We therefore examined whether activation of p38 MAPK in response to V(D)J-mediated DSBs in DN3 thymocytes also leads to nuclear accumulation of this kinase. DN3 and DN4 thymocytes were isolated from wild type mice by cell sorting, stained for p38 MAPK and examined by confocal microscopy. p38 MAPK localized primarily in the nucleus of most DN3 thymocytes, whilst it was excluded from the nucleus in DN4 thymocytes (Fig 5A). Thus, nuclear accumulation of p38 MAPK in DN3 thymocytes correlates with the activation of p38 MAPK by V(D)J-mediated DSBs in this thymocyte population. To demonstrate that the nuclear accumulation of p38 MAPK in DN3 thymocytes was caused by V(D)J-mediated DSBs, we examined the localization of p38 MAPK in DN3 thymocytes isolated from RAG1 deficient mice since they have no V(D)J-mediated DSBs, due to the lack of the RAG1 recombinase. Minimal nuclear accumulation of p38 MAPK was detected in DN3 thymocytes from RAG1 deficient mice compared with that in wild type DN3 thymocytes (Fig 5B and 5C). Unlike RAG1-deficient thymocytes, thymocytes from SCID (Severe Combined Immuno Deficient) mice undergo V(D)J recombination, but they are unable to repair DNA damage due to a deficiency in DNA-PK. As a result, thymocyte development in these mice is also arrested at the DN3 stage, but V(D)J-mediated DNA DSBs are continuously present in these cells since they cannot be repaired. In contrast to DN3 thymocytes from RAG1 mice, most SCID DN3 thymocytes showed p38 MAPK in the nucleus (Fig 5C). Together, these results show that V(D)J-mediated DSBs promote the nuclear accumulation of endogenous p38 MAPK, comparable to the response observed upon ionizing irradiation exposure.
V(D)J-mediated DSBs Induce p38 MAPK Translocation in DN3 Thymocytes. (A) DN3 and DN4 thymocytes purified from wild type mice, were stained with for p38 MAPK (red) and YoYo (green). (B) DN3 and DN4 thymocytes purified from wild type mice and DN3 thymocytes from SCID, and RAG1-/- mice were stained as in (A). A higher magnification view of the insets (marked with white boxes) in the 'merge' panels is shown on the right side. Experiment representative of at least three independent experiments. (C) Graph displaying the percentage of cells demonstrating p38 MAPK nuclear accumulation in different cell populations (total count >150 cells per population, counted from 3 independent areas of visualization). Error bars represent SEM. Data representative of four experiments and multiple fields of view.
Discussion
p38 MAPK has no nuclear localization signal, and it is believed to be diffused throughout the nucleus and the cytoplasm. In this study, we show that p38 MAPK accumulates in the nucleus specifically in response to stimuli that induce DSBs, but not by other stimuli (e.g. TNFα, FasL) that also activate p38 MAPK. The intracellular distribution of p38 MAPK can therefore be determined by the nature of the stimuli and can influence p38 MAPK targets. Thus, DNA damage stimuli promotes nuclear accumulation of p38 MAPK for this kinase to phosphorylate potential nuclear targets (e.g. p53) involved in the induction of cell cycle checkpoints. However, arsenite treatment has been shown to promote cytoplasmic accumulation of p38 MAPK [17].
We also show here that nuclear translocation of p38 MAPK in response to DNA damage stimuli requires phosphorylation of Thr180/Tyr182 by MKK3 and/or MKK6, but it does not require its catalytic activity. This suggests that phosphorylation of these residues may induce a conformational change that unmasks a domain enabling p38 MAPK to interact with a nuclear shuttling chaperone to mediate its re-localization to the nucleus. To date, we cannot conclude whether phosphorylation of one or both residues is necessary for nuclear translocation. The results showing that BIRB 796, but not SB203580, restores nuclear localization of dnp38 MAPK further support the effect of a conformational change. Since phosphorylation of p38 MAPK in response to DNA damage, but not in response to other stimuli, promotes nuclear accumulation, it is possible that this nuclear shuttle is also specifically induced or activated by DNA damage, as described for NBS1 [31] and GADD45 [32, 33]. Thus, selective nuclear transport of p38 MAPK would require both its phosphorylation as well as the active nuclear shuttle to be present. Alternatively, DNA damage signals could also release p38 MAPK from docking molecules that retain p38 MAPK in the cytosol. In this regard, p38 MAPK strongly associates with MAPKAP kinase-2 (MAPKAPK2), a substrate for p38 MAPK which contains both nuclear localization and nuclear export sequences [34]. Binding of phosphorylated MAPKAPK2 to p38 MAPK appears to promote the retention of p38 MAPK in the cytosol [17]. Similarly, binding of TAB-1 (TAK-1 binding protein) to p38 MAPK also directs localization to the cytosol, and induces p38 MAPK autophosphorylation in a MAPKK-independent manner [35, 36]. It is plausible that the conformational change of p38 MAPK upon DNA damage disrupt its interaction with MAPKAPK2 or TAB1, enabling its accumulation in the nucleus.
Several studies have shown a role for p38 MAPK in the induction of a G2/M cell cycle checkpoint in response to DNA damage (ionizing radiation, UV), and phosphorylation of targets involved in this checkpoint such as p53 or Cdc25 [12, 37-39]. p38 MAPK is also activated by V(D)J-mediated DSBs and induces a G2/M cell cycle checkpoint in DN3 thymocytes [30], suggesting that V(D)J-mediated DSBs also represent DNA damage signals. We show here nuclear accumulation of p38 MAPK specifically in DN3 thymocytes undergoing V(D)J recombination (wild type and SCID), but not in DN3 thymocytes from RAG deficient mice. Thus, similar to the DSBs induced by ionizing radiation, V(D)J-mediated DSBs appear to be interpreted as a DNA damage signal. Recent studies have shown that signal ended DNA fragments released during V(D)J recombination of the TCRβ gene can integrate into different chromosomal locations, by either insertion or transposition [40-45], and may lead to genomic instability and potential malignancies. The selective nuclear accumulation of p38 MAPK in response to DNA damage could pose a mechanism to facilitate the phosphorylation of p38 MAPK nuclear targets for the induction of a G2/M cell cycle checkpoint and DNA repair.
In summary, the intracellular localization of p38 MAPK is regulated upon activation of this kinase, and is dependent on the stimuli involved. This most likely reflects the requirement of p38 MAPK to be in close proximity to its targets.
Materials and Methods
Mice
Wild type, RAG1-/-, and SCID mice were purchased from Jackson Laboratories. MKK3-/-and MKK6-/- single mice have been previously reported [46]. MKK3-/-/MKK6+/- were obtained by serial intercrosses of MKK3-/- and MKK6-/- single mice as previously reported [8]. Mice (five to seven weeks of age) were bred and maintained in specific pathogen-free conditions. Procedures involving mice were approved by institutional guidelines for animal care.
Cell Preparation, Transfection, and Stimulation
HEK293T cells were transfected using calcium phosphate. Following exposure to UV (1.5 mins, 15 J/m2), or X-radiation (2.8 Gy) (RadSource 2000, GA) cells were rested at 37ºC for 30 mins. SB203580 (Calbiochem, CA) and BIRB 796 (Axon Medchem) pretreatment (5 μM) was performed at 37ºC for 40 mins, prior to cell stimulation. Cells were additionally stimulated with PMA (Sigma) (10 ng/ml), TNFα (R&D Systems, MN) (10 ng/ml), or Ig-FasL (Alexis Biochemicals, CA) (200 ng/ml) and anti-Flag M2 mouse monoclonal antibody (Sigma) (4 μg/ml) at 37ºC for 30 mins, prior to fixation. DN3 and DN4 thymocytes were purified and stained (CD4, CD8, CD25, and CD44 BD Biosciences), sorted (FacsAria, BD Biosciences), and collected.
Confocal Microscopy and Analysis
Staining of 293T [47] cells and thymocytes [30] was performed as we previously published. Briefly, cells were fixed, permeabilized and stained with primary antibody followed by fluorescently labeled secondary antibody. The primary antibodies used included the rabbit anti-p38 MAPK polyclonal antibody (Santa Cruz, CA), mouse anti-γH2AX monoclonal antibody (Upstate, Millipore), rabbit anti-phospho-p38 MAPK monoclonal antibody #9215 recognizing p38 MAPK dually phosphorylated on Thr180/Tyr182 (Cell Signaling, MA). Secondary antibodies used include Alexa Fluor anti-mouse/anti-rabbit 488/567 highly adsorbed antibodies (Invitrogen). Topro-3 and YoYo were used as nuclear stains. Images of fixed cells were acquired on a Zeiss LSM-510 upright microscope using a 63x Plan Apochromat Ph3 objective lens.
Western Blot
Whole cell extracts were prepared in Triton lysis buffer (TLB) and used for Western blot analysis as we previously described [30] using anti-p38 MAPK (Santa Cruz, CA) and anti-phospho-p38 MAPK (Cell Signaling, MA). Anti-rabbit HRP, and anti-mouse HRP (Santa Cruz, CA) were used as secondary antibodies.
Acknowledgements
We would like to thank Douglas Taatjes and Marilyn Wadsworth at the UVM Microscopy Imaging Center. This work was supported by a NIH R01 A1051454 grant (M.R.).
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Adams R.H, Porras A, Alonso G, Jones M, Vintersten K, Panelli S, Valladares A, Perez L, Klein R, Nebreda A.R. Essential role of p38alpha MAP kinase in placental but not embryonic cardiovascular development. Mol Cell. 2000;6:109-116
2. Allen M, Svensson L, Roach M, Hambor J, McNeish J, Gabel C.A. Deficiency of the stress kinase p38alpha results in embryonic lethality: characterization of the kinase dependence of stress responses of enzyme-deficient embryonic stem cells. J Exp Med. 2000;191:859-870
3. Mudgett J.S, Ding J, Guh-Siesel L, Chartrain N.A, Yang L, Gopal S, Shen M.M. Essential role for p38alpha mitogen-activated protein kinase in placental angiogenesis. Proc Natl Acad Sci U S A. 2000;97:10454-10459
4. Tamura K, Sudo T, Senftleben U, Dadak A.M, Johnson R, Karin M. Requirement for p38alpha in erythropoietin expression: a role for stress kinases in erythropoiesis. Cell. 2000;102:221-231
5. Beardmore V.A, Hinton H.J, Eftychi C, Apostolaki M, Armaka M, Darragh J, McIlrath J, Carr J.M, Armit L.J, Clacher C. et al. Generation and characterization of p38beta (MAPK11) gene-targeted mice. Mol Cell Biol. 2005;25:10454-10464
6. Derijard B, Raingeaud J, Barrett T, Wu I.H, Han J, Ulevitch R.J, Davis R.J. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267:682-685
7. Han J, Lee J.D, Jiang Y, Li Z, Feng L, Ulevitch R.J. Characterization of the structure and function of a novel MAP kinase kinase (MKK6). J Biol Chem. 1996;271:2886-2891
8. Brancho D, Tanaka N, Jaeschke A, Ventura J.J, Kelkar N, Tanaka Y, Kyuuma M, Takeshita T, Flavell R.A, Davis R.J. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003;17:1969-1978
9. Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell research. 2005;15:11-18
10. She Q.B, Chen N, Dong Z. ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J Biol Chem. 2000;275:20444-20449
11. She Q.B, Bode A.M, Ma W.Y, Chen N.Y, Dong Z. Resveratrol-induced activation of p53 and apoptosis is mediated by extracellular-signal-regulated protein kinases and p38 kinase. Cancer Res. 2001;61:1604-1610
12. Bulavin D.V, Higashimoto Y, Popoff I.J, Gaarde W.A, Basrur V, Potapova O, Appella E, Fornace AJJr. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature. 2001;411:102-107
13. Mikhailov A, Shinohara M, Rieder C.L. The p38-mediated stress-activated checkpoint. A rapid response system for delaying progression through antephase and entry into mitosis. Cell Cycle. 2005;4:57-62
14. Kurosu T, Takahashi Y, Fukuda T, Koyama T, Miki T, Miura O. p38 MAP kinase plays a role in G2 checkpoint activation and inhibits apoptosis of human B cell lymphoma cells treated with etoposide. Apoptosis. 2005;10:1111-1120
15. Thornton T.M, Rincon M. Non-classical p38 map kinase functions: cell cycle checkpoints and survival. Int J Biol Sci. 2009;5:44-51
16. Raingeaud J, Gupta S, Rogers J.S, Dickens M, Han J, Ulevitch R.J, Davis R.J. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420-7426
17. Ben-Levy R, Hooper S, Wilson R, Paterson H.F, Marshall C.J. Nuclear export of the stress-activated protein kinase p38 mediated by its substrate MAPKAP kinase-2. Curr Biol. 1998;8:1049-1057
18. Cuenda A, Cohen P, Buee-Scherrer V, Goedert M. Activation of stress-activated protein kinase-3 (SAPK3) by cytokines and cellular stresses is mediated via SAPKK3 (MKK6); comparison of the specificities of SAPK3 and SAPK2 (RK/p38). EMBO J. 1997;16:295-305
19. Waskiewicz A.J, Flynn A, Proud C.G, Cooper J.A. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997;16:1909-1920
20. Zhao M, New L, Kravchenko V.V, Kato Y, Gram H, di Padova F, Olson E.N, Ulevitch R.J, Han J. Regulation of the MEF2 family of transcription factors by p38. Mol Cell Biol. 1999;19:21-30
21. Alvarado-Kristensson M, Melander F, Leandersson K, Ronnstrand L, Wernstedt C, Andersson T. p38-MAPK signals survival by phosphorylation of caspase-8 and caspase-3 in human neutrophils. J Exp Med. 2004;199:449-458
22. Trouillas M, Saucourt C, Duval D, Gauthereau X, Thibault C, Dembele D, Feraud O, Menager J, Rallu M, Pradier L. et al. Bcl2, a transcriptional target of p38alpha, is critical for neuronal commitment of mouse embryonic stem cells. Cell Death Differ. 2008;15:1450-1459
23. Jenner T.J, Fulford J, O'Neill P. Contribution of base lesions to radiation-induced clustered DNA damage: implication for models of radiation response. Radiation research. 2001;156:590-593
24. Rogakou E.P, Pilch D.R, Orr A.H, Ivanova V.S, Bonner W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858-5868
25. Sedelnikova O.A, Rogakou E.P, Panyutin I.G, Bonner W.M. Quantitative detection of (125)IdU-induced DNA double-strand breaks with gamma-H2AX antibody. Radiation research. 2002;158:486-492
26. Kumar S, McDonnell P.C, Gum R.J, Hand A.T, Lee J.C, Young P.R. Novel homologues of CSBP/p38 MAP kinase: activation, substrate specificity and sensitivity to inhibition by pyridinyl imidazoles. Biochem Biophys Res Commun. 1997;235:533-538
27. Pargellis C, Tong L, Churchill L, Cirillo P.F, Gilmore T, Graham A.G, Grob P.M, Hickey E.R, Moss N, Pav S. et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat Struct Biol. 2002;9:268-272
28. Enslen H, Raingeaud J, Davis R.J. Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J Biol Chem. 1998;273:1741-1748
29. Sen J, Kapeller R, Fragoso R, Sen R, Zon L.I, Burakoff S.J. Intrathymic signals in thymocytes are mediated by p38 mitogen-activated protein kinase. J Immunol. 1996;156:4535-4538
30. Pedraza-Alva G, Koulnis M, Charland C, Thornton T, Clements J.L, Schlissel M.S, Rincon M. Activation of p38 MAP kinase by DNA double-strand breaks in V(D)J recombination induces a G2/M cell cycle checkpoint. Embo J. 2006;25:763-773
31. Carney J.P, Maser R.S, Olivares H, Davis E.M, Le Beau M, Yates JR3rd, Hays L, Morgan W.F, Petrini J.H. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477-486
32. Hollander M.C, Alamo I, Jackman J, Wang M.G, McBride O.W, Fornace AJJr. Analysis of the mammalian gadd45 gene and its response to DNA damage. J Biol Chem. 1993;268:24385-24393
33. Zhu N, Shao Y, Xu L, Yu L, Sun L. Gadd45-alpha and Gadd45-gamma utilize p38 and JNK signaling pathways to induce cell cycle G2/M arrest in Hep-G2 hepatoma cells. Mol Biol Rep. 2008 [Epub ahead of print]
34. Engel K, Kotlyarov A, Gaestel M. Leptomycin B-sensitive nuclear export of MAPKAP kinase 2 is regulated by phosphorylation. EMBO J. 1998;17:3363-3371
35. Ge B, Gram H, Di Padova F, Huang B, New L, Ulevitch R.J, Luo Y, Han J. MAPKK-independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science. 2002;295:1291-1294
36. Lu G, Kang Y.J, Han J, Herschman H.R, Stefani E, Wang Y. TAB-1 modulates intracellular localization of p38 MAP kinase and downstream signaling. J Biol Chem. 2006;281:6087-6095
37. Dmitrieva N.I, Bulavin D.V, Fornace AJJr, Burg M.B. Rapid activation of G2/M checkpoint after hypertonic stress in renal inner medullary epithelial (IME) cells is protective and requires p38 kinase. Proc Natl Acad Sci U S A. 2002;99:184-189
38. Chao J.I, Kuo P.C. Role of p53 and p38 MAP kinase in nitric oxide-induced G2/M arrest and apoptosis in the human lung carcinoma cells. Carcinogenesis. 2004;25:645
39. Reinhardt H.C, Aslanian A.S, Lees J.A, Yaffe M.B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer cell. 2007;11:175-189
40. Messier T.L, O'Neill J.P, Finette B.A. V(D)J recombinase mediated inter-chromosomal HPRT alterations at cryptic recombination signal sequences in peripheral human T cells. Hum Mutat. 2006;27:829
41. Murray J.M, O'Neill J.P, Messier T, Rivers J, Walker V.E, McGonagle B, Trombley L, Cowell L.G, Kelsoe G, McBlane F. et al. V(D)J recombinase-mediated processing of coding junctions at cryptic recombination signal sequences in peripheral T cells during human development. J Immunol. 2006;177:5393-5404
42. Chatterji M, Tsai C.L, Schatz D.G. Mobilization of RAG-generated signal ends by transposition and insertion in vivo. Mol Cell Biol. 2006;26:1558-1568
43. Reddy Y.V, Perkins E.J, Ramsden D.A. Genomic instability due to V(D)J recombination-associated transposition. Genes Dev. 2006;20:1575-1582
44. Vanura K, Montpellier B, Le T, Spicuglia S, Navarro J.M, Cabaud O, Roulland S, Vachez E, Prinz I, Ferrier P. et al. In vivo reinsertion of excised episomes by the V(D)J recombinase: a potential threat to genomic stability. PLoS biology. 2007;5:e43
45. Curry J.D, Schulz D, Guidos C.J, Danska J.S, Nutter L, Nussenzweig A, Schlissel M.S. Chromosomal reinsertion of broken RSS ends during T cell development. J Exp Med. 2007;204:2293-2303
46. Tanaka N, Kamanaka M, Enslen H, Dong C, Wysk M, Davis R.J, Flavell R.A. Differential involvement of p38 mitogen-activated protein kinase kinases MKK3 and MKK6 in T-cell apoptosis. EMBO Rep. 2002;3:785-791
47. Hatle K.M, Neveu W, Dienz O, Rymarchyk S, Barrantes R, Hale S, Farley N, Lounsbury K.M, Bond J.P, Taatjes D. et al. Methylation-controlled J protein promotes c-Jun degradation to prevent ABCB1 transporter expression. Mol Cell Biol. 2007;27:2952-2966
Author contact
![]() Correspondence to: Mercedes Rincon, tel: +1 802 656 0937; fax: +1 802 656 3854; email: mercedes.rinconedu
Correspondence to: Mercedes Rincon, tel: +1 802 656 0937; fax: +1 802 656 3854; email: mercedes.rinconedu