Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Transcriptional control of...
PPARs and C/EBPs: the master...
Sterol regulatory element...
Other adipogenic factors
Krüppel-like factors...
Pro-adipogenic KLFs
KLFs as negative regulators
C. elegans fat biology
Role of KLFs in C. elegans fat...
Conclusions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2009; 5(6):622-636. doi:10.7150/ijbs.5.622 This issue Cite
Review
Krüppel-like family of transcription factors: an emerging new frontier in fat biology
Christopher W. Brey1, Mark P. Nelder1, Tiruneh Hailemariam2, Randy Gaugler1, Sarwar Hashmi3, ![]()
1. Center for Vector Biology, Rutgers University, New Brunswick, New Jersey 08901, USA.
2. Laboratory of Molecular parasitology, Lindsley F. Kimball Research Institute, New York Blood Center, New York, New York 10065, USA.
3. Laboratory of Developmental Biology, Lindsley F. Kimball Research Institute, New York Blood Center, New York, New York 10065, USA.
Received 2009-9-11; Accepted 2009-9-28; Published 2009-10-1
Abstract
In mammals, adipose tissue stores energy in the form of fat. The ability to regulate fat storage is essential for the growth, development and reproduction of most animals, thus any abnormalities caused by excess fat accumulation can result in pathological conditions which are linked to several interrelated diseases, such as cardiovascular diseases, diabetes, and obesity. In recent years significant effort has been applied to understand basic mechanism of fat accumulation in mammalian system. Work in mouse has shown that the family of Krüppel-like factors (KLFs), a conserved and important class of transcription factors, regulates adipocyte differentiation in mammals. However, how fat storage is coordinated in response to positive and negative feedback signals is still poorly understood. To address mechanisms underlying fat storage we have studied two Caenorhabditis elegans KLFs and demonstrate that both worm klfs are key regulators of fat metabolism in C. elegans. These results provide the first in vivo evidence supporting essential regulatory roles for KLFs in fat metabolism in C. elegans and shed light on the human counterpart in disease-gene association. This finding allows us to pursue a more comprehensive approach to understand fat biology and provides an opportunity to learn about the cascade of events that regulate KLF activation, repression and interaction with other factors in exerting its biological function at an organismal level. In this review, we provide an overview of the most current information on the key regulatory components in fat biology, synthesize the diverse literature, pose new questions, and propose a new model organism for understanding fat biology using KLFs as the central theme.
Keywords: C. elegans, Ce-KLF-1, Ce-KLF-3, fat storage, Krüppel-like factors, KLF, Obesity, PPAR, C/EBP, SREBP proteins, Transcription factor
Introduction
Urbanization, sedentary life-styles, and an over abundance of high caloric foods in human populations have collectively created conditions in which the primordial metabolic pathways that evolved to extract optimal energy from the environment have begun to function out of control [1]. Energy reserve is a basic property universal to all animals, from the simplest of organisms, such as C. elegans, to the highly complex, such as humans, allowing organisms in their readiness to continue life in fasting and starvation. In mammals, excess fat is stored in adipose tissue and, when needed, is able to provide energy. As a complex, multi-factorial trait driven by natural selection based upon food availability, fat storage is highly regulated and dynamically balanced with energy consumption in physiological settings; its perturbation in either excess (obese) or deficiency (lipodystrophy) has devastating pathologic consequences in the homeostasis and fitness of an organism. The abnormalities caused by excess fat accumulation can result in pathological conditions which are linked to several interrelated diseases, such as cardiovascular disease and obesity. This set of conditions, known as metabolic syndrome, is a global pandemic of enormous medical, economic, and social concern affecting a significant portion of the world's population. Human obesity reflects an imbalance between energy expenditure and caloric intake resulting from an increase in either the number or the size of fat cells, or both, and is induced by the enlargement of adipocytes as well as the generation of new adipocytes from precursor cells [2]. The differentiation of adipocytes consists of two distinctive steps, i.e., determination (fibroblasts to preadipocytes) and commitment (preadipocytes to adipocytes) [2]. Although it is clear that genetics, physiology and environmental components play a major role in the onset of obesity, little is known about how or to what extent each of these factors contributes to it. In addition, there are several other key factors, such as dietary habits and sedentary lifestyles, which are also responsible for excess fat accumulation in the body.
The obese state preconditions insulin resistance and negatively impacts pancreatic β-cell function [3], two hallmarks of type 2 diabetes, a serious metabolic disease and health concern for worldwide populations. Obesity is further a risk factor for the development of cardiovascular disease [4]. As adipose tissue stores fat the amount of adipose tissue which can be altered under the influence of various physiological conditions in an animal may be a determinant of energy homeostasis. In recent years, many labs have endeavored to understand the mechanisms that govern adipose tissue differentiation and function. Investigations into preadipocyte and fibroblast cultured cell lines have produced an abundance of data concerning the transcriptional cascade governing adipogenesis. These observations have been tested in transgenic and knockout mouse. As a result, a complex network of transcription factors has emerged which includes several activators, co-activators, and repressors which coordinate the expression of hundreds of proteins that take part in the development of mature fat cells under the influence of important signaling pathways [5]. Three classes of transcription factors have been identified that directly influence fat cell development. These include members of PPARγ (Peroxisome proliferator-activated receptors), C/EBPα, (CCAAT/enhancer-binding proteins) and the basic-helix-loop-helix protein ADD1/SREBP (Sterol regulatory element binding proteins) families that are essential factors in mammalian adipogenesis [6, 7]. In recent years, several members of the mammalian Krüppel-like factor, KLF, family have been identified to be key players in the transcription network controlling preadipocyte formation, adipogenesis, lipogenesis, and obesity.
Thus far 17 members of the KLF family have been identified and characterized across mammalian systems [8, 9, 10], out of which 7 members of KLF family have definite role in adipogenesis. The question of what the specific regulatory role of this family of genes in overall adipogenesis is, however, remains unclear. Insight about how KLFs influence the development and regulation of fat storage came from studies which used established murine cell lines (3T3-L1, 3T3-F442A) and knockout mice. However, in vivo and in vitro mouse models provide only partial information [11], and the main limitation at present being the existence of instances in which predictions concerning adipose function do not translate well from mouse to human [4]. A genetically tractable model could bolster the speed of research on fat metabolism which can detect new genes and pathways with significant roles in fat metabolism.
One of the most challenging tasks in this line of research is to identify the genetic machinery of fat storage. Many years of development in C. elegans research shows that this microscopic organism can be an excellent eukaryotic genetic model which may enable us to answer many questions concerning fat storage which yet remain unresolved. In the search for possible treatments for obesity, the C. elegans model should be investigated in order to explore the genetic component of fat storage. The free-living roundworm, C. elegans (commonly referred to as the worm), has become an increasingly useful experimental model in the study of genetic manipulations and overall animal physiology. Although a direct comparison between human disease pathology and C. elegans-mutant phenotypes is difficult, C. elegans assays mimic certain pathologies thereby assisting in our understanding of the molecular mechanisms that underlie human metabolic disorders. We provide a brief overview of what is currently known of the key components in fat biology, synthesize the diverse literature, suggest novel avenues of research, and propose a new model organism, for use in the study of fat storage. Over all we expect to exploit the unique features of C. elegans to provide a detailed mechanistic description of fat accumulation, including the identification of additional and as of yet uncharacterized components of the C. elegans fat storage pathway. Understanding this pathway may plausibly lead to the development of new strategies in the treatment of obesity. For example, drugs that block or facilitate the efforts of key proteins involved in fat accumulation can potentially be developed. We present a synopsis of some of the important regulatory factors involved in adipogenesis and lipogenesis (Table 1). This review focuses on the main regulators of fat metabolism with an emphasis on the Krüppel-like factors (KLFs) which govern many distinct developmental and physiological processes and are thought to function via their engagement with different coactivators and repressors.
Roles of key molecular components in lipid biology.
| Term or acronym | Definition | Role(s) |
|---|---|---|
| CPB | p300/cAMP response element-binding protein | A transcriptional co-activator |
| CREB | cAMP response element binding | A transcription factor that regulates gene expression in conjunction with cAMP |
| C/EBPα | CCAAT/enhancer-binding protein alpha | Involved in insulin resistance in cells |
| C/EBPβ | CCAAT/enhancer binding protein beta | Can activate KLF5 transcription |
| C/EBPδ | CCAAT/enhancer binding protein delta | Can activate KLF5 transcription |
| CtBP | C-terminal binding protein | A transcriptional repressor, Repressor of proapoptotic genes |
| DAF | Insulin receptor-like | Insulin-like receptor in worm |
| FAT | Delta-9 fatty acid desaturation enzyme genes | Genes involved in the synthesis of ∆9 desaturase |
| FOXO | A subset of the Forkhead box “FOX” family of transcription factors | Transcription factors involved in a wide array or processes; involved in insulin signaling |
| GLUT4 | Insulin-regulated glucose transporter 4 | Involved in insulin-regulated glucose disposal |
| HDAC3 | Histone deacetylase 3 | A transcriptional repressor |
| HIT-T15 | Cell line derived from human pancreatic islet beta cells | Used as a model system for fat biology |
| KLF | Krüppel-like factor | Transcription factors involved in a wide array of cellular processes; regulation of adipogensis |
| KLF2 | Krüppel-like factor 2. | Negative regulator of adipocyte differentiation and adipogenesis |
| KLF3 | Krüppel-like factor 3 | Negative regulator of adipogenesis |
| KLF4 | Krüppel-like factor 4 | Positive regulation of adipogenesis; can down regulate C/EBPβ levels |
| KLF5 | Krüppel-like factor 5. | Involved in the induction of PPARγ and positive regulation of adipogenesis |
| KLF6 | Krüppel-like factor 6 | Positive regulator of adipogenesis through PPARγ |
| KLF7 | Krüppel-like factor 7 | Negative regulator of adipogenesis through insulin regulation |
| KLF11 | Krüppel-like factor 11 | glucose-induced regulator of the insulin gene |
| KLF15 | Krüppel-like factor 15 | Preadipocyte differentiation to adipocytes, positive regulator of adipogenesis |
| NHR | Nuclear hormone receptor | Regulates fat usage |
| NHR-49 | Nuclear hormone receptor 49 | Control consumption of fat and maintains a normal balance of fatty acids |
| NHR-80 | Nuclear hormone receptor 80 | Isoform of NHR-49 with similar functions |
| HLH | Helix-loop-helix | Transcription factor protein that regulates stearoyl-CoA desaturases |
| Krox20 | Krox20 protein | Protein that promotes expression of C/EBPβ and, with C/EBPβ, facilitates terminal adipogenesis |
| N-CoR | Nuclear receptor corepressor | A transcriptional co-regulatory protein |
| PPARα | Proliferator-activator receptor alpha | Involved with retinoid x receptor proteins to regulate genes involved in fat metabolism |
| PPARγ | Proliferator-activator receptor gamma | Master regulator of adipogenesis; Fatty acid metabolism: lipogenesis and fat storage |
| PPARδ | Proliferator-activator receptor delta | Fatty acid metabolism: catabolism |
| Retinoid Xreceptor | Retinoid X receptor | A type of nuclear receptor activated by 9-cis retinoic acid and involved in regulating genes involved in fat metabolism as a heterodimer with PPARα |
| SCD | Stearoyl-CoA desaturases | An important enzyme in fat metabolism and insulin signaling |
| SCD1 | Stearoyl-CoA desaturases 1 | Involved in inducing adipogenesis |
| SP-1 | Specificity protein 1 | Transcription factor involved in early development |
| SREBP | Sterol regulatory element binding protein | Transcription factor protein that regulates stearoyl-CoA desaturases |
| SREBP-1a | Sterol regulatory element binding protein 1a | Regulator of lipid metabolism |
| SREBP-1c | Sterol regulatory element binding protein 1c | Involved in adipose differentiation and adipogenesis; facilitates PPARγ activity |
| SREBP-2 | Sterol regulatory element binding protein 2 | Induces genes in the cholesterol biosynthesis pathway |
List of genes predicted to participate in fatty acid transport, and β-oxidation (breakdown) pathways.
| C. elegans Genes | Mammalian name |
|---|---|
| FA transport | |
| Dsc-4 | Microsomal triglyceride transfer protein (MTP) |
| vit-2 ( yolk protein70) | Apolipoprotein B (apoB) |
| vit-3( yolk protein) | Apolipoprotein B (apoB) |
| vit-4 ( yolk protein) | Apolipoprotein B (apoB) |
| vit-5( yolk protein) | Apolipoprotein B (apoB) |
| vit-6( yolk protein88 and yp115) | Apolipoprotein B (apoB) |
| Beta oxidation | |
| Acs-1 | Acyl CoA synthase (ACS) |
| Acs-2 | Acyl CoA synthase (ACS) |
| F08A8.1 | Acyl CoA oxidase |
| F08A8.2 | Acyl Co oxidase |
| F44C4.5 | Palmitoyl protein thioesterase |
| T05G5.6 | Trifunctional enzyme (ECH) |
| Cpt-1 | Carnitine palmitoyl transferase |
| Cpt-3 | Carnitine palmitoyl transferase |
| Cpt-6 | Carnitine palmitoyl transferase |
Transcriptional control of adipogenesis
In order to better understand the mechanism underlying adipogenesis the differentiation of adipocytes is used as a model system for the study of terminal cell differentiation. This is partly due to the existence of well characterized cell lines such as the 3T3-L1 preadipocyte that can be differentiated in vitro. The proliferation and differentiation of preadipocytes is characterized by apparent changes in the pattern of gene expression that are achieved by the orderly induction of transcription factors. When exposed to correct hormonal stimuli, 3T3-L1 fibroblasts become fat-loaded adipocytes in approximately 7 days [12, 13, 14]. This transformation is accompanied by the expression of a number of adipocyte-specific factors as well as cell-cycle regulators that cooperatively facilitate the expression of the major transcription factors PPARγ and C/EBPα. The designated cells undergo a terminal differentiation that is marked by both the production of lipid droplets and the appearance of the many metabolic factors unique to a developed fat cell. The current model for adipocyte differentiation suggests that during the entire differentiation process there are several essential molecular interactions that occur among members of C/EBP, the PPAR families and the basic-helix-loop-helix protein ADD1/SREBP1c [6, 7]. C/EBPβ and C/EBPδ induce PPARγ, which in turn initiates the adipogenic program that is required to promote fat cell differentiation [15]. C/EBPα induces PPARγ, and continues to maintain PPARγ levels whilst conferring insulin sensitivity to adipocytes [16].
PPARs and C/EBPs: the master minds of adipogenesis
Both PPARs and C/EBP regulate fat, glucose, and cholesterol homeostasis. Members of the PPARs family, such as PPARγ, PPARα, and PPARδ, are ligand-activated transcription factors belonging to the nuclear hormone receptor (NHR) superfamily. The C/EBP family, whose members include C/EBPα, C/EBPβ, and C/EBPδ, belong to the basic leucine zipper class of transcription factors. PPARγ and PPARα are expressed predominantly in adipose and liver tissue, respectively, while PPARδ is expressed in a variety of tissues, including adipose, brain, and skin tissue [17]. Within the C/EBP family, C/EBPα, C/EBPβ, and C/EBPδ are expressed primarily in adipose tissue and are associated with the maintainance of terminally differentiated adipocytes [18, 19].
The transcriptional cascade leading to adipogenesis involves hormonal stimulation followed by the rapid expression of C/EBPβ and C/EBPδ [9]. Within 48 hrs, the expression levels of these proteins peak and begin to decline, an occurrence which coincides with a rise in C/EBPα and PPARγ levels. PPARγ and C/EBPα induce each other's expression in a positive feedback loop, thereby maintaining the differentiated cell state [20]. Studies using loss-of-function mutants have shown that cells lacking PPARγ in vivo and in vitro express reduced levels of C/EBPα and are unable to undergo adipogenesis [11]. Conversely, cells without C/EBPα are capable of adipocyte differentiation but are insulin resistant, even in the presence of PPARγ [21]. These reports suggest that C/EBPα and PPARγ traverse a codependent path, and that C/EBPα requires PPARγ for normal cell differentiation [20]. Several studies using cell transfection assay as well as mouse mutants implicate PPARγ as the central regulator in adipocyte differentiation. Ectopic expression of PPARγ promotes the differentiation of preadipocytes into adipocytes [22]. In vivo mouse experiments show that cells lacking in PPARγ do not participate in the formation of fat tissue [11]. In vitro, PPARγ-null cells in both stem cells and fibroblasts are unable to undergo adipogenesis [11, 20, 23], and the expression of PPARγ is able to restore adipogenesis in these cells. These experiments demonstrate the critical importance of PPARγ in adipogenesis.
The two N-terminal variants of PPARγ are evidently formed by their use of an alternative promoter and an alternative first exon. Also, PPARγ2 possesses an additional 30 amino acids at its extreme N terminus and its expression is more adipose-specific [24]. In mammals, naturally occurring mutations in PPARγ are associated with severe lipodystrophy [25] and efforts to identify the site at which ligand binding to PPARγ occur have been unsuccessful in pharmaceutical development. There exist varieties of fatty-acid-derived molecules that have been shown to bond to PPARγ, but their binding affinity is too low for them to be true ligands. However, there also exists evidence which shows that the binding and activation of PPARγ by a high affinity endogenous ligand is not necessary for adipogenesis [26]. As an NHR, PPARγ contains a α-helix 3 ligand-binding domain at its C terminus. As a result of a screening of colon cancer cells Sarraf and colleague (1999) identified a mutation in the α-helix 3 ligand-binding domain that caused a glutamine-to-proline shift at residue 286 (Q286P) [27]. They found that without agonist treatment, wild-type PPARγ and mutant PPARγ Q286P were both adipogenic. This would suggest that the activation of these proteins is not dependent on PPARγ agonist ligands under physiological conditions [26]. There has been also report that PPARs play significant roles in both type-2-diabetes and atherosclerosis [28]. It is known that antidiabetic drugs, thiazolidinediones, act as PPARγ-agonists that block vascular smooth muscle cell growth and migration, thus potentially reducing atherosclerosis [29]. Whereas PPARδ has been linked to lipid accumulation in human macrophages that leads to atherosclerosis, arthritis, and neourodegeneration [30], the PPARα acts as a heterodimer with the retinoid X receptor [31], inducing the transcription of target genes involved in fat metabolism. Unsuitable signaling by retinoid X complexes contributes to a host of severe pathologies which includes diabetes, heart disease, and obesity. PPARα is an essential regulator in fatty acid metabolism whose activation results in reduced serum levels of triacylglycerides, free fatty acids, and cholesterol. Consequently, synthetic PPARα ligands are known to be efficient lipid-lowering drugs used in the treatment of diabetes [32]. The discovery of PPARs and C/EBPs has paved the way for new avenues in lipid research by advancing our knowledge about adipogenesis at a molecular-level.
Sterol regulatory element binding proteins (SREBPs): the housekeepers of lipid homeostasis
Aside from core adipogenic factors, PPARs and C/EBP, regulatory factor that impact adipogenesis has been identified. The sterol regulatory element binding proteins (SREBPs) regulate lipid homeostasis by controlling the expression of a range of enzymes required for endogenous cholesterol, fatty acid (FA), triacylglycerol and phospholipid synthesis [33]. SREBP protein is essentially a housekeeping protein responsible for the regulation of the lipid composition of cell membranes, contributing to overall lipid homeostasis. There are three SREBP isoforms, SREBP-1a, SREBP-1c and SREBP-2 known thus far. However, each SREBP isoform possesses exclusive regulation and activation properties that facilitate the co-ordinate regulation of lipid metabolism [33] and is found as a membrane-bound precursor in the endoplasmic reticulum. Membrane lipids (sterols and polyunsaturated fatty acids) control SREBP activities through feedback mechanisms. While SREBP-1a and SREBP-1c selectively induce the expression of genes involved in fatty acid biosynthesis [34], SREBP-1c is additionally involved in adipose differentiation [35]. Kim and colleagues (1998a) have shown that the expression of SREBP-1c increases in response to insulin, in 3T3-L1 adipocytes and in turn putatively controls the regulation of adipogenesis [36]. By analogy, the ectopic expression of a dominant-negative SREBP-1c prevents preadipocyte differentiation [37] and facilitates PPARγ activity through the production of an endogenous ligand [38]. Interestingly, SREBP-2 acts as a putative gene inducer functioning in the cholesterol biosynthesis pathway [33].
The most toxic of the congeners, 2,3,7,8-Tetrachlorodibenzo-p-dioxin, up-regulates SREBP activity in the liver while down-regulating its activity in adipose tissue [39]. Collectively these metabolic changes may be linked to the onset of wasting syndrome. This syndrome results in a disordered distribution of lipids resulting in diabetes-like symptoms and a fatty liver pathology [40]. The importance of tight regulatory activities of SREBPs in the fat storage pathway is evident, we still know little about the effects of SREBPs on overall animal physiology. Because of the lack of a suitable model organism, the majority of physiological insights are generated from in vitro studies. Six years ago, McKay and colleagues [41] investigated C. elegans as a model organism for use in the study of the molecular mechanisms that control adipocyte formation. They took advantage of the complete genome sequence available on the C. elegans data base (www.wormbase.org). The existence of C. elegans homologs of mammalian SREBP and C/EBP led these researchers to examine whether C. elegans SREBP and C/EBP have roles in fat storage. The Ce-SREBP (Y47D3B.7; lpd-1) is exclusively expressed in the intestines while Ce-C/EBP (C48E7.3; lpd-2) is weakly yet extensively expressed in the nervous system (www.wormbase.org). Disruption of either C/EBP or SREBP by RNAi results in pale, skinny, lipid-depleted, and developmentally-arrested worms [41]. Although both SREBP and C/EBP RNAi worms did eat and defecate normally, they did not possess fat granules. This data suggests that the SREBP and C/EBP required for mammalian adipocyte differentiation are also essential for the fat storage in the worms. Interestingly, SREBP and C/EBP regulate the expression of the same lipogenic enzymes in both worms and mammals [41]. The role of SREBPs in adipogenesis and the connection between SREBPs and metabolic disorders remains to be a division of fat biology in which important discoveries continue to be made. The data obtained concerning worm SREBP and C/EBP in relation to their function in fat accumulation has the potential to serve as a model for mammalian fat storage. The C. elegans possesses a clear homolog of C/EBP, of the 284 NHRs encoded by the C. elegans genome, however, none exhibit sequence identity with PPARs. C. elegans possesses several genes which display similarity to PPARγ, making the identification and investigation of a potential PPARγ homolog difficult [42].
Other adipogenic factors
In addition to PPARγ, C/EBPs, and SREBPs, a number of other factors have been recently found to be involved in the regulation of adipogenesis, for example FoxO1. It has been shown that the overexpression of a constitutively active fork-head containing transcription factor FoxO1 in 3T3-L1 preadipocytes blocks adipogenesis by increasing cyclin-kinase inhibitor p21 expression and blocking clonal expansion [43]. Further study has shown that FoxA2 inhibits adipogenesis by inducing the expression of preadipocyte factor-1 (DLK1/Pref-1) [44]. Still, the expression of the third member of the family, FoxC2, is confined to adipose tissue in mice, supporting only the differentiation of brown adipocytes, not white adipocytes [45]. Another adipogenic factor, the zinc finger-containing transcription factor Krox20 (early growth response gene 2, or Egr2) has reported to act early in the adipogenic program of 3T3-L1 cells and seems to contribute to the induction of C/EBPβ expression. This induction may occur by activating another intermediary transcription factor, such as a homeobox family protein. GATA factor family of transcription factor has been known for its importance in many gene regulations in diverse physiological processes. GATA2 suppresses adipogenesis [46]. During cell differentiation, the expression of GATA2 is down-regulated, thus exerting its negative effects on adipogenesis by suppressing pparg2 promoter activity [47]. After extensive search of body of literatures indicates that although many transcription factors have essential regulatory roles in adipose cell differentiation, it is likely that most of these factors interact with PPARγ and the C/EBPs, these two proteins remain the master regulators of adipogenesis.
Krüppel-like factors (KLFs): the new kids on the block
The Krüppel-like factors are an important family of Cys2/His2 zinc-finger DNA-binding proteins homologous to the Drosophila melanogaster segmentation gene product, Krüppel. The C terminus zinc fingers bind to the CACCC/GC/GT-box found in the regulatory regions of genes, controlling various biological processes [48]. The N terminus is highly variable and involved in gene activation, gene repression, or both, as well as in protein-protein interactions [48]. KLFs are known to participate in cell proliferation, differentiation, hematopoiesis [49, 50, 51], and cell cycle progression [52]. All KLFs bind to similar DNA sequences which consist of CACCC or related GC-rich elements in the promoter and enhancer regions, an occurrence which leads to a remarkable overlap in the target genes that KLFs regulate. Despite these common features, the majority of KLFs have unique tissue-specific roles, a fact which explains why the exact function of KLFs in vivo as well as their target specificity is poorly understood. Eight members of mammalian KLFs: KLF2, KLF3, KLF4, KLF5, KLF6, KLF7, KLF-11 and KLF15 have been identified as key components of transcription network responsible for controlling adipocyte differentiation, adipogenesis, and obesity [53-57]. Interestingly, the functional CACCC binding sites are found in the control region of key adipogenic factors, such as C/ebpα and PPARγ. We will focus on these KLFs, the variability in their expression patterns during adipocyte differentiation, and their effects on adipogenesis and gene expression (Table-1).
Pro-adipogenic KLFs
Krüppel-like factor 15 was the first KLF to be found to be involved in adipogenesis. KLF15 participates in adipogenesis through its regulation of PPARγ expression. In addition to this, the overexpression of KLF15 both induces adipocyte differentiation and positively regulates the expression of the insulin-glucose transporter-4 (GLUT4) [54]. In an experiment involving microarray analysis, KLF15 was shown to be up-regulated when induced 3T3-L1 preadipocytes differentiated into adipocytes; it has been shown that blocking the expression of KLF15 by use of either a dominant negative mutant or through RNAi reduces the expression of PPARγ and prevents adipogenesis [55]. Notably, the dominant negative mutant of KLF15 does not affect the expression of C/EBPβ [34]. In NIH 3T3 cells, C/EBPβ and C/EBPδ activate KLF15 and acting together, KLF15 and C/EBPα increase the expression of PPARγ following a decrease in C/EBPβ and C/EBPδ transcript levels [55]. PPARγ is further able to elevate C/EBPα levels, indicating that a positive feedback mechanism exists between the two factors [16]. An additional related factor, KLF4, is an essential early regulator of adipogenesis. KLF4, also known as GKLF/ZIF, is highly expressed in differentiated, post-mitotic cells of the skin and the gastrointestinal tract and possesses a variety of roles as a differentiation-proliferation switch and regulator of the cell cycle [58, 59, 60]. When KLF4 activity was eliminated the KLF4 knockout mice died approximately 12 hours after birth due to defects in skin development coupled with a failure to form a normal basement membrane [61]. The fat layer of the skin of those mice was disrupted, resulting in a malfunctioning of the skin barrier and by a rapid loss of body fluids. These significant alteration in mouse suggest that KLF4 has a critical regulatory role in the development of adipose tissue. Studies in 3T3-L1 cell lines indicated that KLF4 is expressed in these cells within 30 minutes of exposure to a standard adipogenic cocktail. However, elimination of KLF4 activity in 3T3-L1 cells inhibits adipogenesis and down regulates C/EBPβ levels [56], suggesting that KLF4 is an upstream transcriptional regulator of C/EBPβ in the network controlling adipogenesis. Notable is the observation that KLF4 binds directly to the C/EBPβ promoter and, in concert with Krox2 [62], markedly transactivates C/EBPβ expression [56]. Conversely, the knockdown of C/EBPβ increases KLF4 expression while an overexpression of C/EBPβ decreases KLF4 expression, indicating that C/EBPβ reduces klf4 gene expression through negative feedback [56].
Another important member of KLF family, KLF5 also known as basic transcription element-binding (BTEB)2, or intestinal-enriched Krüppel-like factor, is a key element involved in the pathogenesis of cardiovascular disease. KLF5 regulates the proliferation of various cell types, including fibroblasts, smooth muscle cells, white adipose tissue and intestinal epithelial cells. Recently, KLF5 has been noted to be a key player in the transcription factor network which controls adipocyte differentiation. KLF5 is expressed early in adipocyte differentiation at the point at which C/EBPβ/δ binds directly to the KLF5 promoter [63]. KLF5 then acts in concert with early factors, C/EBPβ and C/EBPδ, in facilitating the expression of the late factor, PPARγ2, mediating the early and late stages of adipogenesis in neonatal heterozygous KLF5 knockout mice [63]. There is evidence to support that the SUMOylation of KLF5 acts as a molecular switch in response to PPARδ and the adipogenic signals that can activate or repress the genes involved in lipid synthesis [64]. A PPARδ isoform is expressed in skeletal muscle, blocking its expression decreases the muscle's oxidative capabilities, leading to obesity and glucose intolerance [65]. Conversely, an overexpression of PPARδ protects mice from contracting diet-induced obesity [66]. The reversible nature of SUMOlyation makes it a major regulatory modification moiety which leads to transcription inhibition [67]. Under basal conditions in response to cellular and environmental stress, SUMoylated KLF5's binding to the corepressor N-CoR (nuclear receptor corepressor) and SMRT (silencing mediator of retinoid and thyroid receptor) is increased, while its affinity for CREB binding proteins and PPARδ is decreased and fatty acid metabolism is suppressed [64]. KLF5 further acts as a key regulator, controlling the expression of FASN (fatty acid synthase) through an interaction with SREBP-1. KLF5 binds to SREBP-1 and additionally enhances the SREBP-1-mediated increase in FASN promoter activity [68].
The Krüppel-like factor 6, a tumor suppressor gene, appears to promote adipogenesis by the transcriptional suppression of proto-oncogene delta-like 1 (Dlk1). The DLK1 is a transmembrane protein that inhibits adipocyte differentiation through its interaction with HDAC3 [69]. A cell culture study suggests that HDAC3 deacetylase represses PPARγ function as result of its interaction with hypophosphorylated retinoblastoma (pRb) [70]. Although pRb interacts with a considerable number of growth-promoting transcription factors [71, 72], few studies suggest that pRb phosphorylation is involved in preadipocyte differentiation [73, 74]. Li et al. (2005) [69] demonstrated that KLF6 could, independent of Rb, moderate adipocyte differentiation through the transcriptional activation of adipocyte inducers such as PPARγ, C/EBPα/β, and steroyl-CoA desaturase-1 (SCD1).
KLFs as negative regulators
Other KLFs (i.e., KLF2, KLF3, and KLF7) function to block adipogenesis. For instance, the KLF2 is amply expressed in younger preadipocytes, yet displays reduced expression in mature adipocytes. Constitutive expression of KLF2 results in an inhibition of PPARγ expression but does not have any effect on the upstream regulators C/EBPβ and C/EBPδ [75]. Using a combination of promoter mutational analysis and gel mobility shift assays, a binding site identified within the PPARγ2 promoter has been found to be responsible for the inhibitory effects of KLF2 [75]. Wu et al. (2005) [76] generated tetracycline-responsive lines of 3T3-L1 which expressed physiological levels of KLF2 and were thus able to show that KLF2 prevented preadipocyte differentiation by partially restoring preadipocyte factor-1 (Pref-1). As evidenced by the formation of lipid globules, embryonic cells derived from KLF2-/- lines can differentiate into adipocytes, implying that the presence of KLF2 is not important during the formation of preadipocytes, but rather during later development, when it inhibits preadipocyte maturation into adipocytes. In 3T3-L1 cell line, the forced expression of mammalian KLF-3 blocks adipocyte differentiation as result of a direct association with the C/ebpα promoter, whilst a decrease in KLF-3 levels prevents differentiation [77]. The klf-3 deficient mice created by disrupting klf-3 gene by homologous recombination were smaller than wild-type showed a significant reduction in white adipose tissue [77]. Further analysis of the fat pads in knock out mice indicated that pads contained fewer and smaller cells. Turner and Crossley (1998) observed that KLF3 inhibits transcription by recruiting to its N terminal repression domain the corepressor C-terminal binding protein (CtBP) [78].
The Krüppel-like factor 7, KLF7, has also been implicated as a negative regulator of adipogenesis. Significantly, KLF7 has been linked to the pathogenesis of type 2 diabetes. Kawamura et al (2005) over expressed KLF7 in insulin secreting cell line (HIT-T15 cells) and showed that the modest increase in KLF7 expression dramatically suppressed the glucose-induced secretion of insulin and reduced the expression PPARγ and C/EBPα, thereby blocking adipogenesis [79]. KLF11 is known to be a pancreas-enriched transcription factor. Both in vitro and in vivo studies have shown KLF11 to be a negative regulator of exocrine cell growth. Neve and colleague (2005)[80], in performing detailed functional and genetic analyses of KLF-11, found that KLF11 is a glucose-induced regulator of the insulin gene and that functional KLF11 gene variants possess a significant association with diabetes. Although KLF-11 has been proposed to be a glucose-induced regulator of the insulin gene [80], this protein may also been involved in adipogenesis and metabolic control [81].
A greater understanding of the mechanisms governing adipogenesis will emerge with use of a model organism in which changes in cellular development are associated with observable genetic modifications. Several laboratories have demonstrated that vertebrate KLFs, as well as other regulators, are key players in fat storage pathways [63, 55, 76]. As the majority of these reports have come through biochemical and tissue culture studies, a good genetic model organism is needed for a thorough examination of the fat storage process in vivo during the development of an organism. To better understand how KLFs regulate fat storage, our laboratory uses C. elegans as a model organism. Many of the regulatory genes involved in C. elegans fat metabolism are involved in adipocyte biology [82]. This relatively simple organism provides a powerful system to study the genes with critical roles in human metabolic diseases.
C. elegans fat biology
The worm Caenorhabditis elegans has emerged as a powerful molecular genetic model for use in studying genes which possess vital roles in human health and diseases. A number of studies reported thus far have already shown significant similarities between molecular components of mammalian and C. elegans fat pathways that extend to disease-associated genes. Recent studies have shown that fat regulation in C. elegans engages a complex network of genes with likely functions in food sensation, neuroendocrine signaling [94, 84], uptake, transport [85], and storage [41]. The C. elegans stores fat in intestine, an organ derived from the endodermal layer and composed of several types of gut granules which contain protein, carbohydrate molecules, and lipid fat granules. In contrast, mammalian fat is stored in a derivative of mesodermal tissue which contains a single cell type, the adipocyte. The mammalian adipoctye is dedicated to maintaining a homeostasis in fat metabolism, whereas the worm intestine is responsible for many diverse functions. Although they are clearly distinct, the common factor between C. elegans and mammalian adipocytes is the genetic conservation of the regulation of fat granules [41]. Though C. elegans does not have a dedicated adipocyte, the shared worm and mammalian fat regulatory pathways link to the developmental programs that underlie fat storage capacity. Several of the recently discovered C. elegans fat regulatory pathways play roles resembling those found in mammals. For example, there is evidence to support the existence of neuropeptide, serotonergic, and insulin signaling pathways that regulate fat storage in both nematodes and mammals [86, 87, 88].
It is well established that fat storage is profoundly influenced by differing environmental conditions, it is now possible to uncover the mechanism by which physiological pathways are able to adjust themselves in response to various environmental cues. Thus the questions concerning to what extent and by what mechanism, age, food, and habitat disturb the fat storage networks of C. elegans can be addressed at a molecular level. The C. elegans genome contains several fat regulators, an array which includes various transcription factors, signal transduction modules, nutrient transporters, and biochemical identities and pathways, that are homologous to a wide range of mammalian genes (C. elegans Sequencing Consortium 1998) [89]. Through the use of a genome-wide RNAi worm analysis, Ashrafi et al. (2003) [84] identified 305 C. elegans genes which, when suppressed, decreased body fat, in addition to 112 more genes whose inhibition enhanced fat storage. Furthermore, through an examination into the effects of fat-reducing gene inactivation in various C. elegans insulin, serotonin, tubby and nuclear signaling mutants [90, 91, 92], this group found a new set of fat regulatory genes. Monounsaturated fatty acids (MUFAs) are integral components of both membrane phospholipids and lipid stored triglycerides, playing critical roles in key cellular process such as cell communication and energy storage. C. elegans can synthesize a wide variety of fatty acids using Δ12, Δ3, Δ5, Δ6, three Δ9 desaturases [93], and elongases [94].
The suppression of daf-2 in C. elegans corresponds with insulin resistance and type 2 diabetes in humans. Several C. elegans daf-2 mutants build up fat deposits instead of leading to glucose metabolism, causing metabolic and developmental changes (i.e., increase in life-span) [85]. A related study using a 13C isotope assay to quantify the contribution of dietary fat to de novo synthesis found that alternative alleles of daf-2 do not increase fat synthesis, suggesting that site-specific mutations in the insulin receptor can differentially influence longevity and metabolism [95]. The deletion of a C. elegans nhr-49 caused increased fat accumulation and decreased life span in worms [92]. NHR-49 serves as a key regulator in fat usage, adjusting the pathways that control fat consumption and maintaining a normal balance of fatty acids. A similarity between the biological activity of nhr-49 in C. elegans and that of mammalian PPARs is suggestive of an evolutionarily conserved role for NHRs in modulating fat consumption and composition [64]. These studies illustrate the significance of C. elegans as a model organism for use in fat biology and in advancing our knowledge of whole-organism metabolism and physiology. We highlight recent advances in the role of C. elegans KLFs in fat metabolism as well as their links to human metabolic disorders.
Role of KLFs in C. elegans fat storage
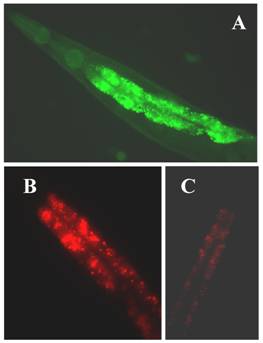
The C. elegans genome predicts three KLFs, which include the gene F56F11.3, referred to as klf-1 [96], gene F53F8.1 (klf-2; a homolog of humanWT1), klf-3a, and klf-3b (mua-1a; F54H5.4 and mua-1b; F54H5.4b; Wormbase; http://www.wormbase.org.) [97]. All three C. elegans KLFs, klf-1, klf-2 and klf-3, share the highest identity with members of mammalian KLFs in terms of their C-terminal C2H2 zinc fingers, despite little homology in their N-terminal regions. Since klf-1 mutant is not available, RNAi was used to suppress klf-1 activity in C. elegans. When KLF-1 functioned normally, the worms had typical fat deposits in their intestines, but when klf-1 activity was suppressed, the worms lost their ability to store appropriate levels of fat and instead accumulated abnormally high amounts of fat in their intestines (Figure 1B). Our findings are therefore consistent with the observed expression profile of klf-1::gfp in C. elegans intestines (Figure 1A). The worm intestine, a major endocrine organ which engages in nutrient sensing and energy metabolism, is positioned close to the sexual organs and a major tissue engaged in nutrient sensing and energy metabolism [98]. In C. elegans, the intestine performs several diverse functions which are executed by many different organs in higher eukaryotes. The key function of C. elegans intestinal cells appears to be digestive as they can be observed to secrete digestive enzymes into the lumen of the intestine and take up the processed material and nutrients. In hermaphrodites, yolk protein is synthesized in the intestine and then transported to the oocytes through the body cavity [99]. Significantly, the intestine serves as the major site for fat storage [100], for fatty acid metabolism, and is a storage depot with a large number of assorted storage granules. We also found that over-expression of klf-1::gfp caused severe egg-laying defects in C. elegans while a suppression resulted in increased cell death. Our results further imply that the inhibition of klf-1activity disrupts cell phagocytosis. An increase in fat accumulation in the absence of Ce-klf-1, its expression pattern in the intestine during larval development, as well as the continued presence in adult worms suggest that C. elegans klf-1plays a critical role in the regulation of fat metabolism; a disturbance in this process may increase cell death and cause the defective phagocytosis of dead cells [96].
(A) The expression of klf-1::gfp. Intense expression of gfp green fluorescent in the intestine of a C. elegans adult hermaphrodite; (B) extensive fat accumulation in klf-1RNAi hermaphrodite; (C) low fat content in wild type adult hermaphrodite. Both wild type and RNAi worms were detected by Nile Red staining. Worms were observed and photographed using Axioskop 2 plus fluorescent microscope (400X magnifications).
Research is underway in our laboratory to define the underlying molecular mechanisms which lead to the increased fat accumulation, cell death, and phagocytosis following KLF-1 misregulation. Fat metabolism is known to be a complex process requiring a number of molecules involved in the absorption, biosynthesis, assembly, transport and catabolism of diverse lipids. The suppression of klf-1 activity may result in a failure in to regulate the specific genes needed in one or more of these processes. The complex and extensive array of metabolic abnormalities that result from excess fat accumulation may also be linked to excessive cell death. Alternatively, cell death in the klf-1 RNAi worms may have occurred as result of a failure to induce the genes required to prevent cell death. To elucidate whether Klf-1 functions in a known regulatory pathway or in a yet unknown new pathway, a thorough analysis of the cellular functions of C. elegans klfs, accompanied by a scrutiny of their interactions with each other and other molecular components of fat regulatory pathways is needed.
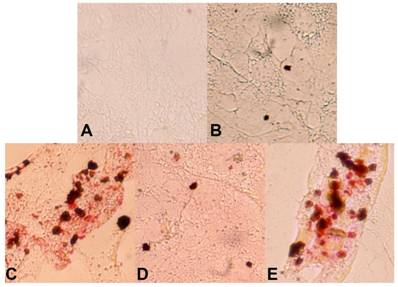
We have continued our studies on C. elegans KLF. Recently we have identified a critical role of Ce-KLF-3 in fat metabolism [97]. In order to gain insight into the mechanisms by which C elegans klfs regulate fat storage, we examined two mutant alleles of C. elegans klf-3. We performed detailed genetic and phenotypic analyses and have thereby shown that the two alleles of klf-3 mutants, klf-3(ok1975) and mua-1(rh160), carry different genomic deletions, with each exhibiting distinctive loss-of-function phenotypes. A partial characterization of a C. elegans mua-1 (rh160) allele suggests a role in the mechanical integrity of the skeletal muscle in the worm [101]. A deletion in mua-1 (rh160) II mutants caused the animals to grow inadequately, the majority failed to reach adulthood. These mutants have defects in the cell matrix attachments that are critical in the transmission of muscle contractions to the cuticle as well as in the attachment of the uterus to the body wall [101]. A detailed molecular analysis of mua-1 (rh160) II mutants confirmed that a more extensive genetic disruption had occurred that affected two other neighboring genes in addition to mua-1. Although mua-1 functions in muscle development, a further analysis of the mua-1 deletion within its own coding sequence is crucial in order to gain insight into the full spectrum of functions performed by mua-1. However, the klf-3(ok1975) allele, characterized by a 1658-bp deletion in the klf-3 gene spanning the 3' end of exon 2 A in the klf-3 coding sequence which causes a loss in klf-3 function, subsequently resulting in severe reproductive defects in mutant worms. Klf-3 is exclusively expressed in the intestine of both larvae and adult worms increases the amounts of fat deposited in the intestine [97]. We extended this study to a mammalian adipocyte model. The mouse 3T3-L1 preadipocyte is a cellular model in which adipogenesis is studied through the use of defined media and hormones. We explored its utility in order to determine whether worm klf-3 would be able to function in this heterologous system and manifest a conserved regulatory role. We tested the expression of worm klf-3 in this cell line via transfection with a Ce-Klf3::gfp plasmid and were able to determine its sub-cellular location. Interestingly, Ce-klf-3 localizes in the nucleus of mouse 3T3-L1 preadipocytes, supporting the assertion that it functions as a transcription factor in gene expression within the nucleus. In order to examine the role of transiently expressed Ce-klf-3 during preadipocyte to adipocyte differentiation, we induced 3T3-L1 cells with a hormone mix supplement. We found that an overexpression of Ce-klf-3 both significantly blocks cell differentiation (Figure 2 D) and down-regulates pro-adipogenic genes PPARγ (Hashmi et al., unpublished) in the presence of the hormone mix. These occurrences resemble the characteristics of overexpression of KLF2 in 3T3-L1 cells [76]. Mammalian KLF2 is known to be a negative regulator in adipocyte differentiation. An Overexpression of KLF2 inhibits PPARγ expression with no effects on the upstream regulators C/EBPβ and C/EBPδ. KLF2-/- cells do not exhibit a significant differences in lipid accumulation when compared with KLF2+/+ after 12 days of differentiation, however, 2 days following confluence (day 0 of differentiation), KLF2-/- cells displayed considerably more lipid deposits (~10-fold) in comparison to those of wild-type cells. At the initial stage of differentiation, i.e., day 1−3, KLF2-/- cells still exhibit about 100% more lipid accumulation than do KLF2+/+ cells [76]. Taken together, this data establishes mouse 3T3-L1 preadipocytes to be feasible heterologous model systems when endeavoring to evaluate the conserved regulatory functions of the worm klf-3 gene.
Over-expression of worm klf-3 in mouse 3T3-L1 preadipocyte cells. A) cell without induction; B) klf-3 transfected cell without induction; C) cell after induction; D) klf-3 transfected cell after induction; E) cell transfected with vector (pEGFP) alone and after induction. Note that klf-3 significantly suppress the formation of fat droplets, probably acting as a negative regulator of adipogenesis.
The dramatic phenotype observed in the klf-3 (ok1975) mutant suggests that klf-3 plays a key role in fat regulation and that its deletion interferes with lipid metabolism and signal transduction associated with fat storage. We recently tested this hypothesis by performing microarray and qRT-PCR using RNA extracted from klf-3 mutant worms and compared this with RNA obtained from wild type worms. We found that a deletion in the klf-3 coding sequence had a profound effect on multiple genes involved in the fatty acid β-oxidation (mitochondrial β-oxidation and peroxisomal β-oxidation) pathways [97](Table-2). Fatty acids in the form of Acyl-CoA molecules are broken down in mitochondria and/or peroxisomes in order to generate Acetyl-CoA. This process requires three major events to occur: i) activation and transport into mitochondria, ii) β-oxidation, and iii) electron transport chain. Fatty acids are first transported through the outer mitochondrial membrane by carnitine-palmitoyl transferase I (CPT-I) and are then dispatched across the inner mitochondrial membrane by carnitine [102]. The action of CPT-I is known to be the rate limiting step in fatty acid oxidation. Once inside the mitochondrial matrix, fatty acids undergo β-oxidation. During this process, two-carbon molecules, contained in acetyl-CoA, are repeatedly cleaved from the fatty acid. Acetyl-CoA is then able to enter the TCA cycle, which produces NADH and FADH. NADH and FADH are subsequently used in the electron transport chain to produce ATP. Fatty acid oxidation also occurs in peroxisomes, which carry out initial oxidation when fatty acid chains become too long (>C-22), and cannot be processed in mitochondria. However, the oxidation of fatty acids in peroxisomes is not coupled with ATP synthesis. Instead, high-potential electrons are transferred to oxygen, yielding hydrogen peroxide. The enzyme catalase, found exclusively in peroxisomes, converts the hydrogen peroxide into water and oxygen. Peroxisomal β-oxidation requires the action of special enzymes specific to peroxisomes and very long fatty acids. Thus the reduced expression of the genes that facilitate the transport and breakdown (Table-2) of fatty acids could plausibly cause the disruption of these processes, ultimately leading to the accumulation of fat typically seen in klf-3 mutants.
Disruption of klf-3 activity in the klf-3 (ok1975) allele contributes to defects in germ cell differentiation and oocyte development. Although the KLF-3 protein is not present in germ cells or in oocytes, it is constantly and highly expressed during larval development, suggesting that its function and regulation in larvae is required in later stage-specific cell proliferation. Reproductive defects could be a result of an increase in fat storage, given that the loss of KLF-3 function would primarily affect the functioning of the intestine. Worm klfs provide a link to the essential negative regulatory mechanisms of fat storage in the intestine, an organ which has been conserved through evolution. The C. elegans intestine presents a useful model for use in future studies which strive to address the positive and negative impact of neuroendocrine signals on lipogenesis and fat deposition [103]. Fat storage is a significant energy investment retained by the natural selection acting upon metazoan evolution. Fat storage offsets food shortage, a constant threat to animal survival (except humans of recent evolution) [104], and is most likely to have first emerged in the gut, the most ancient organ. The intestine-specificity of worm KLFs and their identification as a key factor in fat regulation reinforces the assertion that their role in lipid metabolism and energy homeostasis originated and was adapted early in evolutionary history.
Future areas of research will need to examine the molecular mechanisms employed by worm KLFs that play role (s) in fat metabolism. Exploiting C. elegans genetics, biochemistry, and physiology is needed in order to determine how the regulatory activities of KLFs contribute to normal fat storage and adaptation to nutritional stress. Additional studies must be aimed at addressing the two key questions that are central to understanding the function and regulation of KLFs. These two critical questions are: What is the molecular basis and regulatory specificities of KLF-3 in recruiting protein partners and their binding to target genes to mediate transcription and how this affect fat metabolism? (2) What is the mechanism linking the function of KLF-3 to metabolic and signaling pathways and how disruption in KLF function leads to fat accumulation? By exploiting unique features of C. elegans we hope to develop a detailed mechanistic description of fat accumulation, including identification of additional as yet uncharacterized components of the C. elegans fat metabolism pathway that may suggest new strategies in the treatment of obesity. Our lab is currently endeavoring to better understand the fundamental methods by which fat accumulates following a loss in klf-3 function. Using C. elegans homologs of some mammalian fat metabolism genes, we are performing in depth studies that how the genes and pathways known to be involved in mammalian fat absorption, assembly and transport are being affected in response to a loss in the functioning of klfs.
Conclusions
It is evident from the fact that an inappropriate increase and decrease in fat accumulation level is a characteristic of many human diseases. Fat storage is mediated by a core biochemical pathway that requires the activation of transcriptions factors. Considerable progress has been made in the identification of the transcriptional factors that are involved in the processes controlling the differentiation of preadipocytes into mature fat cells. It is quite clear that we have gone many steps forward in the understanding of the KLFs' role in adipocyte differentiation, however, there is still a large void in our understanding concerning the cascade of events that result in KLFs activation, repression, and interaction with other molecules in exerting their specific functions in fat metabolism. The C. elegans has served as a pioneering model for gene discovery and functional analysis of many important developmental processes. We are using this model organism to study fat storage. We believe that studying KLFs in C. elegans will enable us to perform a greater range of experiments which are not feasible in mammalian models. The identification of C. elegans klfs as being key regulators in fat storage through a genetically tractable experimental model will allow the pursuit of a more comprehensive approach to better understanding the biology of fat. It is possible that many other unidentified worm genes exist that regulate levels of body fat. A thorough genetic analysis of the many mediators of klfs, in concert with an examination of their cellular functions in C. elegans, will allow a more detailed description of the basic regulatory networks conserved between worm and humans. As part of the systematic effort to uncover treatments for obesity and its associated metabolic disorders, it is important to identify components of the known and novel signaling pathways that regulate fat storage. It will be interesting to determine whether worm klfs have similar regulatory functions in fat storage as those of mammalian klfs in adipogenesis and other fat metabolic processes.
Acknowledgements
We thank Jun Zhang and Chuan Yang for technical assistance and Drs. Mohan Narla and Chen Huang for helpful discussion. We thank Sanya Hashmi for her critical reading and editing of the manuscript. We thank C. elegans Genetics Center, University of Minnesota, Minneapolis, MN, for providing rh160 and ok1975 mutant alleles.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Speakman JR. A nonadaptive scenario explaining the genetic predisposition to obesity: the "predation release" hypothesis. Cell Metab. 2007;6:5-12
2. Lane MD, Tang QQ. From multipotent stem cell to adipocyte. Birth Defects Res. A Clin. Mol. Teratol. 2005;73:476-477
3. Muoio DM, Newgard CB. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008;9:193-205
4. Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875-880
5. Farmer SR. Transcriptional control of adipocyte formation. Cell Metab. 2006;4:263-273
6. Rosen ED, Walkey CJ, Puigserver P. et al. Transcriptional regulation of adipogenesis. Genes Dev. 2000;14:1293-1307
7. Rosen ED, Spiegelman BM. Molecular regulation of adipogenesis. Annu. Rev. Cell Dev. Biol. 2000;16:145-171
8. Bieker JJ. Isolation, genomic structure, and expression of human erythroid Krüppel-like factor (EKLF). DNA Cell Biol. 1996;15:347-352
9. Turner J, Crossley M. Mammalian Krüppel-like transcription factors: more than just a pretty finger. Trends Biochem. Sci. 1999;24:236-240
10. van Vliet J, Crofts LA, Quinlan KG. et al. Human KLF17 is a new member of the Sp/KLF family of transcription factors. Genomics. 2006;87:474-482
11. Rosen ED, Saraf P, Troy AE. et al. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell. 1999;4:611-617
12. Green H, Kehinde O. An established preadipose cell line and its differentiation in culture. II. Factors affecting the adipose conversion. Cell. 1975;5:19-27
13. Cowherd RM, Lyle RE, Mcgehee REJr. Molecular regulation of adipocyte differentiation, Semin. Cell Dev Biol. 1999;10:2-10
14. Student AK, Hsu RY, Lane MD. Induction of fatty acid synthetase synthesis in differentiating 3T3-L1 preadipocytes. J. Biol. Chem. 1980;255:4745-4750
15. Barak Y, Nelson MC, Ong ES. et al. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol. Cell. 1999;4:585-595
16. Wu Z, Rosen ED, Brun R. et al. Cross-regulation of C/EBPα and PPARγ controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol. Cell. 1999;3:151-158
17. Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421-424
18. Yeh WC, Cao Z, Classon M. et al. Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes Dev. 1995;9:161-181
19. Mandrup S, Lane MD. Regulating adipogenesis. J. Biol. Chem. 1997;272:5367-5370
20. Rosen ED, Hsu CH, Wang X. et al. C/EBPalpha induces adipogenesis through PPARgamma: a unified pathway. Genes Dev. 2002;16:22-26
21. El-Jack AK, Hamm JK, Pilch PF. et al. Reconstitution of insulin-sensitive glucose transport in fibroblasts requires expression of both PPARγ and C/EBPα. J. Biol. Chem. 1999;274:7946-7951
22. Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006;7:885-896
23. Altiok S, Xu M, Spiegelman BM. PPARgamma induces cell cycle withdrawal: inhibition of E2F/DP DNA-binding activity via down-regulation of PP2A. Genes Dev. 1997;11:1987-1998
24. Tontonoz P, Hu E, Graves RA. et al. mPPARγ2: tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994;8:1224-1234
25. Hegele RA, Cao H, Frankowski C. et al. PPARG F388L, a transactivation-deficient mutant, in familial partial lipodystrophy. Diabetes. 2002;51:3586-3590
26. Walkey CJ, Spiegelman BM. A functional peroxisome proliferator-activated receptor-gamma ligand-binding domain is not required for adipogenesis. J. Biol. Chem. 2008;283:24290-24294
27. Sarraf P, Mueller E, Smith WM. et al. Loss-of-function mutations in PPAR gamma associated with human colon cancer. Mol. Cell. 1999;3:799-804
28. Rosen ED, Spiegelman BM. PPARgamma: a nuclear regulator of metabolism, differentiation, and cell growth. J. Biol. Chem. 2001;276:37731-37734
29. Pastromas S, Sakellariou D, Koulouris S. Biodegradable Polymer Based Particulate Carrier(s) for the Delivery of Proteins and Peptides. Medicinal Chem. 2008;7:217-223
30. Vosper H, Patel L, Graham TL. et al. The peroxisome proliferator-activated receptor delta promotes lipid accumulation in human macrophages. J. Biol. Chem. 2001;276:44258-44265
31. Chawla A, Repa JJ, Evans RM. et al. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294:1866-1870
32. Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649-88
33. Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331-340
34. Tabor DE, Kim JB, Spiegelman BM. et al. Transcriptional activation of the stearoyl-CoA desaturase 2 gene by sterol regulatory element-binding protein/adipocyte determination and differentiation factor 1. J. Biol. Chem. 1998;273:22052-22058
35. Yokoyama C, Wang X, Briggs MR. et al. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell. 1993;75:187-197
36. Kim JB, Sarraf P, Wright M. et al. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J. Clin. Invest. 1998;101:1-9
37. Kim JB, Spiegelman BM. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev. 1996;10:1096-1107
38. Kim JB, Wright HM, Wright M. et al. ADD1/SREBP1 activates PPAR gamma through the production of endogenous ligand. Proc. Natl. Acad. Sci. USA. 1998;95:4333-4337
39. Nishiumi S, Yabushita Y, Furuyashiki T. et al. Involvement of SREBPs in 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced disruption of lipid metabolism in male guinea pig. Toxicol. & Appl. Pharmocol. 2008;229:281-289
40. Lee CC, Yao YJ, Chen HL. et al. Fatty liver and hepatic function for residents with markedly high serum PCDD/Fs levels in Taiwan. J. Toxicol. Environ. 2006;69:367-380
41. McKay RM, McKay JP, Avery L. et al. C. elegans: A model for exploring the genetics of fat storage. Dev. Cell. 2003;4:131-142
42. Sluder A, Mathews S, Hough D. et al. The nuclear receptor superfamily has undergone extensive proliferation and diversification in nematodes. Genome Res. 1999;9:103-120
43. Nakae J, Kitamura T, Kitamura Y. et al. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev. Cell. 2003;4:119-129
44. Wolfrum C, Shih DQ, Kuwajima S. et al. Role of Foxa-2 in adipocyte metabolism and differentiation. J. Clin. Invest. 2003;112:345-356
45. Cederberg A, Gronning LM, Ahren B. et al. FOXC2 is a winged helix gene that counteracts obesity, hypertriglyceridemia, and diet-induced insulin resistance. Cell. 2001;106:563-573
46. Tong Q, Dalgin G, Xu H. et al. Function of GATA transcription factors in preadipocyte-adipocyte transition. Science. 2000;290:134-138
47. Tong Q, Tsai J, Tan G. et al. Interaction between GATA and the C/EBP family of transcription factors is critical in GATA-mediated suppression of adipocyte differentiation. Mol. Cell. Biol. 2005;25:706-715
48. Bieker JJ. Krüppel-like factors: three fingers in many pies. J. Biol. Chem. 2001;276:34355-34358
49. Black AR, Black JD, Azizkhan-Clifford J. Sp1 and Krüppel-like factor family of transcription factors in cell growth regulation and cancer. J. Cell Physiol. 2001;188:143-160
50. Nuez B, Michalovich D, Bygrave A. et al. Defective haematopoiesis in fetal liver resulting from inactivation of the EKLF gene. Nature. 1995;375:316-318
51. Perry C, Soreq H. Transcriptional regulation of erythropoiesis. Fine tuning of combinatorial multi-domain elements. Eur J Biochem. 2002;269:3607-3618
52. Atkins GB, Jain MK. Role of Krüppel-like transcription factors in endothelial biology. Circ. Res. 2007;100:1686-1695
53. Wei D, Kanai M, Huang S. et al. Emerging role of KLF4 in human gastrointestinal cancer. Carcinogenesis. 2006;27:23-31
54. Gray S, Feinberg MW, Hull S. et al. The Krüppel-like factor KLF15 regulates the insulin-sensitive glucose transporter GLUT4. J. Biol. Chem. 2002;277:34322-34328
55. Mori T, Sakaue H, Iguchi H. et al. Role of Krüppel- like transcription factor 15 (KLF15) in transcriptional regulation of adipogenesis. J. Biol. Chem. 2005;280:12867-12875
56. Birsoy K, Chen Z, Friedman J. Transcriptional regulation of adipogenesis by KLF4. Cell Metab. 2005;7:339-347
57. Feinberg MW, Wara AK, Cao Z. et al. The Krüppel-like factor KLF4 is a critical regulator of monocyte differentiation. EMBO J. 2007;26:4138-4148
58. Garrett-Sinha L, Eberspecher H, Seldin M. et al. A gene for a novel zinc-finger protein expressed in differentiated epithelial cells and transiently in certain mesenchymal cells. J. Biol. Chem. 1996;271:31384-31390
59. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663-676
60. Yoon H, Chen X, Yang V. Krüppel-like factor 4 mediates p53-dependent G1/S cell arrest in response to DNA damage. J. Biol. Chem. 2003;278:2101-2105
61. Segre J, Bauer C, Fuchs E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat. Genet. 1999;22:356-360
62. Chen Z, Torrens JI, Anand A. et al. Krox20 stimulates adipogenesis via C/EBP b-dependent and - independent mechanisms. Cell Metab. 2005;1:93-106
63. Oishi Y, Manabe I, Tobe K. et al. KLF5 is a key regulator of adipocyte differentiation. Cell Metab. 2005;1:27-39
64. Oishi Y, Manabe I, Tobe K. et al. SUMOylation of Krüppel-like transcription factor 5 acts as a molecular switch in transcriptional programs of lipid metabolism involving PPAR-δ. Nature Med. 2008;14:656-666
65. Schuler M, Faisal A, Chambon C. et al. PGC1α expression is controlled in skeletal muscles by PPARβ, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006;4:407-414
66. Wang YX, Zhang CL, Yu RT. et al. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004;2:1532-5139
67. Pascual G, Fong AL, Ogawa S. et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature. 2005;437:759-763
68. Lee MY, Moon JS, Park S. et al. KLF5 enhances SREBP-1 action in androgen-dependent induction of fatty acid synthase in prostate cancer cell line. Biochem J. 2009;417:313-322
69. Li D, Yea S, Li S. et al. Krüppel-like factor-6 promotes preadipocyte differentiation through histone deacetylase 3-dependent repression of DLK1. J. Biol. Chem. 2005;280:26941-26952
70. Fajas L, Egler V, Reiter R. et al. The retinoblastoma-histone deacetylase 3 complex inhibits PPARgamma and adipocyte differentiation. Dev Cell. 2002;3:903-910
71. Ferreira R, Naguibneva I, Pritchard LL. et al. The Rb/chromatin connection and epigenetic control. Oncogene. 2001;20:3128-3133
72. Wade PA. Transcriptional control at regulatory checkpoints by histone deacetylases: molecular connections between cancer and chromatin. Hum. Mol. Genet. 2001;10:693-698
73. Hansen LH, Madsen B, Teisner B. et al. Characterization of the inhibitory effect of growth hormone on primary preadipocyte differentiation. Mol. Endocrinol. 1998;12:1140-1149
74. Classon M, Kennedy BK, Mulloy R. et al. Opposing roles of pRB and p107 in adipocyte differentiation. Proc. Natl. Acad. Sci. USA. 2000;97:10826-10831
75. Banerjee SS, Feinberg MW, Watanabe M. et al. The Krüppel-like factor KLF2 inhibits peroxisome proliferators-activated receptor-gamma expression and adipogenesis. J. Biol. Chem. 2003;278:2581-2584
76. Wu J, Srinivasan SV, Neumann JC. et al. The KLF2 transcription factor does not affect the formation of preadipocytes but inhibits their differentiation into adipocytes. Biochemistry. 2005;44:11098-11105
77. Nancy S, Jack BHA, Eaton SA. et al. Targeted disruption of the basic Krüppel-like factor gene (Klf3) reveals a role in adipogenesis. Mol. & Cell. Bio. 2008;28:3967- 3978
78. Turner J, Crossley M. Cloning and characterization of mCtBP2, a co-repressor that associates with basic Krüppel-like factor and other mammalian transcriptional regulators. EMBO J. 1998;17:5129-5140
79. Kawamura Y, Tanaka Y, Kawamori R. et al. Over-expression of Krüppel-like factor 7 (KLF7) regulates adipocytokine gene expressions in human adipocytes, and inhibits glucose-induced insulin secretion in pancreatic beta cell line. Mol. Endocrinol. 2005;20:844-856
80. Neve B, Fernandez-Zapico ME, Ashkenazi-Katalan V. et al. Role of transcription factor KLF11 and its diabetes-associated gene variants in pancreatic beta cell function. Proc. Natl. Acad. Sci. U S A. 2005;102:4807-4812
81. Cao S, Fernandez-Zapico ME, Jin D. et al. KLF11-mediated repression antagonizes Sp1/sterol-responsive element-binding protein-induced transcriptional activation of caveolin-1 in response to cholesterol signaling. J Biol. Chem. 2005;280:1901-1910
82. Schlegel A, Stainier DY. Lessons from “Lower” Organisms: What Worms, Flies, and Zebrafish Can Teach Us about Human Energy Metabolism. PLOS Genetics. 2007;3:2037-2048
83. Jeong PY, Jung M, Yim YH. et al. Chemical structure and biological activity of the Caenorhabditis elegans dauer-inducing pheromone. Nature. 2005;433:541-545
84. Ren P, Lim CS, Johnsen R. et al. Control of C. elegans larval development by neuronal expression of a TGF-beta homolog. Science. 1996;274:1389-1391
85. Ashrafi K, Chang FY, Watts JL. et al. Genome-wide RNAi analysis of Caenorhabditis elegans fat regulatory genes. Nature. 2003;421:268-272
86. Kimura KD, Tissenbaum HA, Yanxia L. et al. Daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942-946
87. Leibowitz S, Alexander J. Hypothalamic serotonin in control of eating behavior, meal size and body weight. Biol. Psychiatry. 1998;44:851-862
88. Sze JY, Victor M, Loer C. et al. Food and metabolic signaling defects in a Caenorhabditis elegans serotonin-synthesis mutant. Nature. 2000;403:560-564
89. Caeanorhabdidit elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998;282:2012-2018
90. Mukhopadhyay A, Deplancke B, Walhout AJM. et al. C. elegans tubby regulates life span and fat storage by two independent mechanisms. Cell Metab. 2005;2:35-42
91. Ogg S, Paradis S, Gottlieb S. et al. The fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994-999
92. Van Gilst MR, Hadjivassiliou H, Jolly A. et al. Nuclear hormone receptor NHR-49 controls fat consumption and fatty acid composition in C. elegans. PLoS Biol. 2005;3:301-312
93. Watts JL, Browse JA. A palmitoyl-CoA-specific Δ9 fatty acid desaturase from Caenorhabditis elegans. Biochem. Biophy. Res. Commun. 2000;272:263-269
94. Watts JL, Browse JA. Genetic dissection of polyunsaturated fatty acid synthesis in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002;99:5854-5859
95. Perez CL, Van Gilst MR. A 13C Isotope Labeling Strategy Reveals the Influence of Insulin signaling on Lipogenesis in C. elegans. Cell Metab. 2008;8:266-274
96. Hashmi S, Ji Q, Zhang J. et al. A Krüppel-like factor in Caenorhabditis elegans with essential roles in fat regulation, cell death and phagocytosis. DNA Cell Biol. 2008;27:545-551
97. Zhang J, Yang C, Brey C. et al. Mutation in Caenorhabditis elegans Krüppel-like factor, KLF-3 results in fat accumulation and alters fatty acid composition. Exp Cell Res. 2009;315:2568-2580
98. White J. The nematode Caenorhabditis elegans. Cold Spring Harbor, New York, NY: Cold Spring Harbor Laboratory. 1988
99. Kimble J, Sharrock WJ. Tissue-specific synthesis of yolk proteins in Caenorhabditis elegans. Dev Biol. 1983;96:189-196
100. Brock TJ, Browse J, Watts JL. Genetic regulation of unsaturated fatty acid composition in C. elegans. PLoS Genetics. 2006;2:997-1005
101. Plenefisch JD, Zhu X, Hedgecock EM. et al. Fragile skeletal muscle attachments in dystrophic mutants of Caenorhabditis elegans: isolation and characterization of the mua genes. Development. 2000;127:1197-1207
102. Ramsay RR. The role of the carnitine system in peroxisomal fatty acid oxidation. Am. J. Med. Sci. 1999;318:28-35
103. Murphy KG, Bloom SR. Gut hormones and the regulation of energy homeostasis. Nature. 2006;444:854-859
104. Stöger R. The thrifty epigenotype: an acquired and heritable predisposition for obesity and diabetes? BioEssay. 2008;30:156-166
Author contact
![]() Correspondence to: Sarwar Hashmi, Laboratory of Developmental Biology, Lindsley F. Kimball Research Institute, New York Blood Center, 310 East 67th Street, New York, NY 10065, USA. E-mail: shashmiorg; Tel: 212-570-3254; Fax: 212-570-3121
Correspondence to: Sarwar Hashmi, Laboratory of Developmental Biology, Lindsley F. Kimball Research Institute, New York Blood Center, 310 East 67th Street, New York, NY 10065, USA. E-mail: shashmiorg; Tel: 212-570-3254; Fax: 212-570-3121