Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2010; 6(1):96-106. doi:10.7150/ijbs.6.96 This issue Cite
Research Paper
Overexpression of bacterial ethylene-forming enzyme gene in Trichoderma reesei enhanced the production of ethylene
Xi Chen1, 2, Yong Liang1, Jing Hua1, Li Tao1, Wensheng Qin2, Sanfeng Chen1, ![]()
1. State Key Laboratory for Agrobiotechnology and College of Biological Sciences, China Agricultural University, Beijing 100193, P. R. China
2. Biorefining Research Initiative and Department of Biology, Lakehead University, Thunder Bay, ON, P7B 5E1, Canada
Received 2009-12-28; Accepted 2010-2-3; Published 2010-2-6
Abstract
In order to efficiently utilize natural cellulose materials to produce ethylene, three expression vectors containing the ethylene-forming enzyme (efe) gene from Pseudomonas syringae pv. glycinea were constructed. The target gene was respectively controlled by different promoters: cbh I promoter from Trichoderma reesei cellobiohydrolases I gene, gpd promoter from Aspergillus nidulans glyceraldehyde-3-phosphate dehydrogenase gene and pgk I promoter from T. reesei 3-phosphoglycerate kinase I gene. After transforming into T. reesei QM9414, 43 stable transformants were obtained by PCR amplification and ethylene determination. Southern blot analysis of 14 transformants demonstrated that the efe gene was integrated into chromosomal DNA with copy numbers from 1 to 4. Reverse transcription polymerase chain reaction (RT-PCR) analysis of 6 transformants showed that the heterologous gene was transcribed. By using wheat straw as a carbon source, the ethylene production rates of aforementioned 14 transformants were measured. Transformant C30-3 with pgk I promoter had the highest ethylene production (4,012 nl h-1 l-1). This indicates that agricultural wastes could be used to produce ethylene in recombinant filamentous fungus T. reesei.
Keywords: Trichoderma reesei, ethylene-forming enzyme, promoter, wheat straw, overexpression.
Introduction
Ethylene is an important chemical industry feedstock, which can be used to manufacture plastics, rubber, textiles, packaging materials and chemical machinery. It has also been widely applied to transportation, catering services, and other industries. In nature, ethylene is an important plant hormone, which plays a significant role in the regulation of many physiological processes.
Higher plants produce ethylene from methionine via 1-aminocyclopropane-1-carboxylic acid [1], while microorganisms produce ethylene via two pathways which are different from that of higher plants. The majority of microorganisms produce trace amounts of ethylene from methionine via 2-keto-4-methyl-thiobutyric acid using an NADH:Fe(III)EDTA oxidoreductase [2]. However, some pathovars of Pseudomonas syringae and a few species of fungi such as Penicillium digitatum efficiently produce ethylene from 2-oxoglutarate by an ethylene-forming enzyme (EFE) [3,4]. The ethylene-forming enzyme (efe) gene from P. syringae pv. glycinea ICMP2189 has been cloned and expressed in Escherichia coli at a high level [5]. Recently this gene was introduced into Saccharomyces cerevisiae and fermented in well-controlled reactors using glucose as a carbon source. It resulted in a total yield of 890±160 μg ethylene/g glucose [6]. In order to make the biological conversion of carbon dioxide into ethylene, this gene was expressed in Cyanobacterium Synechococcus sp. PCC 7942, and the recombinant cyanobacterial strains produced ethylene, albeit with the yield was low [7].
The filamentous fungus of Trichoderma reesei produces a complete set of cellulases, including cellobiohydrolases (CBH I and CBH II), endoglucanases (EG I, EG II, and EG III), and β-glucosidase, which act synergistically to hydrolyze crystalline cellulose to glucose [8]. The major cellulase, cellobiohydrolase I (CBH I), produced from a single-copy gene, represents ~50% of protein secreted and corresponded to roughly 25% of total protein synthesized by the fungus in cellulose-inducing growth conditions [9,10]. T. reesei has been used as a host for production of homologous and heterologous proteins because of its excellent protein secretion capability [11]. Several mutants secrete up to 40 g l-1 proteins, most of which are cellulases [12].
In our study, we constructed three expression vectors (pPcbh1-efe-hph, pPgpd-efe-hph and pPpgk1-efe-hph) in which the efe gene was controlled by frequently used promoters: cellobiohydrolases I (cbh I) promoter from T. reesei, glyceraldehyde-3-phosphate dehydrogenase (gpd) promoter from Aspergillus nidulans and 3-phosphoglycerate kinase I (pgk I) promoter from T. reesei. The linearized plasmids were respectively transformed into T. reesei QM9414 which was cultured in the inducing medium using wheat straw as a sole carbon source. By developing successful ethylene-producing T. ressei strains, we can make full use of abundant renewable agricultural wastes to produce ethylene as chemical industry feedstock.
Materials and Methods
Microorganisms and plasmids
E. coli DH5α was used as a host for DNA cloning. Bacterial strain P. syringae pv. glycinea ICMP2189 was obtained from Plant Protection Institute of Chinese Academy of Agricultural Science. The fungal strain T. reesei QM9414 [13], which is a hyperactive cellulases mutant that contains all the identified T. reesei cellulases, was used as a recipient for transformations. The pTRIL which carries T. reesei cbh I promoter and terminator was used for constructing expression vectors. It was kindly provided by Dr. Tianhong Wang, Shandong University, China [14]. A. nidulans gpd promoter was amplified from plasmid pAN7-1 [15]. The promoter of pgk I was amplified from the genomic DNA of T. reesei QM9414. The hygromycin B resistance cassette was obtained from plasmid pCSN44 [16] which was kindly provided by Dr. Qun He, China Agricultural University. The pBluescript SK II(+) was used for constructing the expression vector pPgpd-efe-hph.
Culture conditions
The wild type strain T. reesei QM9414 was maintained on potato dextrose agar (PDA) [17] in test tube at 30 ℃. The recombinant T. reesei was cultivated in minimal medium (MM) [18] with dextrose as a carbon source. The dextrose in the minimal medium was replaced with cellulose (Sigma Chemicals Co., USA), lactose, carboxymethyl cellulose sodium (CMC) (Sigma Chemicals Co., USA) or wheat straw to make different inducing media, which were adopted to fermenting ethylene. All liquid cultivations were incubated in a rotary shaker at 150 rpm and 30 ℃.
Construction of recombinant plasmids
Construction of pPcbh1-efe-hph
The vector pPcbh1-efe-hph was constructed for EFE overexpression. A 1,071 bp fragment containing the entire efe coding region, was amplified from P. syringae pv. glycinea ICMP2189 using PCR with P1 primer (5'-CCGTCGACATGACCAACCTACAGACTT-3'), including an engineered Sal I site; P2 primer (5'-TATGGATCCAACTCATGAGCCTGTCGCG-3'), including an engineered BamH I site. The PCR fragment was digested with Sal I and BamH I, and subsequently ligated into pTRIL vector digested with the same restriction enzymes. After transforming the ligated DNAs into E. coli DH5α, a recombinant pTRIL-efe was obtained. Furthermore, a hygromycin B resistance cassette fragment was amplified from pCSN44 with P3 primer (5'-GGCGCATGCTAATACGACTCACTATAG-3'), including an engineered Sph I site; P4 primer (5'-ATTGCATGCTCTAGAACTAGTGGATCC-3'), including an engineered Sph I site. The PCR fragment was digested by Sph I and ligated into the plasmid pTRIL-efe digested with the same restriction enzyme. Finally the efe gene was driven by the cbh I promoter in this recombinant vector pPcbh1-efe-hph.
Construction of pPgpd-efe-hph
The recombinant plasmid pPgpd-efe-hph contained a gpd promoter from Aspergillus nidulans, the efe gene, a cbh I terminator and a hygromycin B resistance cassette. The efe gene, obtained by PCR amplification with P9 primer (5'-GGCGGAATTCATGACCAACCTACAGACTTTC-3'), including an engineered EcoR I site; P2 primer mentioned above. This fragment digested by EcoR I and BamH I, was inserted into pBluescript SK II(+). The constructed plasmid was named pBlue-efe. A 2,313 bp gpd promoter, Hind III/EcoR I fragment, amplified from pAN7-1 with P5 primer (5'-CCGAAGCTTCCCTTGTATCTCTACACACAG-3'), including an engineered Hind III site; P6 primer (5'-CGCGAATTCGAGTTCAGGCATGGTGATGT-3'), including an engineered EcoR I site, was cloned into the plasmid pBlue-efe by Hind III and EcoR I digestion. Thus, the pBlue-Pgpd-efe was constructed and subsequently cut with Hind III and BamH I. A 3,384 bp Pgpd-efe fragment was released from pBlue-Pgpd-efe. A Hind III/BamH I fragment removed the cbh I promoter and the efe gene from pTRIL-efe was obtained by cutting with Hind III and BamH I. Finally, the recombinant expression vector pPgpd-efe-hph was constructed by ligating the 3,384 bp Pgpd-efe fragment, the released Hind III/BamH I fragment from pTRIL-efe and a hygromycin B resistance cassette together.
Construction of pPpgk1-efe-hph
In the expression vector pPpgk1-efe-hph, the efe gene was controlled by a strong constitutive pgk I promoter from T. reesei [19,20]. This promoter was amplified from the genomic DNA of T. reesei QM9414 using PCR with P7 primer (5'-ACAGCATGCGATGATGGAGGATATACGCGA-3'), including an engineered Sph I site; P8 primer (5'-ACGGTCGACGTTAGACAGAGACATTTTGGC-3'), including an engineered Sal I site. The 967 bp Sph I/Sal I fragment of pgk I promoter substituted for the cbh I promoter of pTRIL-efe. Then a Sph I hygromycin B resistance cassette fragment was inserted into the resulting plasmid.
T. reesei QM9414 protoplast preparation and transformation
Protoplasts of T. reesei QM9414 were prepared by a method modified from [21]. The linear recombinant plasmid was mixed with protoplasts by adding solution I (60% PEG4000, 50 mM CaCl2, 10 mM Tris-HCl, pH7.5), incubated on ice for 30 min and followed by 20 min at room temperature. Subsequently solution I was added again. After incubating for 5 min, the protoplasts were suspended thoroughly in solution II (1 M sorbitol, 10 mM CaCl2, 10 mM Tris-HCl, pH7.5). The hygromycin B resistant transformants were selected on the medium containing 100 μg ml-1 hygromycin B and 1 M sorbitol as an osmotic stabilizer. Transformants were purified to uninuclear clones by plating single spores on selective minimal medium. The fragment of efe was amplified using PCR; the genomic DNA was prepared from the transformant as a template and the above-mentioned primers P1 and P2 were used in the reaction. Then the positive transformants were incubated on PDA containing 100 μg ml-1 hygromycin B. After five generations, stable transformants were obtained.
All stable single clones were cultured in minimal medium for two days and transferred to the inducing medium. Ethylene production was then measured with an HP6890 gas chromatograph as described by Chalutz E [22].
Confirmation of transformants
Southern blot analysis
The entire efe gene fragment was recovered from the recombinant plasmid pPcbh1-efe-hph and labeled using DIG-DNA Labeling and Detection Kit purchased from Roche (Germany). The chromosomal DNA of T. reesei QM9414 and 14 high yielding ethylene transformants were isolated using the CTAB procedure as described by Tel-zur N [23] with some modifications and digested thoroughly by SalⅠ. Southern blot hybridization was carried out using the labeled efe gene as a probe.
RT-PCR confirmation
Approximately 106 spores of transformants were inoculated in 50 ml of lactose inducing media, and then grown in shake flasks for two days. Next, mycelia samples were collected by filtration and centrifugation. Then total RNAs were extracted from the fresh mycelia samples of positive transformants and the parental strain, using the Trizol reagent (Invitrogen) according to the manufacturer's protocol. The cDNA was amplified by using oligo-dT primers in the RT-PCR following the instructions of AMV reverse transcriptase. The efe gene was amplified using the cDNA as a template with P1 and P2 primers.
Ethylene production and cellulase activities in transformants
Determination of ethylene production
The spore suspensions were prepared by washing 7-day-old PDA cultures from all positive transformants with 9 g l-1 sterile NaCl solution. 1 ml spore suspension of each transformant (106 spores ml-1) was inoculated into 100 ml flasks with 30 ml of minimal medium. After 48 h incubation, mycelia were collected and washed twice to remove the medium. Then the mycelia were transferred into 2 ml of wheat straw inducing medium in 12×120 mm test tube sealed with a rubber stopper. All were grown by shaking at 150 rpm at 30 °C for an additional 24 h. Subsequently, 0.1 ml air samples were collected using a gas tight syringe and injected into an HP6890 gas chromatograph. Ethylene production rate was calculated as nanoliters of ethylene produced in 1 l of inducing medium for 1 h (nl h-1 l-1) [22].
Unless otherwise stated, all treatments were performed in five replicates and all experiments were repeated thrice.
Effect of different inducers and nitrogen compounds on ethylene production
In order to obtain a higher ethylene production rate, dextrose carbon source in minimal medium was replaced with 2% (w/v) cellulose, 2% (w/v) CMC, 2% (w/v) lactose or 2% (w/v) wheat straw. Nitrogen compounds such as 0.2% (w/v) peptone and 0.2% (w/v) yeast extract were added respectively in these media. Following that, the ethylene production rate was determined by the aforementioned method.
Measurement of cellulase activities and extra-cellular protein concentration
Filter paper activity (FPA) (total cellulase activity) and cellobiohydrolase (exoglucanase) activity were measured according to the modified Ghose's method [24]; filter paper and absorbent cotton were substrates of these reactions. In addition, the released sugar was measured by the dinitrosalicylic acid (DNS) method [25]. Enzyme activity was defined as the amount of enzyme required to liberate 1 μmol of dextrose per gram protein per min at 50 °C (U mg-1).
The extra-cellular total protein concentration was determined by the dye-binding method of the Bradford assay [26] using bovine serum albumin (BSA) fraction V (Sigma) as the standard.
Results
Expression of efe gene in T. reesei QM9414
A 1,071 bp fragment including the entire efe coding region was amplified by PCR from P. syringae pv. glycinea ICMP2189 using primers designed according to the known sequence of efe gene of P. syringae. Three vectors were constructed: the efe gene was controlled by a strong inducible promoter of cbh I in pPcbh1-efe-hph, a strong constitutive promoter of gpd in pPgpd-efe-hph and another strong constitutive promoter of pgk I in pPpgk1-efe-hph. A 2,500 bp hygromycin B resistance cassette derived from pCSN44 was cloned into these constructs.
More than 200 clones on high osmotic media containing 100 ug ml-1 hygromycin B were purified to uninuclear clones. After five generations they were further identified by PCR amplification using primers P1 and P2 based on the efe gene sequence. The results of PCR analysis concluded that 14 clones of pPcbh1-efe-hph, 16 of pPgpd-efe-hph and 13 of pPpgk1-efe-hph contained their corresponding gene sequences. Nevertheless no amplified fragment was detected in the control parental strain T. reesei QM9414. It was proved that the heterologous efe gene was stably integrated into the genomes of transformants. After fermentation in the inducing media and measurement of the capacities of ethylene production, 14 transformants were selected to perform Southern blot analysis and six transformants were tested in RT-PCR detection.
Southern blot and RT-PCR analysis
Southern blot analysis was performed on 14 stable high yielding transformants and their transgenic nature was confirmed. Nine of 14 transformants, A55-9, A33-2, A55-5, B2-7, B12-3, B2-6, C30-10, C1-3, C2-5 had a single copy of the efe gene; A10-6 and C30-3 had two copies; B38-2 and B38-4 had three copies; A41-4 had four copies, whereas no hybridization signal was observed in the control parental strain genome (Fig.1).
Southern blot analysis of genomic DNA isolated from 14 transformants. (a) Lane 1: DNA molecular weight marker (λDNA/HindⅢ+EcoRⅠ); 2: transformant A10-6 (pPcbh1-efe-hph); 3: transformant A33-2 (pPcbh1-efe-hph); 4: transformant A41-1 (pPcbh1-efe-hph); 5: transformant A55-5 (pPcbh1-efe-hph); 6: transformant A55-9 (pPcbh1-efe-hph); 7: transformant B2-6 (pPgpd-efe-hph); 8: transformant B2-7 (pPgpd-efe-hph); 9: transformant B12-3 (pPgpd-efe-hph); (b) Lane 1: DNA molecular weight marker (λDNA/HindⅢ+EcoRⅠ); 2: transformant B38-2 (pPgpd-efe-hph); 3: transformant B38-4 (pPgpd-efe-hph); 4: transformant C1-3 (pPpgk1-efe-hph); 5: transformant C2-5 (pPpgk1-efe-hph); 6: transformant C30-3 (pPpgk1-efe-hph); 7: transformant C30-10 (pPpgk1-efe-hph); Lane 8: genomic DNA from non-transformed host strain T. reesei QM9414; Lane 9: DNA from the whole coding sequence of ethylene-forming enzyme gene. Genomic DNA digested by SalⅠand hybridized with a digoxigenin-labelled whole coding sequence of the efe gene.
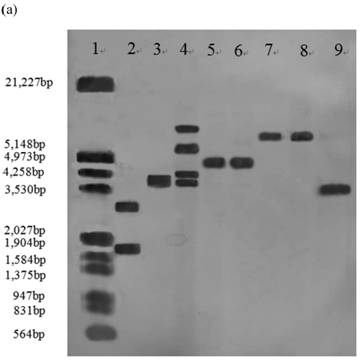
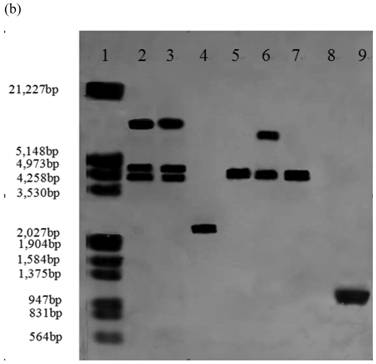
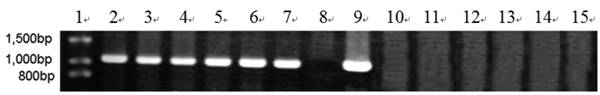
To further determine expression of the heterologous efe gene, six transformants (A10-6, A55-5, B12-3, B38-4, C1-3 and C2-5) from above 14 Southern hybridization confirmed transformants, were selected for RT-PCR. T. reesei QM9414 was used as a control. A 1,071 bp efe fragment was detected in all six transformants, whereas no signal was obtained in the parental strain. There was no fragment amplified by PCR using total RNAs treated with DNase I as the template. This indicated that the PCR products from aforementioned six transformants were due to the amplification of reverse transcribed cDNA, rather than trace amount of genomic DNA (Fig. 2). This result showed that the heterologous efe gene from P. syringae pv. glycinea was transcribed in positive transformants of filamentous fungus T. reesei.
RT-PCR analysis of EFE mRNA in 6 transformants. Lane 1: DNA molecular weight marker (λDNA/HindⅢ+EcoRⅠ); 2: transformant A10-6 (pPcbh1-efe-hph); 3: transformant A55-5 (pPcbh1-efe-hph); 4: transformant B12-3 (pPgpd-efe-hph); 5: transformant B38-4 (pPgpd-efe-hph); 6: transformant C1-3 (pPpgk1-efe-hph); 7: transformant C2-5 (pPpgk1-efe-hph); 8: the non-transformed parental strain T. reesei QM9414; 9: PCR production from plasmid pPcbh1-efe-hph as a positive control; 10 to 15: PCR amplified with total RNAs of aforementioned 6 transformants treated by DNaseⅠas a negative control. No signals were detected in the negative controls illustrated that the corresponding fragments from lane 2 to lane 7 were amplified with cDNAs of transformants as templates rather than low levels of genomic DNAs.
Comparison of ethylene production and cellulase activities in transformants
The effect of carbon sources and nitrogen compounds on ethylene production
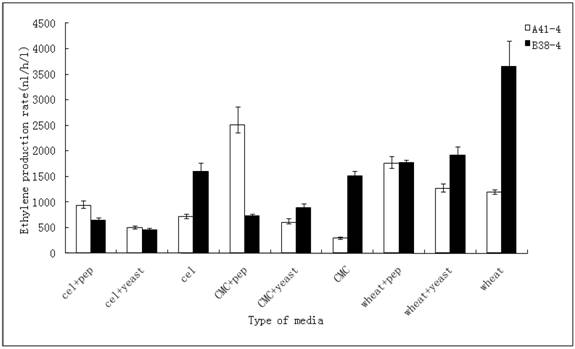
The strong inducible promoters, together with high capacity of secretory protein have made T. reesei an attractive host for protein production. The protein production of this fungus is strictly controlled by the carbon and nitrogen sources available, ranging from several tens of grams per liter of native hydrolytic enzymes to completely shutting down the heterologous protein expression [27,28]. The 106 spores of two positive transformants A41-4 and B38-4 were inoculated to different media in which cellulose, CMC, lactose, wheat straw and dextrose were used as a carbon source. The transformant A41-4 with the efe gene controlled by the inducible cbh I promoter yielded more ethylene when wheat straw, CMC and cellulose were used rather than lactose and dextrose. This result was slightly different from the conclusion of de Faria's, that the lactose was the better inducer than the other carbon sources when the hetetologous xyn I was expressed under the control of cbh I promoter [29]. Transformant A41-4 produced ethylene at the lowest level using dextrose. B38-4 with the efe gene driven by the gpd promoter produced more ethylene when inoculated in media containing wheat straw as carbon source rather than CMC, lactose, cellulose and dextrose.
Subsequently, the ethylene production rates using three inducers (wheat straw, CMC and cellulose) with peptone or yeast extract in these media respectively were determined. The result, represented in Figure 3, revealed that the ethylene production rate of A41-4 reached its highest when the transformant was incubated in the media containing CMC and peptone. Besides this medium, the effects of wheat straw inducing media (adding peptone or yeast extract or not) were better than the other media. The production rate of B38-4 reached its highest level in the wheat straw inducing media compared to other media.
Effect of different carbon resources and nitrogen compounds on ethylene production in random-selecting transformants A41-4 and B38-4. The results shown are averages (± the standard deviation [±SD]) from five parallel cultures.
Comparison of ethylene production of three expression vectors
Cellulase gene, especially cbh I controlled by cbh I promoter can be induced efficiently by sophorose, cellulose and other carbon sources in T. reesei [8], so the expression vector pPcbh1-efe-hph was constructed. In transformants A10-6, A33-2, A41-4, A55-5 and A55-9, the heterologous efe gene was driven by the inducible promoter of cbh I in pPcbh1-efe-hph. In transformants B2-6, B2-7, B12-3, B38-2 and B38-4, the efe gene was controlled by gpd promoter in the vector pPgpd-efe-hph. In transformants C1-3, C2-5, C30-3 and C30-10, the target gene was under the control of pgk I promoter in pPpgk1-efe-hph. In order to compare the effect of three different promoters, 14 transformants with different copy numbers of efe gene were incubated in wheat straw inducing media for two days and then the ethylene production rates were determined (Table 1). The parental stain was used as a negative control. It was shown that C30-3, C30-10 and C1-3 whose production rates were 4,012, 2,593 and 2,467 nl h-1 l-1 produced ethylene more efficiently than the other transformants, followed by B38-2, B38-4 and A41-4, whereas the production rates of A10-6, B2-6 and C2-5 were the lowest. No ethylene production was detected in T. reesei QM9414 when cultured by the same condition (Fig. 4).
Comparison of ethylene production of 14 transformants using different promoter controlling the efe gene and their corresponding copy number(s).
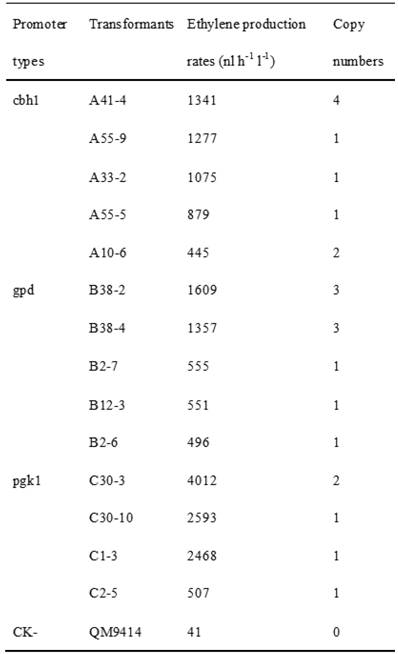
Comparison of ethylene production of 14 stable transformants and the parental strain T. reesei QM9414. A10-6, A32-3, A41-4, A55-5 and A55-9 were different transformants introduced the expression vector pPchb1-efe-hph; B2-6, B2-7, B12-3, B38-2 and B38-4 were transformed by pPgpd-efe-hph; C1-3, C2-5, C30-3 and C30-10 were transformants of pPpgk1-efe-hph; T. reesei QM9414 was used as a negative control.
Growth condition, extra-cellular protein concentration and cellulases assay of transformants from three constructs
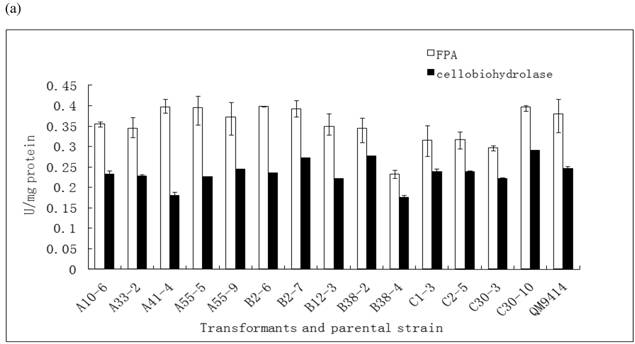
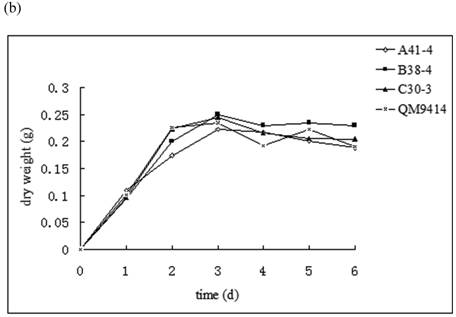
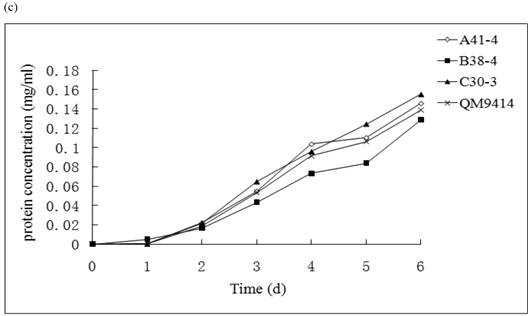
In order to illustrate the effect of heterologous gene integration on the filamentous fungus, the growth behavior, extra-cellular total protein concentration and the cellulase (especially cellobiohydrolase) activities of transformants were analyzed. The wild type T. reesei QM9414 was used as a control. When spores were cultured in wheat straw inducing media for four days, the FPA and cellobiohydrolase activities reached their peak (data not shown). The protein concentrations and cellulase activities in the fourth day were the same level except for B38-4. The transformant B38-4 had the lowest total cellulase and cellobiohydrolase activities and the lowest protein concentration, whereas the dry weight was slightly higher (Fig. 5). It suggested that the heterologous efe gene may integrate into the cellulase gene loci in T. reesei genome and affect the expression of cellulase gene.
(a) Comparison of FPA and cellobiohydrolases activities in 14 tansformants and the host strain cultured in wheat straw inducing media in the fourth day. (b) Growth curves for 3 transformants respectively came from three expression vectors and the non-tranformed T. reesie QM9414. (c) Change of extracellular protein concentration in 3 transformants and T.reesei QM9414.
Discussion
Filamentous fungi such as Aspergillus and Trichoderma species are commonly used as cell factories for industrial production of proteins. However, the production of heterologous proteins is less efficient than endogenous proteins, especially genes which came from phylogenetically distant species [30,31]. In order to improve heterologous protein production, several strategies have been developed, including introduction of multiple copies of gene of interest; constructing expression vector with the target gene driven by a strong homologous promoter and fused with a gene encoding part or whole well-secreted protein; and selection of protease-deficient mutant strains [11].
In our study, we selected three strong promoters of cbh I, gpd and pgk I. The cbh I promoter is one of the most efficient promoters in T. reesei, which could strongly promote expression of many homologous and heterologous genes [29,32]. The gpd is a housekeeping gene which plays a central role in glycolysis and gluconeogenesis [33]; the promoter was strong and constitutive. In the presence of some chemicals, the mRNA of the heterologous gene controlled by the gpd promoter was increased 20-fold [34]. The 3-phosphoglycerate kinase I (PGK) is a key enzyme in ATP generation by glycolysis. In the yeast Saccharomyces cerevisiae, the PGK protein constitutes about 1% of the total cellular proteins, and its mRNA is correspondingly abundant. Vectors carrying the pgk I promoter are among the most efficient expression systems developed for yeast [35]. The transformants had remarkably higher ethylene yields in the wheat straw inducing media than in the minimal medium containing dextrose as a carbon source. This indicates that wheat straw materials could induce ethylene production. However, in the same wheat straw inducing media, the transformants with the efe gene controlled by pgk I promoter had higher ethylene production than that driven by cbh I promoter which is one of the strongest promoters in T. reesei. This may be due to some limitations in transcriptional or (post) translational levels [36] or due to some factors in the media being more effective on one promoter than another. To increase the ethylene production, in future research we may use other renewable resources like forest biomass or pulp/paper mill sludge as inducers and other chemical such as glutamate, ammonium or nitrate to facilitate induction.
In earlier reports, people succeeded in enhancing protein yields by increasing the copy number of target genes, particularly in Aspergillus and Trichoderma species. Nevertheless a significantly growing number of results showed that protein yields cannot be elevated further conceivably because of insufficient supply of regulation proteins or other essential transcription factors [11,37]. It is a complex process to yield heterologous proteins in filamentous fungi, with a number of limitations such as transcription, protein modification and degradation. Our analysis of Southern blot and the ethylene production illustrated that copy number of the target gene and the ethylene production did not have a linear correlation.
In this research, the ethylene productions of 43 stable positive transformants from three constructs were measured (data not shown). Different clones transformed by the same vector had very different yields. The differences between the transformants were likely due to the position effects of the heterologous gene in the genome [38]. In the absence of stable autonomously replicating plasmids in filamentous fungi, the target gene in the expression vector needs to be integrated into the special loci in the genome. Random insertion more commonly occurred than homologous recombination [39]. Our results showed that cellulase (especially cellobiohydrolase) activities of the transformants with the efe gene between the cbh I promoter and terminator were similar to that of the parental strain. It indicated that the heterologous gene was possibly integrated into an ectopic chromosomal location other than the cbh I locus. The cellulases activities of B38-4 were much lower than the other transformants and the parental strain may due to the insertion into some cellulase genes loci.
The optimal growth for T. reesei is in aerobic condition. Also, oxygen is essential for ethylene production catalyzed by ethylene-forming enzyme (EFE) from P. syringae. The overall reaction for EFE from P. syringae is shown by the following: 3C5H6O5+3O2 + C6H14N4O2 → 2C2H4 + 7CO2 + C4H6O4 +3H2O + CH5N3 + C5H7NO2 [40]. Thus, the optimal growth and the most effective ethylene-production for T. reesei transformants carrying efe gene from P. syringae are also in aerobic condition. R. Lejeune and G. V. Baron reported that the growth and enzyme activity of CMCase, xylanase and endoglucanase of T. reesei were the most affected by agitation [41]. In this study, the parental strain and all the transformants were incubated under the aerobic condition for 48h and then they were transferred to the test tubes and sealed with the rubber stoppers for collection of the ethylene gas. From those researches, we deduced that if the transformants were continually cultured aerobically, the observed ethylene production level would be higher than which if they were measured with current methods. In future studies, the transformants will be cultured in a bioreactor in which oxygen or air can be continuously pumped in to facilitate T. reesei growth and ethylene can easily be collected. The ethylene production in the bioreactor should be much higher than the results demonstrated in this study.
In the previous study by our lab in China Agricultural University, overexpression of ethylene-forming enzyme gene driven by chb I promoter was performed in T. rivide, however the ethylene production was low and the transformant was not stable [42]. The cost of the microbial ethylene production was high, so it was not feasible to apply in large-scale industrial production. In this work, we used wheat straw as a carbon source for T. reesei to produce ethylene, and the production rate of transformant C30-3 reached 4,012 nl h-1 l-1 by using this substrate. There is great potential to convert natural cellulose materials to ethylene as chemical industry feedstock by engineered T. reesei. We will continue to optimize the inducing condition by using different renewable resources as carbon sources in order to further enhance the yield of ethylene.
Acknowledgements
This work was supported by China National Fundamental Fund of Personnel Training (Grant No. J0730639).
We thank Miranda Maki for critically reading the manuscript, and for her helpful comments and improvement of the text.
Conflict of interests
The authors have declared that they have no conflict of interest exists.
References
1. Hall MA, Smith AR. Ethylene and the responses of plants to stress. Bulgarian Journal Of Plant Physiology. 1995;21:71-79
2. Weingart H, Volksch B, Ullrich MS. Comparison of Ethylene Production by Pseudomonas syringae and Ralstonia solanacearum. Phytopathology. 1999;89:360-365
3. Nagahama K, Ogawa T, Fujii T, Tazaki M, Tanase S, Morino Y, Fukuda H. Purification and properties of an ethylene-forming enzyme from Pseudomonas syringae pv. phaseolicola PK2. J Gen Microbiol. 1991;137:2281-2286
4. Volksch B, Weingart H. Comparison of ethylene-producing Pseudomonas syringae strains isolated from kudzu (Pueraria lobata) with Pseudomonas syringae pv. phaseolicola and Pseudomonas syringae pv. glycinea. European Journal of Plant Pathology. 1997;103:795-802
5. Katsuya I, Masayoshi M, Yorinao I, Sumio T, Takahira O, Hideo F. Overexpression and in vitro reconstitution of the ethylene-forming enzyme from Pseudomonas syringae. Journal of Fermentation and Bioengineering. 1995;79:205-211
6. Pirkov I, Albers E, Norbeck J, Larsson C. Ethylene production by metabolic engineering of the yeast Saccharomyces cerevisiae. Metab Eng. 2008;10:276-280
7. Takahama K, Matsuoka M, Nagahama K, Ogawa T. Construction and analysis of a recombinant cyanobacterium expressing a chromosomally inserted gene for an ethylene-forming enzyme at the psbAI locus. J Biosci Bioeng. 2003;95:302-305
8. Ilmen M, Saloheimo A, Onnela ML, Penttila ME. Regulation of cellulase gene expression in the filamentous fungus Trichoderma reesei. Appl Environ Microbiol. 1997;63:1298-1306
9. Keranen S, Penttila M. Production of recombinant proteins in the filamentous fungus Trichoderma reesei. Curr Opin Biotechnol. 1995;6:534-537
10. Kubicek CP, Mikus M, Schuster A, Schmoll M, Seiboth B. Metabolic engineering strategies for the improvement of cellulase production by Hypocrea jecorina. Biotechnol Biofuels. 2009;2:19
11. Nevalainen KM, Te'o VS, Bergquist PL. Heterologous protein expression in filamentous fungi. Trends Biotechnol. 2005;23:468-474
12. Durand H, Clanet M, Tiraby G. Genetic improvement of Trichoderma reesei for large scale cellulase production. Enzyme and Microbial Technology. 1988;10:341-346
13. Mandels M, Weber J, Parizek R. Enhanced cellulase production by a mutant of Trichoderma viride. Appl Microbiol. 1971;21:152-154
14. Wang TH, Wu ZH, Liu SL, Qu YB. Construction of heterogeneous genes expression system of filamentous fungus Trichoderma reesei. Chinese Journal of Biochemistry and Molecular Biology. 2003;19:736-742
15. Mullaney EJ, Hamer JE, Roberti KA, Yelton MM, Timberlake WE. Primary structure of the trpC gene from Aspergillus nidulans. Mol Gen Genet. 1985;199:37-45
16. Staben C, Jensen B, Singer M, Pollock J, Schechtman M, Kinsey J, Selker EU. Use of a bacterial hygromycin B resistance gene as a dominant marker in Neurospora crassa transformation. Fungal Genet Newslett. 1989;36:79-81
17. Adsul MG, Bastawde KB, Varma AJ, Gokhale DV. Strain improvement of Penicillium janthinellum NCIM 1171 for increased cellulase production. Bioresour Technol. 2007;98:1467-1473
18. Sun T, Liu BH, Liu DM, Li ZH. Effect of elevated temperature on Trichoderma viride SL-1 in solid state fermentations. Biotechnol Lett. 1997;19:171-174
19. Vanhanen S, Saloheimo A, Ilmen M, Knowles JK, Penttila M. Promoter structure and expression of the 3-phosphoglycerate kinase-encoding gene (pgk1) of Trichoderma reesei. Gene. 1991;106:129-133
20. Vanhanen S, Penttila M, Lehtovaara P, Knowles J. Isolation and characterization of the 3-phosphoglycerate kinase gene (pgk) from the filamentous fungus Trichoderma reesei. Curr Genet. 1989;15:181-186
21. Penttila M, Nevalainen H, Ratto M, Salminen E, Knowles J. A versatile transformation system for the cellulolytic filamentous fungus Trichoderma reesei. Gene. 1987;61:155-164
22. Chalutz E, Lieberman M. Methionine-induced Ethylene Production by Penicillium digitatum. Plant Physiol. 1977;60:402-406
23. Tel-zur N, Abbo S, Myslabodski D, Mizrahi Y. Modified CTAB procedure for DNA isolation from epiphytic cacti of the genera Hylocereus and Selenicereus. Plant Molecular Biology Reporter. 1999;17:249-254
24. Ghose TK. Measurement of cellulase activities. Pure Appl Chem. 1987;59:257-268
25. Miller G. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Analytical Chemistry. 1959;46:426-428
26. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248-254
27. el-Gogary S, Leite A, Crivellaro O, Eveleigh DE, el-Dorry H. Mechanism by which cellulose triggers cellobiohydrolase I gene expression in Trichoderma reesei. Proc Natl Acad Sci U S A. 1989;86:6138-6141
28. Sternberg D, Mandels GR. Induction of cellulolytic enzymes in Trichoderma reesei by sophorose. J Bacteriol. 1979;139:761-769
29. de Faria FP, Te OV, Bergquist PL, Azevedo MO, Nevalainen KM. Expression and processing of a major xylanase (XYN2) from the thermophilic fungus Humicola grisea var. thermoidea in Trichoderma reesei. Lett Appl Microbiol. 2002;34:119-123
30. Archer DB, Peberdy JF. The molecular biology of secreted enzyme production by fungi. Crit Rev Biotechnol. 1997;17:273-306
31. Gouka RJ, Punt PJ, van den Hondel CA. Efficient production of secreted proteins by Aspergillus: progress, limitations and prospects. Appl Microbiol Biotechnol. 1997;47:1-11
32. Heli Haakana AM-O, Vesa Joutsjoki, Arja Mäntylä, Pirkko Suominen, Jari Vehmaanperä. Cloning of cellulase genes from Melanocarpus albomyces and their efficient expression in Trichoderma reesei. Enzyme and Microbial Technology. 2004;34:159-167
33. Punt PJ, Dingemanse MA, Jacobs-Meijsing BJ, Pouwels PH, van den Hondel CA. Isolation and characterization of the glyceraldehyde-3-phosphate dehydrogenase gene of Aspergillus nidulans. Gene. 1988;69:49-57
34. Alves AM, Record E, Lomascolo A, Scholtmeijer K, Asther M, Wessels JG, Wosten HA. Highly efficient production of laccase by the basidiomycete Pycnoporus cinnabarinus. Appl Environ Microbiol. 2004;70:6379-6384
35. Holland MJ, Holland JP. Isolation and identification of yeast messenger ribonucleic acids coding for enolase, glyceraldehyde-3-phosphate dehydrogenase, and phosphoglycerate kinase. Biochemistry. 1978;17:4900-4907
36. Stricker AR, Mach RL, de Graaff LH. Regulation of transcription of cellulases- and hemicellulases-encoding genes in Aspergillus niger and Hypocrea jecorina (Trichoderma reesei). Appl Microbiol Biotechnol. 2008;78:211-220
37. Verdoes JC, Punt PJ, Schrickx JM, van Verseveld HW, Stouthamer AH, van den Hondel CA. Glucoamylase overexpression in Aspergillus niger molecular genetic analysis of strains containing multiple copies of the glaA gene. Transgenic Res. 1993;2:84-92
38. Valkonen M, Penttila M, Saloheimo M. The ire1 and ptc2 genes involved in the unfolded protein response pathway in the filamentous fungus Trichoderma reesei. Mol Genet Genomics. 2004;272:443-451
39. Ilmen M, Onnela ML, Klemsdal S, Keranen S, Penttila M. Functional analysis of the cellobiohydrolase I promoter of the filamentous fungus Trichoderma reesei. Mol Gen Genet. 1996;253:303-314
40. Fukuda H, Ogawa T, Tazaki M, Nagahama K, Fujii T, Tanase S, Morino Y. Two reactions are simultaneously catalyzed by a single enzyme: the arginine-dependent simultaneous formation of two products, ethylene and succinate, from 2-oxoglutarate by an enzyme from Pseudomonas syringae. Biochem Biophys Res Commun. 1992;188:483-489
41. Hayward TK, Hamilton J, Tholudur A, McMillan JD. Improvements in titer, productivity, and yield using Solka-floc for cellulase production. Appl Biochem Biotechnol. 2000;84:859-874
42. Tao L, Dong HJ, Chen X, Chen SF, Wang TH. Expression of ethylene-forming enzyme (EFE) of Pseudomonas syringae pv. glycinea in Trichoderma viride. Appl Microbiol Biotechnol. 2008;80:573-578
Author contact
![]() Corresponding author: Sanfeng Chen, State Key Laboratory for Agrobiotechnology and College of Biological Sciences, China Agricultural University, Beijing 100193, P. R. China. Tel./Fax: +86 10 62731551. E-mail address: chensfedu.cn
Corresponding author: Sanfeng Chen, State Key Laboratory for Agrobiotechnology and College of Biological Sciences, China Agricultural University, Beijing 100193, P. R. China. Tel./Fax: +86 10 62731551. E-mail address: chensfedu.cn