Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2010; 6(4):327-332. doi:10.7150/ijbs.6.327 This issue Cite
Research Paper
Generation of Fgfr3 Conditional Knockout Mice
Nan Su1*, Xiaoling Xu2*, Cuiling Li2, Qifen He1, Ling Zhao1, Can Li1, Siyu Chen1, Fengtao Luo1, Lingxian Yi1, Xiaolan Du1, Haiyang Huang1, Chuxia Deng2 ![]() , Lin Chen1
, Lin Chen1 ![]()
1. State Key Laboratory of Trauma, Burns and Combined Injury, Center of Bone Metabolism and Repair, Trauma Center, Institute of Surgery Research, Daping Hospital, Third Military Medical University, Chongqing 400042, China
2. Genetics of Development and Disease Branch, National Institute of Diabetes, Digestive and Kidney Diseases, US National Institutes of Health, 10 Center Drive, Bethesda, Maryland 20892, USA
*These authors contributed equally to this work
Received 2010-2-23; Accepted 2010-6-13; Published 2010-6-15
Abstract
Fibroblast growth factor receptor 3 (FGFR3), highly conserved in both humans and murine, is one of key tyrosine kinase receptors for FGF. FGFR3 is expressed in different tissues, including cartilage, brain, kidney, and intestine at different development stages. Conventional knockout of Fgfr3 alleles leads to short life span, and overgrowth of bone. In clinic, human FGFR3 mutations are responsible for three different types of chondrodysplasia syndromes including achondroplasia (ACH), hypochondroplasia (HCH) and thanatophoric dysplasia (TD). For better understanding of the roles of FGFR3 in different tissues at different stages of development and in pathological conditions, we generated Fgfr3 conditional knockout mice in which loxp sites flank exons 9-10 in the Fgfr3 allele. We also demonstrated that Cre-mediated recombination using Col2a1-Cre, a Cre line expressed in chondrocyte during bone development, results in specific deletion of the gene in tissues containing cartilage. This animal model will be useful to study distinct roles of FGFR3 in different tissues at different ages.
Keywords: FGFR3, conditional knock out, Cre-Loxp, gene targeting
Introduction
Fibroblast growth factor receptor 3 (FGFR3) is one of the four membrane-spanning receptor tyrosine kinases that serve as high affinity receptors for multiple fibroblast growth factors [1]. Fgfr3 is mainly expressed in cartilage, and negatively regulates bone growth [2, 3]. Gain-of-function mutations in human FGFR3 lead to three types of chondrodysplasia syndromes including achondroplasia (ACH), hypochondroplasia (HCH) and thanatophoric dysplasia (TD) [4-9]. Recently partial loss-of-function mutation of FGFR3 was found to cause camptodactyly, tall stature, scoliosis, and hearing loss (CATSHL) syndrome [10]. The expression of Fgfr3 is also detected in other organs such as kidney, intestine, brain, spinal cord and so on [11-14], which indicates that FGFR3 could regulate the development and physiological and pathological functions of these cells.
To explore the roles of FGFR3 in development and diseases, mouse models with genetic modification of FGFR3 have been generated [1-3, 15-18]. Deng and Colvin, etc have independently generated conventional Fgfr3 knock out mouse models [2, 3] or model with deficiency in a specific isoform [18], which have been used to study the role of FGFR3 in a variety of developmental and disease processes [2, 19-21]. In these conventional knock out mice, Fgfr3 is disrupted in all cells, which prevents the effort for further studying of the role of FGFR3 in specific types of cells and their corresponding cellular and molecular mechanisms. For example, it is difficult to know if the decreased bone mass in Fgfr3 null mice [20] is secondarily related to the altered chondrogenesis and/or directly related to changes in osteogenesis [2, 3], because both chondrogenesis and osteogenesis were found changed in Fgfr3 knock out mice [16, 20]. Furthermore, in certain genetic background, the lifespan of Fgfr3 null mice is relatively shorter than their normal littermates [20], which prevents us from studying the role of FGFR3 in later development and diseases in adult stages such as aging related diseases. In addition, in conventional Fgfr3 knock out mouse models, the morphology structure of tissue has significant changes during early development stage. It is impossible to exclude the potential role of already changed tissue in the pathogenesis of regeneration and healing. To overcome these disadvantages, we have generated a Fgfr3 conditional null allele, the Fgfr3flox allele.
The mouse Fgfr3 gene contains 19 exons and spans about 25 kb on chromosome 5. The protein product of FGFR3 is encoded by exons 2-18 [22]. As for FGFR3, alternative splicing of the second half of the third Ig domain produces two isoforms, IIIb (encoded by exons 7 and 8) and IIIc (encoded by exons 7 and 9) [23]. The transmembrane domain is encoded by exon 10. Deletion of exons 9 and 10 is expected to cause loss of function of both IIIb and IIIc isoform of FGFR3.
Materials and Methods
Generation of the Fgf3floxneo allele
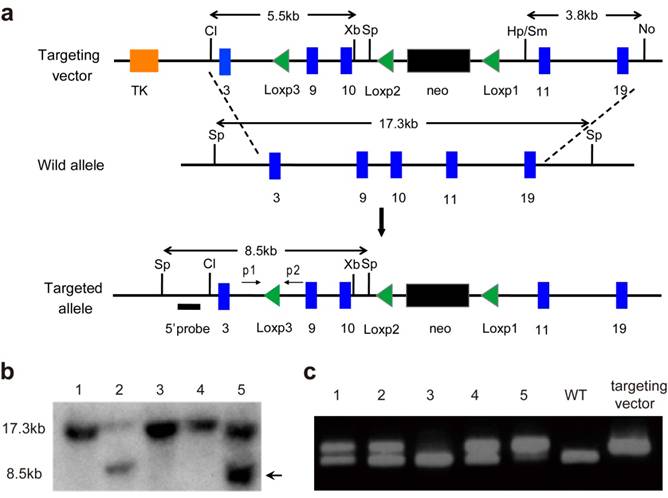
The targeting vector was constructed using Fgfr3 genomic DNA isolated previously [3]. The ploxPneo vector was described previously [24]. Briefly, in the targeting vector, the 3.8-kb fragment including intron 10 to part of exon 19 was inserted into ploxPneo vector using Not I and Sma I to generate the 3' homology recombination arm. The 5' homology recombination arm in the targeting vector was generated by inserting the 5.5-kb Cla I-Xba I fragment including part of intron 2 to intron 10 and the third Loxp was inserted into intron 8 of the Fgfr3 locus. The finished targeting construct, Fgfr3floxneo, is shown in Figure 1a.
Generation of the Fgfr3floxneo allele. (a) Strategy for generating the Fgfr3 targeting vector and Fgfr3floxneo (targeted) allele. Blue boxes represent exons. The 5' external probes for Southern Blot is indicated by thick lines. The predicted length of Southern fragments are indicated with double arrow lines. Cl, Cla I; No, Not I; Sm, Sma I; Sp, Spe I; Xb, Xba I; Hp, Hpa I; p, primer. (b) Targeted events were identified by Southern analysis of Spe I- digested genomic ES cell DNAs with a 5'flanking probe. (c) The third Loxp in targeted allele was confirmed by PCR.
Electroporation of ES cells and generation of germline chimeras
TC1 embryonic stem (ES) cells [3] were transfected with Not I-digested ploxPneoFgfr3 by electroporation and selected using G418 and -(2-deoxy-2-fluoro-β-D-avabinofuranosyl-S-iodouracil (FIAU). The homologous recombination events within the Fgfr3 locus from both G418- and FIAU-resistant ES clones were analyzed by Southern blot using the 5' external probe with Spe I-digested DNA. The 5′ probe is a 1.9-kb BamH I-Sac I fragment specific to the Fgfr3 sequence. The third Loxp was identified by PCR using primer p1 (5'-GATGCCTCAACAATACTGGTAGCCC-3') and p2 (5'-CCAGACAGATGGATGGACAGGAA-3'). Cells heterozygous for the targeted mutation were microinjected into blastocysts from C57/B6 mice to obtain germ-line transmission following standard procedures.
Genotype analysis
The chimeric males were bred to C57BL/6 females, and F1 agouti offspring were analyzed by PCR for the presence of the Fgfr3floxneo allele. Genotypes of the mice bearing the Fgfr3neofloxed, Fgfr3flox, Fgfr3null and wild-type allele were determined by PCR analysis as illustrated in Figure 2 and 3. The primers are p3 (5'-GCTCCCTGTCCTGCCTCGTG-3'), p4(5'-CAGCTCATTCCTCCCACTCATGAT-3') and p5 (5'-TGTAAAAGGGGTGGGGTGGTAG-3').
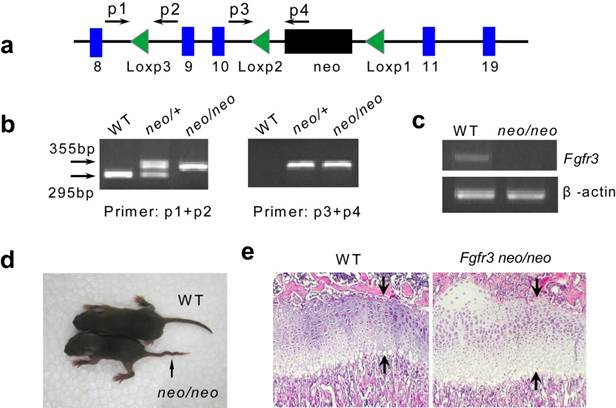
Generation of Fgfr3floxneo/floxneo mice. (a) Map of Fgfr3floxneo allele. The position of primers was marked in the map. (b) Genotype of Fgfr3floxneo/floxneo mice was identified by PCR (primer p1, p2, p3 and p4). (c) There is no expression of Fgfr3 in Fgfr3floxneo/floxneo mice (d) Fgfr3floxneo/floxneo mice showed kinky tails, which is also found in Fgfr3 knock out mice. (e) Increased expansion of proliferating and hypertrophic chondrocytes in the growth plate in Fgfr3floxneo/floxneo mice on P15.
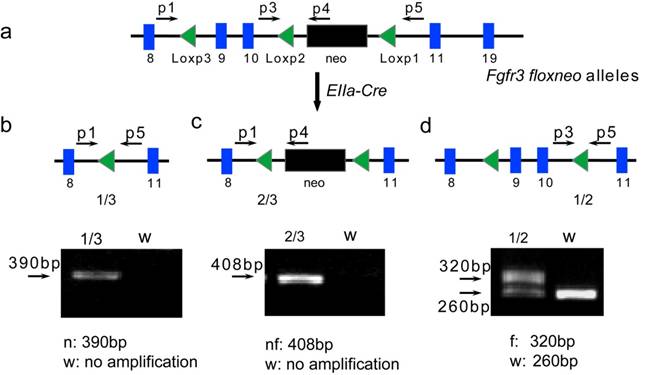
Validation of exons 9-10 and neo gene in Fgfr3floxneo alleles deleted by Cre recombinase. (a) Mice containing Fgfr3floxneo alleles were crossed with EIIa-Cre transgenic mice resulting in three kinds of deletion between three Loxp sites (b-d). Position of primers was also marked. (b) Primer p1 and p5 amplify fragments of 390 bp from Fgfr3null allele with deletion exons 9-10 and neo between Loxp1 and 3 (1/3), no amplification for the wild type (WT). (c) Primer p1 and p4 amplify fragments of 408 bp from Fgfr3neofloxed allele with deletion exons 9-10 between Loxp2 and 3 (2/3), no amplification for WT. (d) Primer p3 and p5 amplified fragments of 320 bp from Fgfr3flox allele with deletion neo between Loxp1 and 2 (1/2), but only 260bp from WT. n, Fgfr3null allele; nf, Fgfr3neofloxed allele; f, Fgfr3flox allele; w, wild type.
Skeletal preparation and histology
Mice at P15 were sacrificed. The femurs were fixed overnight in 4% paraformaldehyde at 4°C and rinsed in PBS and decalcified in 15% EDTA (pH 7.4) for 10-15 days before embedding in paraffin as described previously. Six-micron thick sections were used for H.&E. staining. High-resolution X-ray of femurs from 3-month-old mice were obtained using a Faxitron MX20.
RNA extraction and RT-PCR
RNA was extracted from brain of mice using Trizol (Invitrogen) reagent according to the manufacturer's instructions. Single strand cDNA was synthesized from 1 μg of total RNA using RNA reverse transcriptase kit (Takara). The mRNA level of Fgfr3 was detected by primer FGFR3-F(5'-CCACCGACAAGGAGCTAGAGG-3') and FGFR3-R (5'-CGGTGACAGGCTTGGCAGTA-3'). The β-actin gene detected by primer β-actin-F (5'-TTGTTACCAACTGGGACGACATGG-3') and β-actin-R (5'-GATCTTGATCTTCATGGTGCTAGG-3') was an internal control.
Results and Discussion
To construct the Fgfr3floxneo allele, we have made a targeting vector that contains two Loxp sites flanking the entire exons 9 and 10. In this way, existence of Cre recombinase would lead to deletion of exons 9 and 10, and subsequent inactivation of FGFR3. We inserted a 5.5-kb fragment including exons 3-10 and the third Loxp (located in intron 8) into ploxPneo vector [24] as the 5' homology recombination arm, then put a 3.8-kb fragment containing exons 10 to 19 into ploxPneo to form 3' homology recombination arm (Fig. 1a). The Not I-linearized targeting vector Fgfr3floxneo was transfected into TC1 ES cells by electroporation. Two targeted ES clones were detected by Southern blot using a 5' flanking external probe (Fig. 1a and b). The third Loxp in the ES clones was identified by PCR (Fig. 1a and c), which confirmed that Fgfr3floxneo allele in the two targeted ES clones containing the third Loxp. These two positive ES clones were microinjected into blastocysts harvested from C57/BL6 mice according to standard protocols.
We first assessed if the recombination of Fgfr3 in mice is correct. Since we previously found in the Fgfr3neoG369C/+ mice that the presence of Neomycin gene (neo) at the same site of intron 10 blocked the normal splicing of the Fgfr3 mRNA, and homozygous Fgfr3neoG369C/neoG369C mice showed phenotypes similar to those of Fgfr3-null (Fgfr3-/-) mice [16]. We generated Fgfr3floxneo/floxneo mice by crossing Fgfr3floxneo heterozygous mice. PCR was used to identify Fgfr3floxneo/floxneo mice (Fig. 2a and b). RT-PCR analysis revealed that there was no Fgfr3 expression in Fgfr3floxneo/floxneo mice (Fig. 2c). We also found that Fgfr3floxneo/floxneo mice indeed showed skeleton phenotype including crooked tail and increased height of growth plates (Fig. 2d and e), which is similar to the phenotype of Fgfr3-/- mice [2, 3]. These results indirectly demonstrated the correct recombination of the targeting construct.
To test whether neo gene and exons 9-10 in Fgfr3floxneo allele can be deleted by the Cre recombinase, heterozygous (Fgfr3floxneo/+) mice were crossed with EIIa-Cre transgenic mice that express Cre recombinase in germline [25, 26]. The offspring were identified by PCR with primers shown in Figure 3a. As expected, we found that this crossing breeding generated three types of alleles due to Cre-mediated recombination among three Loxp sites. A 390bp fragment was amplified from Fgfr3null allele with deletion of neo gene and exons 9-10 between Loxp1 and Loxp3 (confirmed by sequencing) (Fig. 3b). Fgfr3neofloxed allele with deletion of exons 9-10 was detected by producing an amplified 408bp fragment (Fig. 3c). The PCR-amplified 320 fragment indicated the excision of the neo gene, which produced a Fgfr3flox allele (conditional knockout allele) in Fgfr3flox/+ heterozygous mice (Fig. 3d).
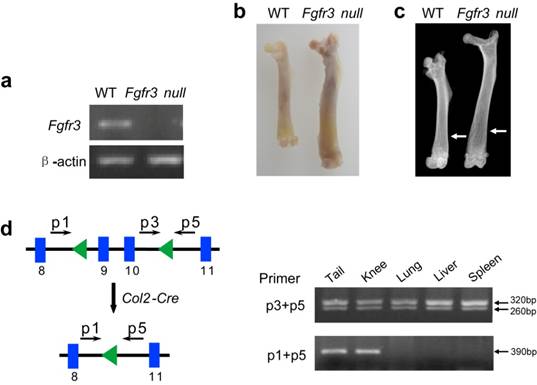
To further determine the correction of Fgfr3 recombination in mice, we generated homozygous Fgfr3 null mice. RT-PCR analysis showed that there was no expression of Fgfr3 in the brain (Fig. 4a). These mice showed increased length of long bone and decreased bone mineral density (Fig. 4b and c), which is also similar to the phenotypes of Fgfr3 knock out mice [3, 20]. Finally we crossed the Fgfr3flox/+ mice with mice that carry a Col2a1-Cre transgene to assess the activity of Fgfr3 conditional knockout allele. The result indicated that the Cre-mediated deletion occurs in several tissues that contain cartilage, including the tail, and knee joint, but not in other tissues where the Col2a1-Cre transgene is not expressed, such as the lung, liver, and spleen (Fig. 4d). These data indicate that the Fgfr3flox allele can be recombined to delete exons 9-10 in a tissue-specific fashion, thus the Fgfr3 conditional mouse model should be very useful for studying Fgfr3 functions during later stages of development.
Identification of Fgfr3 null mice and validation of Fgfr3 conditional knockout allele. (a) There was no expression of Fgfr3 in brain RNA of Fgfr3 null mice (homozygous mice with both exons 9-10 and neo deleted in Fgfr3). (b) The length of femur in 3-month-old Fgfr3 null mice was longer than that in wild-type mice. (c) X-ray analysis showed increased length and decreased bone mineral density of femur from Fgfr3 null mice (Arrows). (d) Tissue-specific inactivation of the Fgfr3 conditional allele by a Col2a1-Cre transgene in tissues containing cartilage was revealed by PCR analysis using primer pair p3/p5, which amplifies about 320 bp from the unrecombined allele, and primer pair p1/p5, which amplifies about 390 bp from the recombined allele.
Conclusion
The results indicate that we successfully generated a mouse model for conditional deletion of Fgfr3, which will help to explore the role of FGFR3 in development and the related skeletal diseases.
Acknowledgements
We thank Xiao Yang for EIIa-Cre and Col2a1-Cre transgenic mice. The work was supported by the Special Funds for Major State Basic Research Program of China (973 program) (No.2005CB522604), National Natural Science Foundation of China (No.30425023, No.30530410, No.30901527).
Conflict of Interest
All the authors declare the absence of conflicts of interests and the absence of financial interests.
References
1. Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16:1446-1465
2. Colvin JS, Bohne BA, Harding GW. et al. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12:390-397
3. Deng C, Wynshaw-Boris A, Zhou F. et al. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911-921
4. Shiang R, Thompson LM, Zhu YZ. et al. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell. 1994;78:335-342
5. Rousseau F, Bonaventure J, Legeai-Mallet L. et al. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature. 1994;371:252-254
6. Tavormina PL, Shiang R, Thompson LM. et al. Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat Genet. 1995;9:321-328
7. Bonaventure J, Rousseau F, Legeai-Mallet L. et al. Common mutations in the fibroblast growth factor receptor 3 (FGFR 3) gene account for achondroplasia, hypochondroplasia, and thanatophoric dwarfism. Am J Med Genet. 1996;63:148-154
8. Naski MC, Ornitz DM. FGF signaling in skeletal development. Front Biosci. 1998;3:d781-794
9. Su N, Du X, Chen L. FGF signaling: its role in bone development and human skeleton diseases. Front Biosci. 2008;13:2842-2865
10. Toydemir RM, Brassington AE, Bayrak-Toydemir P. et al. A novel mutation in FGFR3 causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome. Am J Hum Genet. 2006;79:935-941
11. Chellaiah AT, McEwen DG, Werner S. et al. Fibroblast growth factor receptor (FGFR) 3. Alternative splicing in immunoglobulin-like domain III creates a receptor highly specific for acidic FGF/FGF-1. J Biol Chem. 1994;269:11620-11627
12. Pringle NP, Yu WP, Howell M. et al. Fgfr3 expression by astrocytes and their precursors: evidence that astrocytes and oligodendrocytes originate in distinct neuroepithelial domains. Development. 2003;130:93-102
13. Patstone G, Pasquale EB, Maher PA. Different members of the fibroblast growth factor receptor family are specific to distinct cell types in the developing chicken embryo. Dev Biol. 1993;155:107-123
14. Vidrich A, Buzan JM, Ilo C. et al. Fibroblast growth factor receptor-3 is expressed in undifferentiated intestinal epithelial cells during murine crypt morphogenesis. Dev Dyn. 2004;230:114-123
15. Wang JM, Du XL, Li CL. et al. [Gly374Arg mutation in Fgfr3 causes achondroplasia in mice.]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2004;21:537-541
16. Chen L, Adar R, Yang X. et al. Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. J Clin Invest. 1999;104:1517-1525
17. Li C, Chen L, Iwata T. et al. A Lys644Glu substitution in fibroblast growth factor receptor 3 (FGFR3) causes dwarfism in mice by activation of STATs and ink4 cell cycle inhibitors. Hum Mol Genet. 1999;8:35-44
18. Eswarakumar VP, Schlessinger J. Skeletal overgrowth is mediated by deficiency in a specific isoform of fibroblast growth factor receptor 3. Proc Natl Acad Sci U S A. 2007;104:3937-3942
19. Valverde-Franco G, Binette JS, Li W. et al. Defects in articular cartilage metabolism and early arthritis in fibroblast growth factor receptor 3 deficient mice. Hum Mol Genet. 2006;15:1783-1792
20. Valverde-Franco G, Liu H, Davidson D. et al. Defective bone mineralization and osteopenia in young adult FGFR3-/- mice. Hum Mol Genet. 2004;13:271-284
21. Puligilla C, Feng F, Ishikawa K. et al. Disruption of fibroblast growth factor receptor 3 signaling results in defects in cellular differentiation, neuronal patterning, and hearing impairment. Dev Dyn. 2007;236:1905-1917
22. Wuchner C, Hilbert K, Zabel B. et al. Human fibroblast growth factor receptor 3 gene (FGFR3): genomic sequence and primer set information for gene analysis. Hum Genet. 1997;100:215-219
23. L'Hote CG, Knowles MA. Cell responses to FGFR3 signalling: growth, differentiation and apoptosis. Exp Cell Res. 2005;304:417-431
24. Xu X, Qiao W, Li C. et al. Generation of Fgfr1 conditional knockout mice. Genesis. 2002;32:85-86
25. Lakso M, Pichel JG, Gorman JR. et al. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996;93:5860-5865
26. Xu X, Li C, Garrett-Beal L. et al. Direct removal in the mouse of a floxed neo gene from a three-loxP conditional knockout allele by two novel approaches. Genesis. 2001;30:1-6
Author contact
![]() Corresponding author: Lin Chen, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Institute of Surgery Research, Daping Hospital, Third Military Medical University, Chongqing 400042, China. Tel: 86-23-68702991; Fax: 86-23-68702991; E-mail: linchen70com. Chuxia Deng, Genetics of Development and Disease Branch, National Institute of Diabetes, Digestive and Kidney Diseases, US National Institutes of Health, 10 Center Drive, Bethesda, Maryland 20892, USA. E-mail:chuxiadniddk.nih.gov
Corresponding author: Lin Chen, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Institute of Surgery Research, Daping Hospital, Third Military Medical University, Chongqing 400042, China. Tel: 86-23-68702991; Fax: 86-23-68702991; E-mail: linchen70com. Chuxia Deng, Genetics of Development and Disease Branch, National Institute of Diabetes, Digestive and Kidney Diseases, US National Institutes of Health, 10 Center Drive, Bethesda, Maryland 20892, USA. E-mail:chuxiadniddk.nih.gov