Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2010; 6(4):396-406. doi:10.7150/ijbs.6.396 This issue Cite
Research Paper
Sustained Expression of TDP-43 and FUS in Motor Neurons in Rodent's Lifetime
Cao Huang1, Pedro Yuxing Xia2, Hongxia Zhou1 ![]()
1. Department of Pathology, Anatomy & Cell Biology, Thomas Jefferson University, 1020 Locust Avenue, Philadelphia, PA 19107, USA
2. A volunteer student from Lower Merion High School, PA, USA
Received 2010-6-21; Accepted 2010-7-3; Published 2010-7-4
Abstract
TAR DNA-binding protein (TDP-43) and fused in sarcoma (FUS) are two highly conserved ribonucleoproteins. Pathogenic mutations of the TDP-43 or the FUS gene are all linked to amyotrophic lateral sclerosis (ALS) that is characterized by progressive degeneration of motor neurons. To better understand the correlation of ALS disease genes with the selectivity of chronic motor neuron degeneration, we examined the longitudinal expression of the TDP-43 and the FUS genes in C57BL6 mice and in Sprague-Dawley rats. TDP-43 and FUS were robustly and ubiquitously expressed in the postnatal mice and rats, but were markedly decreased in the adult rodents. In adulthood, TDP-43 and FUS proteins were even undetectable in peripheral organs including skeletal muscles, liver, and kidney, but were constantly expressed at substantial levels in the central nervous system. Motor neurons expressed the TDP-43 and the FUS genes at robust levels throughout rodent's lifetime. Moreover, TDP-43 and FUS were accumulated in the cytoplasm of motor neurons in aged animals. Our findings suggest that TDP-43 and FUS play an important role in development and that constant and robust expression of the genes in motor neurons may render the neurons vulnerable to pathogenic mutation of the TDP-43 or the FUS gene. To faithfully model the pathology of TDP-43- or FUS gene mutations in rodents, we must replicate the expression patterns of the TDP-43 and the FUS gene in animals.
Keywords: TDP-43, FUS, Mice, rats, motor neurons, amyotrophic lateral sclerosis
1. Introduction
TAR DNA-binding protein (TDP-43) is a highly conserved ribonucleoprotein [1, 2]. The primary transcript of the mammalian TDP-43 gene can be alternatively spliced to generate 11 mRNA molecules, with the major molecule containing all six exons and encoding TDP-43 protein [1]. TDP-43 is a member of heterogeneous ribonucleoprotein (hnRNP) family, which is characterized by the ability to bind RNA and DNA sequences through a common nucleotide-binding domain known as RNA recognition motif [3-5]. Deletion of the TDP-43 gene in mice causes an arrest to embryonic development [6-8], indicating that TDP-43 plays a critical role in development. While the physiologic functions of TDP-43 remain to be elucidated, pathogenic mutation of the TDP-43 gene is definitely linked to amyotrophic lateral sclerosis (ALS) and to motor neuron disease associated with frontotemporal lobe degeneration [9-13], indicating that mutant forms of TDP-43 are neurotoxic. Indeed, overexpression of human TDP-43 with pathogenic mutations causes neurodegeneration in drosophila, mice, and rats [14-19]. Interestingly, TDP-43 is hyper-phosphorylated, ubiquitinated, and aggregated in affected cells in sporadic ALS and in a subset of frontotemporal lobe degeneration (FTLD) [20, 21]. The TDP-43 proteinopathy implies that aberrant accumulation and modification of normal TDP-43 is toxic to vulnerable neurons. This notion is supported by accumulating evidence. Clinical studies showed that variation in the 3'-untranslated region of the TDP-43 gene is associated with increased expression of the TDP-43 gene in ALS and FTLD [22, 23]. Overexpression of the normal human TDP-43 gene can induce neurodegeneration in transgenic animals [15, 17, 19]. Previous studies collectively suggest that TDP-43 plays important roles in development and incurs toxicity to vulnerable neurons when the TDP-43 gene is pathogenically mutated or is aberrantly increased in gene expression.
Similar to TDP-43, fused in sarcoma (FUS) also is a conserved ribonucleoprotein and its mutant forms are also linked to ALS [24-29]. FUS is initially reported to translocate and fuse with one of several other genes to form chimeric oncogenes in leukemia and liposarcoma [30, 31]. FUS mainly resides in the nucleus [32], but increasing evidence showed that FUS shuttles between the nucleus and the cytoplasm [33-36]. As a ribonucleoprotein, FUS participates in gene regulation [34, 35, 37, 38]. Deletion of the FUS gene causes postnatal death in inbred mice [39, 40], suggesting an important role for FUS in cell survival. Like TDP-43 proteinopathy, FUS-positive inclusion is a feature of sporadic ALS and FTLD [41, 42]. Findings in the FUS gene suggest an important role for FUS in development and in neuron survival.
Both TDP-43 and FUS are linked to ALS that is characterized by progressive degeneration of motor neurons [43-48]. While loss of the TDP-43 or the FUS gene causes arrest to development, point-mutations of the genes incur selectively chronic toxicity to motor neurons. To better understand why these two ALS genes incur chronic toxicity selective to motor neurons, we examined expression of the TDP-43 and FUS genes in the mice and rats at varying ages. Our findings demonstrated that expression of the TDP-43 and FUS genes was robust and ubiquitous during postnatal development but was markedly decreased in adulthood. TDP-43 and FUS proteins were maintained at substantial levels in the motor neurons throughout rodent's lifetime. Our results suggest that constant and robust expression of the TDP-43 and FUS genes in motor neurons may be related to the selectivity of the neurotoxicity.
2. Materials and Methods
Animal experiments
Animal use followed NIH guidelines and animal use protocols were approved by the Institutional Animal Care and Use Committees at Thomas Jefferson University. C57BL6/J mice and Sprague-Dawley rats were used in this study. At each defined age, three mice and rats were used for analyses. For protein and RNA extraction, anesthetized mice and rats were killed and their tissues were dissected, frozen in powdered dry ice, and stored in a -80°C freezer until analysis. For histology, anesthetized animals were transcardially perfused with 4% paraformaldehyde in phosphate buffer (pH 7.4) and their brains and lumbar spinal cords were dissected and fixed in the same fixative until further use.
Immunohistochemistry
Animal tissues were fixed 4% paraformaldehyde, cryopreserved in 30% sucrose, and cut into three series of consecutive sections (20 μm) at a Cryostat. Each set of tissue sections was immunostained for TDP-43, FUS, or ChAT. For immunohistochemistry, tissue sections were incubated with rabbit polyclonal antibody against TDP-43 (ProteinTech Group) or FUS (Bethyl Laboratories: A300-292A). For double-fluorescence labeling, cross sections of lumbar spinal cord were incubated with goat anti-ChAT (Millipore) plus rabbit anti-TDP-43 or rabbit anti-FUS antibodies. Immunohistochemistry for TDP-43 and FUS was done with biotinylated goat anti-rabbit IgG (1:500; Vector Laboratories) and peroxidase-conjugated avidin-biotin complex (ABC kit; Vector Laboratories). After thorough washing, bound antibodies were visualized by addition of diaminobenzidine (Vector Laboratories). Immunostained cells were observed under a Nikon microscope and were documented with a Nikon digital camera. Immunofluorescence staining for ChAT (green) and TDP-43 (red) or FUS (red) was visualized and documented with a confocal microscope.
Immunoblotting
Animal tissues were mechanically homogenized in phosphate buffer (pH 7.4) supplied with protease inhibitors (Sigma) and tissue lysates were cleared of debris by centrifugation at 16,000 × g for 10 minutes. Protein content in cleared lysate was determined by BCA assay (BioRad). Total protein of 30 μg in each tissue lysate was resolved on 4-20% gradient SDS-PAGE and blotted onto GeneScreen Plus membrane (Perkin Elmer). Rodent's FUS protein is about 68kDa and is much bigger than the other two proteins examined: TDP-43: 42kDa; GAPDH: 25kDa. Blotting membranes were cut at the molecular marker of 55kDa. The upper part of blotting membrane was used for detecting FUS immunoreactivity and the low part was used for detecting TDP-43 and GAPDH immunoreactivity. FUS immunoreactivity was detected with a rabbit polyclonal antibody (Bethyl Laboratories: A300-293A). TDP-43 immunoreactivity was detected with a polyclonal antibody purchased from ProteinTech Group (10782-2-AP). After TDP-43 antibodies were stripped off blotting membrane, immunoreactivity to GAPDH was detected by incubating the membrane with a mouse monoclonal antibody to GAPDH (Abcam). GAPDH immunoreactivity was used as a loading standard. Immunoreactivity signal was developed with Super Signal kit (Pierce).
Quantitative PCR
Frozen lumbar spinal cord was cut into sections of 100 µm on a Cryostat and twenty consecutive sections were collected from each animal. Tissue sections were mounted on RNase-free heat-treated slides and stained with 1% toluidine blue in 70% ethanol for 5 min at -20 °C, followed by several washes in 70% ethanol at -20 °C to remove excess dye. The ventral horn of spinal cord was dissected under microscope and collected in 1ml Trizol (Sigma) for each animal. Total RNA was extracted from pooled ventral horn and cleared of DNA contamination with RNA Easy kit and RNase-free DNase (Qiagen). One microgram of purified RNA from each sample was reversely transcribed to cDNA with oligo-dT primer (RT kit, Invitrogen). Relative levels of TDP-43 and FUS mRNA in individual samples were estimated by quantitative PCR with the following gene-specific primers: 5'-GAGGATTTCCCAGTGGAGGT-3' (forward) and 5'-CCTTACACTGGTTGCATTCA-3' (reverse) for the FUS gene; 5'-GCAATCTGGTCTATGTTGTC-3' (forward) and 5'-ACCCAACACTATGAGGTCAG-3' (reverse) for the TDP-43 gene; and 5'-TGGTTCGCTACTCCCTTGAC-3' (forward) and 5'-CTTGATGGCCTGGGCAGTT-3' (reverse) for the L17 gene. The mRNA level of the L17 gene was used as an internal control for PCR quantification.
The mRNA levels in each sample were estimated by quantitative PCR with SYBR green kit per manufacturer's instruction (Qiagen). Primers were used at a concentration of 500 nM. Cycling conditions were 15 min at 95°C followed by 40 cycles of 15 seconds at 94°C, 30 seconds at 60°C, and 20 seconds at 72°C. Aliquots of the amplified products were separated on 3% agarose gels to ensure amplification of the specific products at the predicted length. The threshold cycle number (Ct) for TDP-43 or FUS was normalized to the Ct for L17 and the relative mRNA levels of TDP-43 or FUS was determined and expressed as a ratio relative to the mRNA level in 10-day-old rats.
3. Results
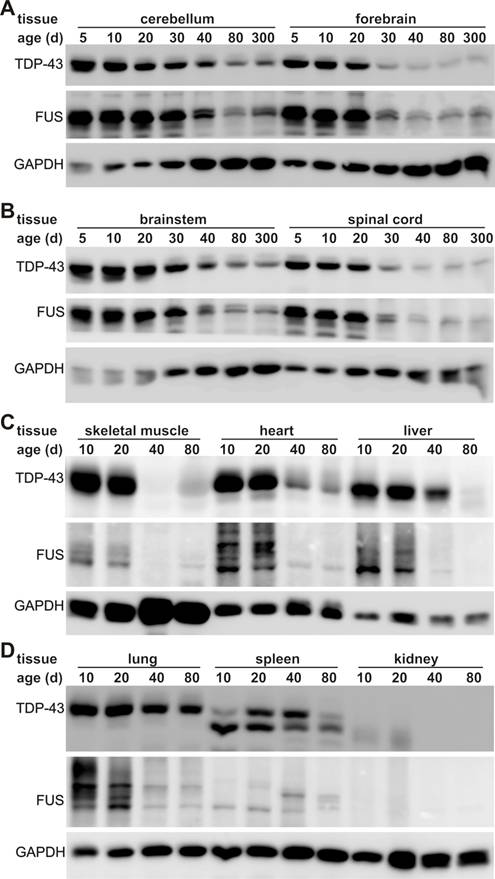
TDP-43 and FUS are two ALS genes and their inactivation causes developmental arrest in inbred mouse strains [6-13, 24-29, 39, 40]. To better understand the correlation of ALS disease genes with selective motor neuron degeneration, we examined the robustness of TDP-43 and FUS gene expression in laboratory mice during postnatal development and in adulthood. We chose C57BL6 mice because this strain is commonly used in laboratories and appears sensitive to ALS disease gene [49-51]. In the postnatal mice, the TDP-43 and the FUS genes were robustly and ubiquitously expressed in all the tissues examined (Figure 1). As the mice grew, TDP-43 and FUS proteins were globally and markedly decreased. By the age of 80 days, these two proteins were undetectable by immunoblotting in some peripheral organs including skeletal muscle, liver, and kidney (Figure 1C-D). In adulthood, TDP-43 and FUS were significantly decreased in the lung though to a lesser extent than in the other peripheral organs (Figure 1D). In contrast, sustainable levels of TDP-43 and FUS proteins were detected in mouse central nervous system (CNS) including brain, cerebellum, and spinal cord (Figure 1A). Substantial and constant levels of TDP-43 and FUS proteins were detected in the CNS of the mice at the ages of 40, 80, or 300 days (Figure 1A), implying that TDP-43 and FUS proteins are critical to neuron survival. We examined three animals at each defined age and obtained similar results. Robust and global expression of the TDP-43 and FUS genes in postnatal mice suggests that these two genes play an important role in development.
Expression of TDP-43 and FUS is markedly decreased in adult mice. A-D, Immunoblotting showing that expression of TDP-43 and FUS was gradually but markedly reduced in mouse tissues during postnatal development. Tissues were dissected from C57BL6 mice at varying ages (day: d) and 30 μg of total proteins in each tissue lysate was resolved on gradient SDS-PAGE. Blotting membranes were probed sequentially with antibodies against FUS, TDP-43, and GAPDH.
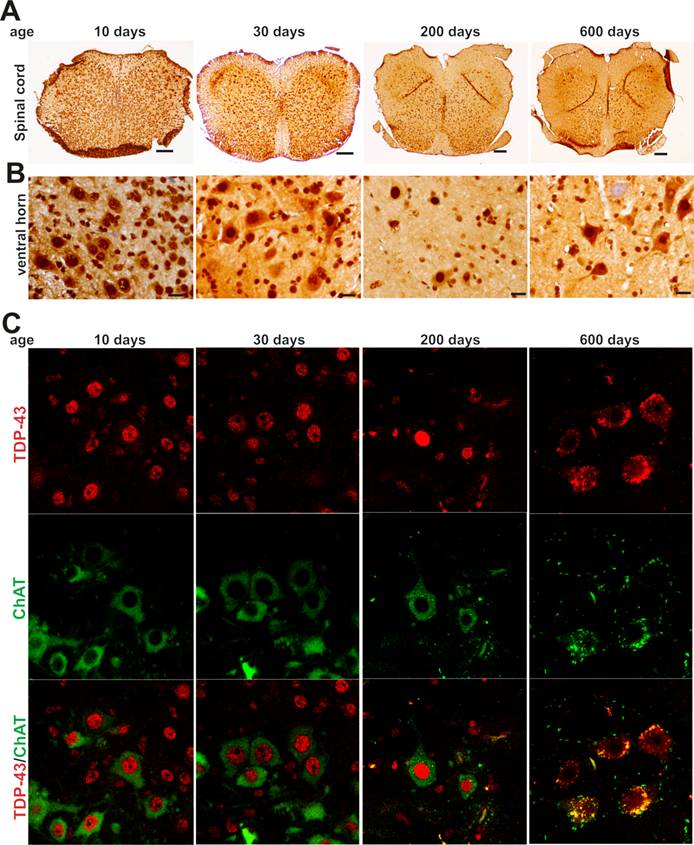
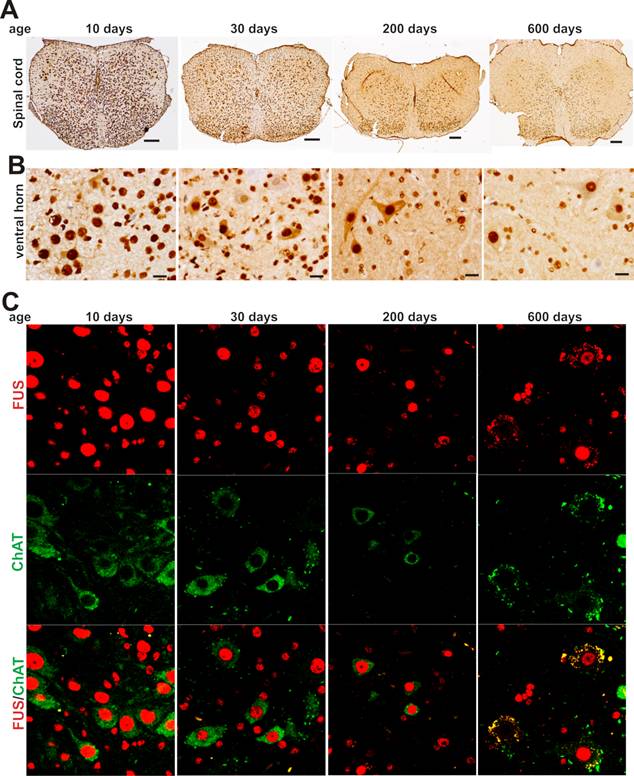
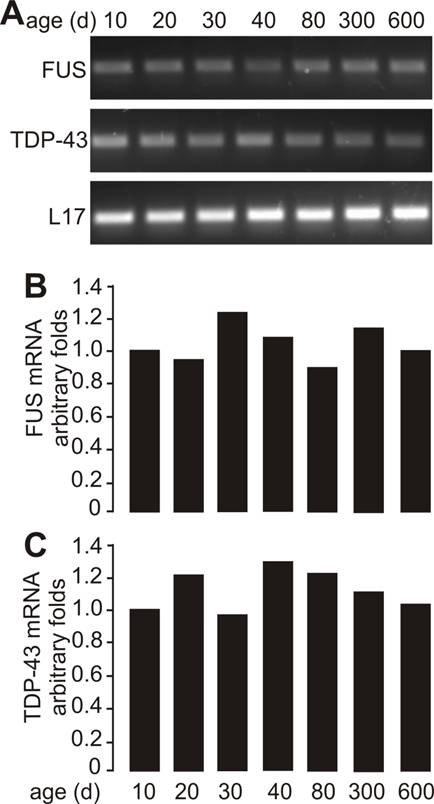
As motor neurons are the primary targets of ALS disease genes, we further examined whether motor neurons express TDP-43 and FUS at constant and substantial levels in adulthood. The intensity of TDP-43 and FUS immunostaining and the density of immunostained cells were much higher in postnatal mice by age of 10 or 30 days than in adult mice by age of 200 or 600 days (Figure 2A-B and Figure 3A-B). Groups of large cells in the ventral horn of spinal cord constantly expressed TDP-43 and FUS at substantial levels throughout mouse lifetime (Figures 1 and 2). To determine whether those large cells were motor neurons, we used double-fluorescence labeling to identify TDP-43- and FUS-expressing cells in mouse spinal cords. Indeed, motor neurons identified by immunostaining for ChAT (a marker of motor neurons) expressed TDP-43 and FUS at substantial and constant levels in adult mice (Figures 2C and 3C). TDP-43 and FUS proteins mainly resided in the nuclei of motor neurons in young mice, but were accumulated in the cytoplasm of motor neurons in aged mice (Figures 2C and 3C). By quantitative PCR analysis, we detected that the levels of TDP-43 and FUS mRNA remained constant in the ventral horn of spinal cord in mouse lifetime (Figure 4). Neurons in the primary motor cortex also expressed TDP-43 and FUS at substantial levels throughout mouse lifetime (Figure 5). Our findings suggest that aging is a factor contributing to cytoplasmic translocation of TDP-43 and FUS.
Expression of TDP-43 in spinal motor neurons remains sustainable in mouse lifetime. A, B, Immunohistochemistry showing that large cells in the ventral horn of spinal cord expressed TDP-43 at sustainable levels while the population of TDP-43-expressing cells was markedly decreased in aged mice. Lumbar spinal cords were dissected from C57BL6 mice at varying ages and cut into transverse sections on a Cryostat. Tissue sections were immunostained with an antibody against TDP-43. Scale bars: A, 100 μm; B, 20 μm. C, Immunofluorescence staining showing that the motor neurons of lumbar spinal cord expressed TDP-43 at sustainable levels in mouse lifetime.
Expression of FUS in spinal motor neurons remains sustainable in mouse lifetime. A, B, Immunohistochemistry showing that large cells in the ventral horn of spinal cord expressed FUS protein at sustainable levels while the population of FUS-expressing cells was markedly decreased in aged mice. Lumbar spinal cords were dissected from C57BL6 mice at varying ages and cut into transverse sections on a Cryostat. Tissue sections were immunostained with an antibody against FUS. Scale bars: A, 100 μm; B, 20 μm. C, Immunofluorescence staining showing that the motor neurons of lumbar spinal cord expressed FUS at sustainable levels in mouse lifetime.
Sustainable levels of FUS and TDP-43 mRNA in the ventral horn of spinal cord. A, Regular PCR analysis showing that the mRNA levels of FUS and TDP-43 were sustainable in the ventral horn of spinal cord throughout mouse lifetime. Lumbar spinal cords were dissected from C57BL6 mice at varying ages and cut into transverse sections. The ventral horns were dissected from the tissue sections and pooled for RNA extraction. B, C, Quantitative PCR analysis showing that the FUS and TDP-43 genes were sustainably expressed in the ventral horn of spinal cord in mouse lifetime. The detection thresholds for FUS and TDP-43 were first normalized to those for L17 mRNA and were then calculated relative to those in the mice at the age of 10 days. Data were the means averaged from three mice at each defined age.
Expression of FUS and TDP-43 remains sustainable in the motor cortex in mouse lifetime. A-J, Immunohistochemistry showing that FUS (A-E) and TDP-43 (F-J) were constantly expressed in the primary motor cortex in mouse lifetime.
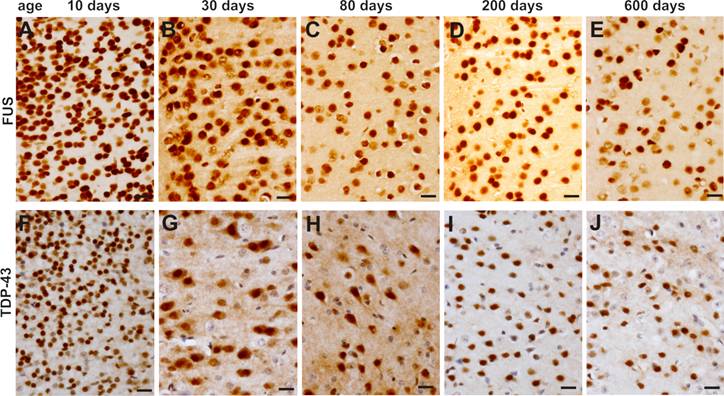
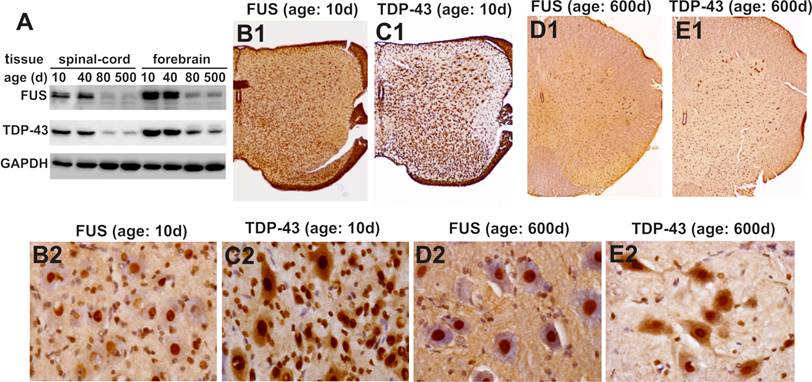
To validate our findings in the mice, we examined age-related expression of the TDP-43 and FUS genes in laboratory rats. Similar to the mice, Sprague-Dawley rats robustly expressed TDP-43 and FUS proteins in the CNS at postnatal ages (Figure 6A). Expression of the TDP-43 and FUS genes was markedly decreased to a constant level in adulthood (Figure 6). While the density of TDP-43- and FUS-expressing cells was largely decreased in the spinal cord, large cells in the ventral horn constantly expressed TDP-43 and FUS at substantial levels throughout rat's lifetime (Figure 6B-E). Our findings in the mice and in the rats consistently showed that motor neurons expressed TDP-43 and FUS at substantial levels in rodent's lifetime while the number of TDP-43- and FUS-expressing cells was gradually and markedly decreased during development.
Expression of FUS and TDP-43 remains sustainable in the motor neurons in rat's lifetime. A, Immunoblotting showing that expression of FUS and TDP-43 in rat's brain and spinal cord was markedly decreased during postnatal development. B-E, Immunohistochemistry showing that spinal motor neurons sustainably expressed FUS and TDP-43 while the population of FUS- and TDP-43-expressing cells was markedly decreased in aged rats. Lumbar spinal cords were dissected from SD rats at varying ages and cut into transverse sections on a Cryostat. Tissue sections were immunostained with antibodies against FUS or TDP-43 and were counterstained with haematoxylin. Micrographs (B1-E1) showed the left halves of lumbar spinal cords and micrographs (B2-E2) showed the ventral horns of the corresponding lumbar spinal cords shown in B1-E1.
4. Discussion
Like other neurodegenerative diseases, ALS primarily affects a subset of neurons in the CNS [43-48]. It remains largely unknown why motor neurons selectively degenerate in a chronic process in ALS. TDP-43 and FUS are identified as novel genes linked to ALS and their mutant forms segregate with some sporadic and familial ALS [9-13, 24-29]. We examined the longitudinal expression of the TDP-43 and FUS genes in the mice and rats. TDP-43 and FUS were globally and robustly expressed during postnatal development, but their expression was markedly decreased in adult animals and was even quiescent in some peripheral organs in adulthood. Strikingly, motor neurons expressed the TDP-43 and FUS genes at constant and substantial levels in rodent's lifetime. As aging, TDP-43 and FUS proteins were accumulating in the cytoplasm of motor neurons. Our findings suggest that high load of TDP-43 and FUS proteins in motor neurons may be correlated with selective degeneration of motor neurons in the TDP-43- or FUS-linked ALS.
Lifetime-long robust expression of the TDP-43 and FUS genes in motor neurons suggests that homeostasis of TDP-43 and FUS proteins is critical to motor neuron survival. Lasting alternation in the functions of these genes may be harmful to motor neurons. Aggregation of hyper-phosphorylated and ubiquitinated TDP-43 is a hallmark of sporadic FTLD and sporadic and non-SOD1-linked familial ALS [20, 21]. Formation of TDP-43 aggregates may reduce the availability of functional proteins, lead to generation of harmful intermediate products, or indicate an excess of functional or malfunctional TDP-43 proteins. Although the consequences of protein aggregation remain to be elucidated, TDP-43 inclusions definitely are pathology [20, 21]. Some disease genes are constantly expressed in vulnerable neurons and an increase in gene expression is sufficient to induce neurodegeneration. Alpha-synuclein is linked to Parkinson's disease, an age-related neurodegenerative disease that primarily results from degeneration of dopaminergic neurons in the midbrain [52-54]. During the process of aging, dopaminergic neurons preserve a high level of alpha-synuclein protein and oxidative modification of alpha-synuclein increases the protein level [55]. As aging compromises cellular protection mechanisms, high load of disease protein may sensitize the cells to internal or external risk factors. Intake of 1-methyl-4-phenyl-1, 2, 3, 4-tetrahydropyridine (MPTP) induces selective loss of dopaminergic neurons in human and in animals [56, 57]. While overexpression of alpha-synuclein sensitizes dopaminergic neurons to MPTP toxicity in transgenic mice [58], deletion of the alpha-synuclein gene protects dopaminergic cells against MPTP toxicity in the knockout mice [59-62]. Similar to alpha-synuclein [55], TDP-43 and FUS retain a high level in vulnerable neurons throughout a lifetime (Figures 1-6). Disturbance of TDP-43 and FUS homeostasis may incur toxicity to motor neurons. Increased expression of the TDP-43 gene is observed in ALS patients [22, 23], and overexpression of the human TDP-43 gene induces motoneuron degeneration in transgenic mice [15]. Increased expression of the TDP-43 gene indeed is neurotoxic [15, 63]. TDP-43 and FUS are two ribonucleoproteins and are all linked to ALS. Increased expression of the FUS gene may also be harmful to motor neurons though convincing evidence is under search.
On the other hand, reduction of functional TDP-43 and FUS proteins could be harmful to motor neurons. Large quantities of TDP-43 and FUS proteins may be ubiquitously required for cell differentiation and division during development, but may be required only for the survival of few cell types such as motor neurons in adulthood. Expression patterns of the TDP-43 and FUS genes may explain why deletion of the TDP-43 or the FUS gene causes arrest to mouse development [6-8, 39, 40]. As ribonucleoproteins, TDP-43 and FUS shuttle between the nucleus and the cytoplasm [33-36, 64]. Disturbance of protein trafficking may result in accumulation of TDP-43 and FUS in a cellular compartment. Cellular stress or aging may induce aggregation of TDP-43 and FUS, reducing the availability of functional proteins. In adulthood, reduction of TDP-43 or FUS protein may induce degeneration of vulnerable neurons or sensitize the affected neurons to stress stimulation. Although the natures of pathogenic mutations in the TDP-43 and the FUS genes remain to be determined, pathogenic mutations may alter the function of the genes and induce chronic toxicity to vulnerable cells. Substantially constant expression of the TDP-43 and the FUS genes in motor neurons may render the cells vulnerable to pathogenic mutation of the genes.
Taken together, ubiquitous, robust gene expression in postnatal rodents suggests an important role for TDP-43 and FUS in development. In adulthood, expression of these two genes was markedly reduced in the most tissues, but was maintained at substantial levels in the motor neurons, implying that TDP-43 and FUS may continuously play a critical role in motor neurons. Such expression patterns may be related to the selective neurotoxicity of pathogenic mutations in the TDP-43 and the FUS genes. To faithfully reproduce the pathology of TDP-43 or FUS gene mutations in rodents, we must replicate the expression patterns of the genes in animals by using sophisticated genetics approaches such as gene knockin. To avoid a disturbance of rodent's development, temporal or spatial gene deletion may be required to determine the functions of TDP-43 and FUS in motor neurons. Our findings provide guidance to the development of rodent models for TDP-43- or FUS-caused pathology.
Acknowledgements
We thank Ms. Dian Wang and Ms. Nina Wei for technical assistance.
Conflict of Interest
The authors declare no conflicts of interest.
References
1. Wang HY, Wang IF, Bose J. et al. Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics. 2004;83:130-139
2. Ayala YM, Pantano S, D'Ambrogio A. et al. Human, Drosophila, and C.elegans TDP43: nucleic acid binding properties and splicing regulatory function. J Mol Biol. 2005;348:575-588
3. Buratti E, Brindisi A, Giombi M. et al. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem. 2005;280:37572-37584
4. Abhyankar MM, Urekar C, Reddi PP. A novel CpG-free vertebrate insulator silences the testis-specific SP-10 gene in somatic tissues: role for TDP-43 in insulator function. J Biol Chem. 2007;282:36143-36154
5. Bose JK, Wang IF, Hung L. et al. TDP-43 overexpression enhances exon 7 inclusion during the survival of motor neuron pre-mRNA splicing. J Biol Chem. 2008;283:28852-28859
6. Wu LS, Cheng WC, Hou SC. et al. TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis. 2010;15:15
7. Kraemer BC, Schuck T, Wheeler JM. et al. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010;119:409-419
8. Sephton CF, Good SK, Atkin S. et al. TDP-43 is a developmentally-regulated protein essential for early embryonic development. J Biol Chem. 2010;285:6826-6834
9. Kabashi E, Valdmanis PN, Dion P. et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572-574
10. Sreedharan J, Blair IP, Tripathi VB. et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668-1672
11. Van Deerlin VM, Leverenz JB, Bekris LM. et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409-416
12. Rutherford NJ, Zhang YJ, Baker M. et al. Novel Mutations in TARDBP (TDP-43) in Patients with Familial Amyotrophic Lateral Sclerosis. PLoS Genet. 2008;4:e1000193
13. Benajiba L, Le Ber I, Camuzat A. et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Annals of Neurology. 2009;65:470-474
14. Wegorzewska I, Bell S, Cairns NJ. et al. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A: 2009 Nov. 2003;2106(2044):18809-18814
15. Wils H, Kleinberger G, Janssens J. et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A 2010: 2010 Feb. 2023;2107(2018):3858-2063
16. Zhou H, Huang C, Chen H. et al. transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLoS Genet. 2010Mar26;6(3):e1000887
17. Li Y, Ray P, Rao EJ. et al. A Drosophila model for TDP-43 proteinopathy. Proc Natl Acad Sci U S A. 2010;107:3169-3174
18. Ritson GP, Custer SK, Freibaum BD. et al. TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J Neurosci. 2010;30:7729-7739
19. Ash PE, Zhang YJ, Roberts CM. et al. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum Mol Genet. 2010 [Epub ahead of print]
20. Neumann M, Sampathu DM, Kwong LK. et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130-133
21. Arai T, Hasegawa M, Akiyama H. et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602-611
22. Gitcho MA, Baloh RH, Chakraverty S. et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63:535-538
23. Gitcho MA, Bigio EH, Mishra M. et al. TARDBP 3'-UTR variant in autopsy-confirmed frontotemporal lobar degeneration with TDP-43 proteinopathy. Acta Neuropathol. 2009 [Epub ahead of print]
24. Vance C, Rogelj B, Hortobagyi T. et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208-1211
25. Kwiatkowski TJJr, Bosco DA, Leclerc AL. et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205-1208
26. Drepper C, Herrmann T, Wessig C. et al. C-terminal FUS/TLS mutations in familial and sporadic ALS in Germany. Neurobiol Aging. 2009;15:15
27. Blair IP, Williams KL, Warraich ST. et al. FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. J Neurol Neurosurg Psychiatry. 2009;3:3
28. Damme PV, Goris A, Race V. et al. The occurrence of mutations in FUS in a Belgian cohort of patients with familial ALS. Eur J Neurol. 2009;13:13
29. Corrado L, Del Bo R, Castellotti B. et al. Mutations of FUS Gene in Sporadic Amyotrophic Lateral Sclerosis. J Med Genet. 2009;26:26
30. Perez-Losada J, Pintado B, Gutierrez-Adan A. et al. The chimeric FUS/TLS-CHOP fusion protein specifically induces liposarcomas in transgenic mice. Oncogene. 2000;19:2413-2422
31. Rabbitts TH, Forster A, Larson R. et al. Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat Genet. 1993;4:175-180
32. Calvio C, Neubauer G, Mann M. et al. Identification of hnRNP P2 as TLS/FUS using electrospray mass spectrometry. RNA. 1995;1:724-733
33. Fujii R, Grossenbacher-Zinchuk O, Jamari I. et al. TLS-GFP cannot rescue mRNP formation near spines and spine phenotype in TLS-KO. Neuroreport. 2009;20:57-61
34. Fujii R, Okabe S, Urushido T. et al. The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr Biol. 2005;15:587-593
35. Fujii R, Takumi T. TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J Cell Sci. 2005;118:5755-5765
36. Wang X, Arai S, Song X. et al. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454:126-130
37. Belly A, Moreau-Gachelin F, Sadoul R. et al. Delocalization of the multifunctional RNA splicing factor TLS/FUS in hippocampal neurones: exclusion from the nucleus and accumulation in dendritic granules and spine heads. Neurosci Lett. 2005;379:152-157
38. Yoshimura A, Fujii R, Watanabe Y. et al. Myosin-Va facilitates the accumulation of mRNA/protein complex in dendritic spines. Curr Biol. 2006;16:2345-2351
39. Hicks GG, Singh N, Nashabi A. et al. Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat Genet. 2000;24:175-179
40. Kuroda M, Sok J, Webb L. et al. Male sterility and enhanced radiation sensitivity in TLS(-/-) mice. EMBO J. 2000;19:453-462
41. Deng HX, Zhai H, Bigio EH. et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol. 2010;67:739-748
42. Seelaar H, Klijnsma KY, de Koning I. et al. Frequency of ubiquitin and FUS-positive, TDP-43-negative frontotemporal lobar degeneration. J Neurol. 2009;28:28
43. Roberts BR, Tainer JA, Getzoff ED. et al. Structural characterization of zinc-deficient human superoxide dismutase and implications for ALS. J Mol Biol. 2007;373:877-890
44. Ermilova IP, Ermilov VB, Levy M. et al. Protection by dietary zinc in ALS mutant G93A SOD transgenic mice. Neurosci Lett. 2005;379:42-46
45. Tydlacka S, Wang CE, Wang X. et al. Differential activities of the ubiquitin-proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J Neurosci. 2008;28:13285-13295
46. Friedman MJ, Shah AG, Fang ZH. et al. Polyglutamine domain modulates the TBP-TFIIB interaction: implications for its normal function and neurodegeneration. Nat Neurosci. 2007;10:1519-1528
47. Gifondorwa DJ, Robinson MB, Hayes CD. et al. Exogenous delivery of heat shock protein 70 increases lifespan in a mouse model of amyotrophic lateral sclerosis. J Neurosci. 2007;27:13173-13180
48. Boston-Howes W, Williams EO, Bogush A. et al. Nordihydroguaiaretic acid increases glutamate uptake in vitro and in vivo: therapeutic implications for amyotrophic lateral sclerosis. Exp Neurol. 2008;213:229-237
49. Tucci V, Achilli F, Blanco G. et al. Reaching and grasping phenotypes in the mouse (Mus musculus): a characterization of inbred strains and mutant lines. Neuroscience. 2007;147:573-582Epub 2007 Jun 2015
50. Heiman-Patterson TD, Deitch JS, Blankenhorn EP. et al. Background and gender effects on survival in the TgN(SOD1-G93A)1Gur mouse model of ALS. J Neurol Sci. 2005;236:1-7
51. Wong PC, Pardo CA, Borchelt DR. et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105-1116
52. Polymeropoulos MH, Lavedan C, Leroy E. et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045-2047
53. Kruger R, Kuhn W, Muller T. et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106-108
54. Zarranz JJ, Alegre J, Gomez-Esteban JC. et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164-173
55. Li W, Lesuisse C, Xu Y. et al. Stabilization of alpha-synuclein protein with aging and familial parkinson's disease-linked A53T mutation. J Neurosci. 2004;24:7400-7409
56. Langston JW, Ballard P, Tetrud JW. et al. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979-980
57. Nicklas WJ, Vyas I, Heikkila RE. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 1985;36:2503-2508
58. Song DD, Shults CW, Sisk A. et al. Enhanced substantia nigra mitochondrial pathology in human [alpha]-synuclein transgenic mice after treatment with MPTP. Experimental Neurology. 2004;186:158-172
59. Fountaine TM, Venda LL, Warrick N. et al. The effect of alpha-synuclein knockdown on MPP+ toxicity in models of human neurons. Eur J Neurosci. 2008;28:2459-2473
60. Drolet RE, Behrouz B, Lookingland KJ. et al. Mice lacking alpha-synuclein have an attenuated loss of striatal dopamine following prolonged chronic MPTP administration. Neurotoxicology. 2004;25:761-769
61. Dauer W, Kholodilov N, Vila M. et al. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci USA. 2002;99:14524-14529
62. Robertson DC, Schmidt O, Ninkina N. et al. Developmental loss and resistance to MPTP toxicity of dopaminergic neurones in substantia nigra pars compacta of gamma-synuclein, alpha-synuclein and double alpha/gamma-synuclein null mutant mice. J Neurochem. 2004;89:1126-1136
63. Kabashi E, Lin L, Tradewell ML. et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet. 2009;19:671-683
64. Winton MJ, Igaz LM, Wong MM. et al. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem. 2008;283:13302-13309
Author contact
![]() Corresponding author: Hongxia Zhou, Department of Pathology, Anatomy & Cell Biology, Thomas Jefferson University, 508 JAH, 1020 Locust Avenue, Philadelphia, PA 19107, USA. Phone: 215-503-9153; Fax: 215-923-3808; E-mail: hongxia.zhouedu
Corresponding author: Hongxia Zhou, Department of Pathology, Anatomy & Cell Biology, Thomas Jefferson University, 508 JAH, 1020 Locust Avenue, Philadelphia, PA 19107, USA. Phone: 215-503-9153; Fax: 215-923-3808; E-mail: hongxia.zhouedu