Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2010; 6(5):428-442. doi:10.7150/ijbs.6.428 This issue Cite
Research Paper
Increased invasiveness and aggressiveness in breast epithelia with cytoplasmic p63 expression
Yi-Hsuan Hsiao1,2, Yan A. Su3, Horng-Der Tsai2, Jeffrey T. Mason4, Ming-Chih Chou1 ![]() , Yan-gao Man4
, Yan-gao Man4 ![]()
1. Institute of Medicine, Chung Shan Medical University, Taiwan
2. Department of Obstetrics and Gynecology, Changhua Christian Hospital, Taiwan;
3. Department of Gene and Protein Biomarkers, GenProMarkers, Inc. Rockville, MD, USA;
4. Armed Forces Institute of Pathology and American Registry of Pathology, Washington DC, USA
Received 2010-7-16; Accepted 2010-8-5; Published 2010-8-8
Abstract
Our previous studies revealed that pregnancy associated breast cancer (PABC) had significantly reduced nuclear p63 expression in myoepithelia, while intense cytoplasmic p63 expression in associated epithelia. Our current study assessed these epithelia using immunohistochemistry with a panel of aggressiveness and invasiveness related markers and comparative genomic hybridization (array-CGH) with over 30,000 DNA probes. These epithelia showed several unique alterations, including (1) immunohistochemical and morphological resemblance to invasive cancer, (2) significant gain in copy numbers of DNA coding genes for morphogenesis, angiogenesis, and metastasis, and (3) significant loss in copy numbers of DNA coding genes for tumor suppressors, cell adhesion, and macromolecular complex assembly or intra-cellular trafficking. Detected array-CGH alterations correlated well with in vivo expression of a number of corresponding proteins tested. These findings suggest that aberrant sub-cellular localization of p63 expression in normal or hyperplastic appearing epithelial cells may significant contribute to increased invasiveness and aggressiveness of these cells.
Keywords: breast epithelia, pregnancy associated breast cancer, p63 expression, invasiveness and aggressiveness.
Introduction
Pregnancy-associated breast cancer (PABC), which occurs during pregnancy or within one year of delivery, is the second most common cancer in pregnant women [1-3]. A normal full-term pregnancy is believed to be one of the most effective means to decrease the lifetime-risk of breast cancer, whereas PABC is associated with the most aggressive clinical course and the highest mortality rate among breast malignancies [1-3]. These seemingly conflicting impacts are believed to result from aberrant proliferation and differentiation of stem cells, and pathological alterations of the microenvironment and extracellular matrix [3,4]. Neither the molecular or cellular mechanisms, nor the specific molecules linking to aggressiveness and invasiveness of PABC, however, have been identified.
Promoted by the fact that the myoepithelium is the sole source of several tumor suppressors, including maspin, Wilms' tumor 1 (WT-1), p63, and p73, which show significant paracrine inhibition on tumor cell growth and invasion in vitro [5-10], our recent studies compared the expression of these tumor suppressors in PABC and the stage, grade, and age matched non-PABC. PABC were found to have several unique alterations, including (1) the absence of p63 and WT-1 expression in a vast majority of myoepithelial cells, and (2) intense cytoplasmic p63 expression in a subset of normal or hyperplastic epithelial cells, which are arranged as segregated clusters or lobules [11].
Our findings appear to have significant scientific and clinical implications for several reasons. First, as the p63 gene encodes multiple protein isoforms that regulate p53, p73, and other tumor suppressors [12,13], aberrant p63 expression may result in aberrant expression of these tumor suppressors. Second, as p63 is required for normal development of the breast and other organs [12,13], aberrant p63 expression may result in structural defects of breast tissues, making them more acceptable to external or internal insults that promote carcinogenesis. Third, as cytoplasmic p63 expression is associated with poorer patient survival in lung cancer and tumor grade in meningiomas [14,15], aberrant p63 expression may have oncogenic properties that contribute to tumor aggressiveness and invasiveness.
Thus, our current study attempted to further elucidate the cellular and molecular profiles of these normal or hyperplastic appearing epithelia with cytoplasmic p63 expression, to assess: (1) whether they harbor aberrant expression of other tumor suppressors or malignancy-associated changes, and (2) whether they differ substantially in DNA copy numbers compared to morphologically similar counterparts without cytoplasmic p63 expression.
Materials and Methods
Formalin-fixed, paraffin-embedded tissue blocks from 20 cases of PABC used in our previous studies [11] were used for our current study. All cases were diagnosed during the pregnancy or within 3-months of postpoterm. No clinical following-up data, however, were available for these patients. For comparison, 20 cases of stage, grade, and age matched non-PABC were selected from the files of our institute and our own tissue bank. Consecutive sections at 4-5 μm thickness were prepared and placed on positively charged slides. From each tissue block, the first and last sections were stained with hematoxylin and eosin (H&E) for morphological classification using our published criteria [16].
To assess whether these normal or hyperplastic acini harbored aberrant expression of other tumor suppressors and malignancy associated alterations, two technical approaches were used. First, sets of six consecutive sections were immuno-stained for known tumor suppressors, including maspin (clone:EAW24), WT-1 (clone: 6F-H2), p63 (clone: 4A4), p53 (clone:DO-7), p73 (clone:24) (Novocastra Laboratories Ltd, Newcastle, UK), and proliferation marker Ki-67 (clone:MM1; Dako, Carpinteria, CA). The expression of these markers in myoepithelial and associated epithelial cells of the same acini at different sections were digitally photographed and reviewed under high magnification in a standard computer. Second, the physical distribution of these acini was examined to determine whether they were exclusively or preferentially associated or shared a similar morphological or immunohistochemical profile, with invasive lesions.
Immunohistochemistry for all antibodies was carried out using protocols provided by manufacturers. Double immunohistochemistry was carried out using our published protocol [17]. To assess the specificity of immunostaining, three technical approaches were used. First, different controls, including (1) the substitution of the primary antibody with normal serum, (2) the omission of the secondary antibody from the immunostaining sequences, and (3) serial dilutions of the primary antibodies, were used. Second, the same immunostaining protocol was used on the same cases, but substituting with a different detection system and chromogen. Third, the immunostaining procedure was repeated at least twice using the same protocol and under the same condition and immunostained sections were independently evaluated by two investigators. A given cell was considered immunoreactive when distinct immunoreactivities were seen consistently in its membrane, cytoplasm, or nucleus, while all negative controls were devoid of immunostaining.
To assess whether they differ substantially in DNA copy numbers compared to their morphologically similar counterparts without cytoplasmic p63 expression, normal or hyperplastic lobules with uniform cytoplasmic p63 expression in acinar cells from 4 PABC cases and morphologically comparable lobules (the same histological type, grade, and similar architecture and size) without cytoplasmic p63 expression from 4 non-PABC cases were microdissected with a 30G1/2 needle, as previously described [18]. Microdissected cells were subjected to DNA extraction and array-comparative genomic hybridization (array-CGH).
Array-CGH was carried out using previously published protocols [19,20] with minor modifications. Briefly, genomic DNAs were extracted and purified using a QIAamp Kit (Cat. No. 56404, QIAGEN, Valencia, CA, USA) following the manufacturer's instructions. The purity of each DNA sample was indicated by 1.8 of the A260/A280 ratio. Two micrograms of purified genomic DNA were labeled with fluorescent Cy3-dUTP using a DNA Labeling Kit (Cat. No. 42671, Enzo Life Sciences, Farmingdale, NY, USA) following the manufacturer's instructions. Hybridization was carried out using a Human OneArray DNA microarray (Cat. # HOA004, Phalanx Biotech Group, Palo Alto, CA, USA) that contains 30,968 human genome probes and 1,082 experimental control probes. All microarray experiments for each DNA sample were performed in triplicate.
Prior to data analysis, identification numbers, symbols and names of all genes on commercial chips were updated according to the Human UniGene Build #218 (ftp://ftp.ncbi.nih.gov/repository/UniGene/, January 5, 2009) using the probe ID and ENSG number came with the gene chips, since the original commercial annotation was based on the Human UniGene Build #163 (Cat. # HOA004, Phalanx Biotech Group). Gene ontology, chromosomal band regions, and phenotypes were compiled from Entrez (ftp://ftp.ncbi.nlm.nih.gov/gene) and DAVID Bioinformatics Resources 2009 (http://david.abcc.ncifcrf.gov).
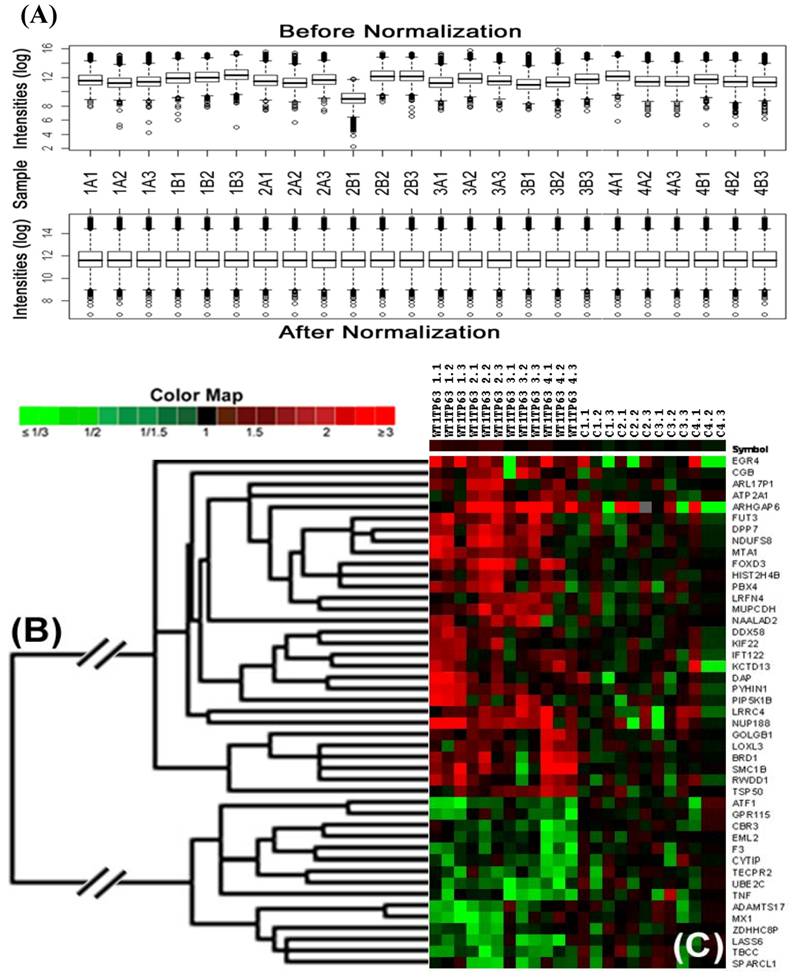
The loss or gain of DNA copy numbers in PABC was calculated by comparison with those in non-PABC as previously described [19,20]. The array-CGH database was constructed using the FileMaker software (FileMaker Pro, Inc., Santa Clara, CA, USA). Data filtering and selection were performed as described previously [19]. The normalization method [21] in software R version 2.7.1 (The R Foundation for Statistical Computing) was used to normalize microarray data. The normalized data was used to compute the ratios of genomic DNA probes coding for each gene between PABC and controls, and to cluster genes based on similarities in gain or loss of DNA probes by using software Cluster version 3.0 [22]. Cluster tree and heatmap reflecting gain, loss, or no change in DNA probes of genes were visualized using software MapleTree (http://rana.lbl.gov/EisenSoftware.htm).
Statistical calculations were performed on triplicate using XLSTAT 2006 (XLSTAT, New York, NY, USA). Gain or loss of DNA copy numbers were identified arbitrarily by >1.2-fold change in the averaged ratios of the background-subtracted mean intensities between PABC and non-PABC. Moderated t-statistic, raw p-values, log2 fold changes in DNA copy numbers, and false discovery rate (FDR) on multiple statistical testing were calculated using the LIMMA package in software R/Bioconductor (version 2.7.1, the R Foundation for Statistical Computing) [23]. To reveal biological functions of altered genes, gene ontology analysis was conducted to group genes based on their functions. The p-values for genes with functional annotation report were calculated using a modified Fisher's Exact Test (DAVID Bioinformatics Resources 2009; http://david.abcc.ncifcrf.gov/). Statistical significance was set at a p-value <0.05 and FDR ≤ 5%.
As it is very difficult, if not impossible, to quantitatively measure the expression of mRNA or protein levels in formalin-fixed and paraffin-embedded tissues, validation of the array-CGH results was conducted using immunohistochemistry with specific antibodies to assess whether alterations detected by array-CGH correlated with in vivo protein expression in adjacent tissue sections. As a number of altered genes detected by array-CGH are essential for morphogenesis, angiogenesis, adhesion, and macromolecular complex assembly or intra-cellular trafficking [24-31], our study compared the vascular density, expression of cell adhesion molecules, and immunoreactivities of luminal secretory products between PABC and non-PABC with antibodies to Tenascin (clone:T2H5), E-cadherin (clone:36B5) (Lab Vision, Fremont, CA), D2-40 (clone:D2-40;Signet, Dedham, MA), CD34 (clone: QBEbd/10), CD45 (clone: 2B11+PD7/26), and ß-catenin (clone: 17C2) (Dako, Carpinteria, CA).
Results
All selected PABC and non-PABC cases contained normal, hyperplastic, and neoplastic components. The overall expression pattern and frequency of all antibodies were consistent with the description of the manufactures and those of our previous studies [9-11]. All negative controls were consistently devoid of immunoreactivities to any of these antibodies. Of 20 PABC, 8 harbored a large number of epithelial cells arranged as lobules or large acinar clusters, in which all or near all cells uniformly expressed high levels of cytoplasmic p63, and 12 harbored variable numbers of cytoplasmic p63 expressing cells blended with cells without p63 expression. None of 20 non-PABC harbored epithelial cells with cytoplasmic p63 expression.
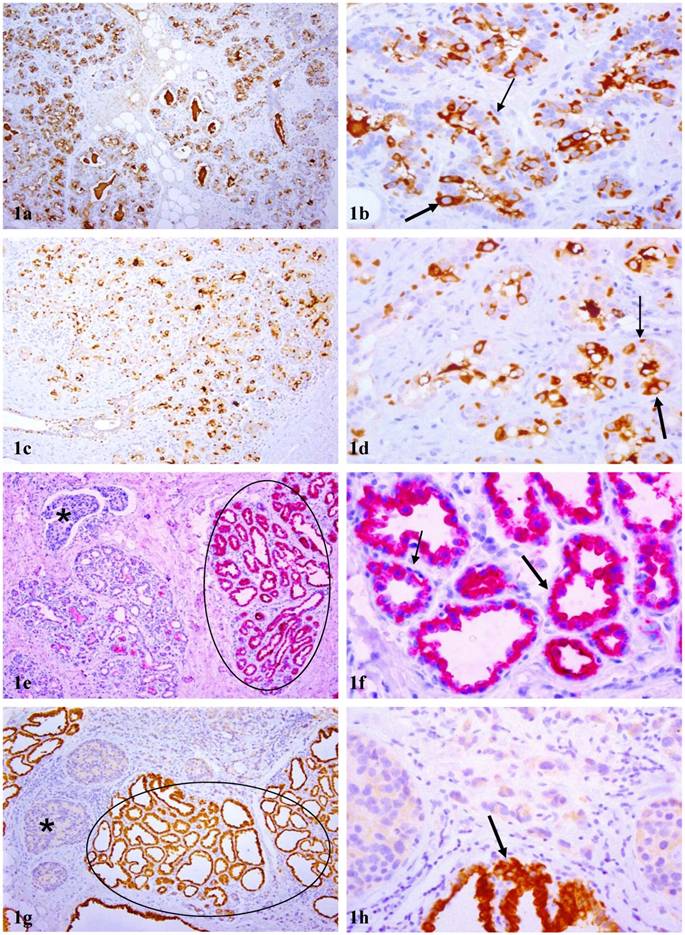
A vast majority of these lobules or acinal clusters with cytoplasmic p63 expression were segregated from their counterparts by inter-lobular stromal tissues with a distinct boundary. In tissues that were not associated with, or distant from, malignant lesions, cytoplasmic p63 expression was seen in about 20-50% of the acinar cells, and nuclear p63 expression was seen in morphologically distinct myoepithelial cells (Fig 1a-d). In tissues associated with or adjacent to malignant lesions, cytoplasmic p63 expression was seen in all or nearly all normal or hyperplastic acinar cells, as well as adjacent cancer cells, while nuclear p63 expression was absent in all or nearly all morphologically distinct myoepithelial cells (Fig 1e-h).
Acinar cell clusters or lobules with cytoplasmic p63 expression. Sections from paraffin embedded tissues of PABC were immunostained for p63 (brown or red). Circles identify normal appearing lobules in which all acinar cells show cytoplasmic p63 expression. Thin and thick arrows identify cells with nuclear and cytoplasmic p63 expression, respectively. Asterisks identify invasive tissue components. In tissues without or distant from malignant lesions, both cytoplasmic and nuclear p63 expression cells are seen (a-d), whereas in tissues harbored or adjacent to malignant lesions (e-h), all or nearly all cells within a given lobule uniformly show high levels of cytoplasmic p63 expression. a, c, e, and g: 100X; b, d, f, and h: a higher (300X) magnification of a, c, e, and g, respectively.
Compared to their morphologically similar counterparts in non-PABC, these normal or hyperplastic lobules with cytoplasmic p63 expression showed the following unique alterations:
1. Simultaneous reduction or loss of expression of multiple tumor suppressors
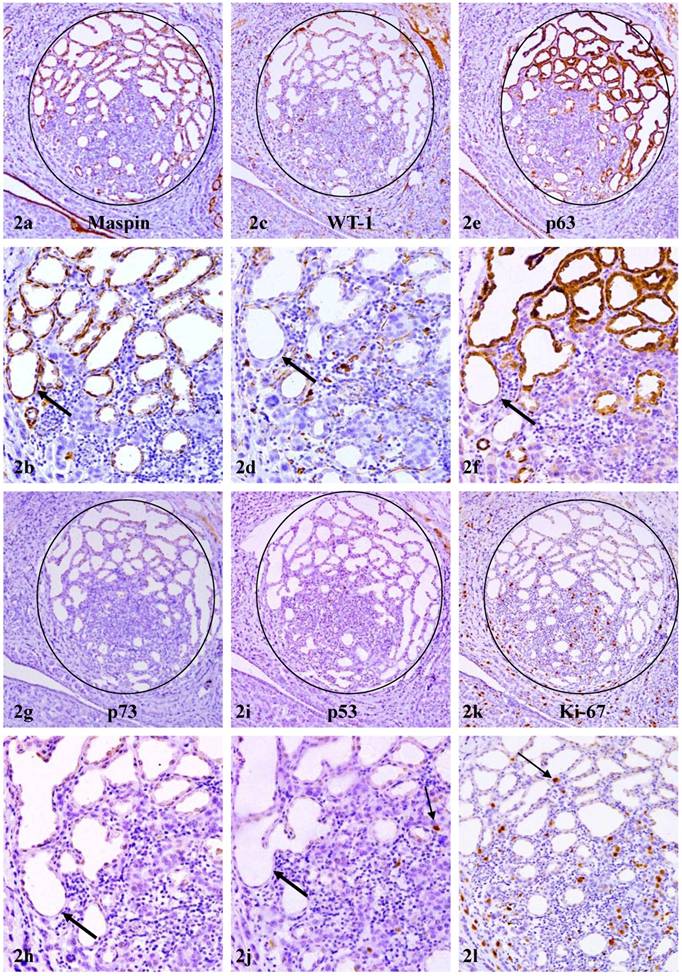
In consecutive sections immunostained for different tumor suppressors, strong immunoreactivities to maspin were consistently seen in all or nearly all myoepithelial cells (Fig 2a-b). In contrast, only 10% to 30% of maspin positive cells showed WT-1, p63, or p73 expression (Fig 2c-h).
Reduced expression of other tumor suppressors in myoepithelium of PABC. A set of 6 consecutive sections of PABC were immunostained for maspin, WT-1, p63, p73, p53, and Ki-67, respectively. Circles identify normal appearing lobules in which all or nearly all acinar cells show cytoplasmic p63 expression. Thick arrows identify the myoepithelial cell layer in different sections. Thin arrows identify isolated tumor cells with p53 and Ki-67 expression. Note that all or nearly all the myoepithelial cells uniformly express maspin, whereas only about 10-30% of maspin positive cells show WT-1, p63, or p73 expression. Also note that these normal appearing lobules harbor some isolated p53 positive tumor cells and have an elevated cell proliferation index. a, c, e, g, I, and k: 80X; b, d, f, h, j, and l: a higher magnification (200X) of a, c, e, g, I, and k, respectively.
2. Morphological and immunohistochemical resemblance to invasive cancer cells
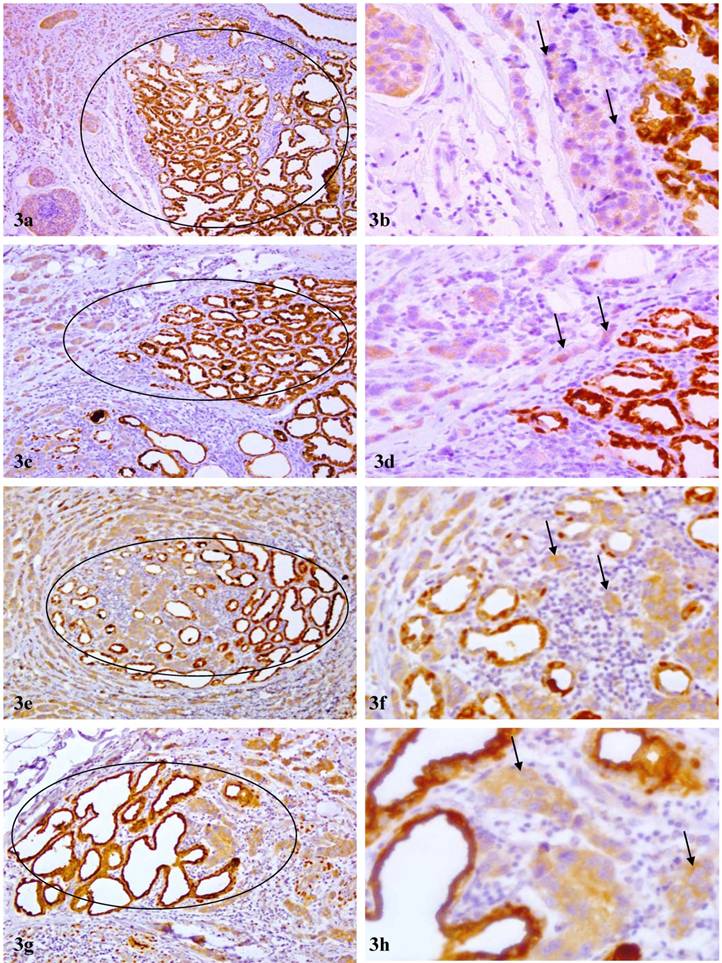
These normal or hyperplastic lobules adjacent to invasive lesions consistently harbored isolated cells or cell clusters that were strongly immunoreactive to p53 and Ki-67 (Fig 2i-l). These cells were exclusively located at the site immediately adjacent to invasive lesions and shared a very similar immunohistochemical and morphological profile with invasive counterparts, which consistently showed diffuse cytoplasmic p63 expression (Fig 3a-d). In addition, some isolated and morphologically distinct malignant cells with diffuse cytoplasmic p63 expression were consistently seen within these normal or hyperplastic lobules (Fig 3e-h).
Morphological and immunohistochemical resemblance to invasive cancer cells. Sections from 4 PABC cases were immunostained for p63 (brown). Circles identify normal or hyperplastic lobules in which all or nearly all cells show high levels of cytoplasmic p63 expression. Arrows identify adjacent invasive cancer cells. Note that some cells within these normal or hyperplastic appearing lobules and their adjacent invasive counterparts share a very similar immunohistochemical or morphological profile. a, c, e, and g: 100X; b, d, f, and h: a higher (300) magnification of a, c, e, and g, respectively.
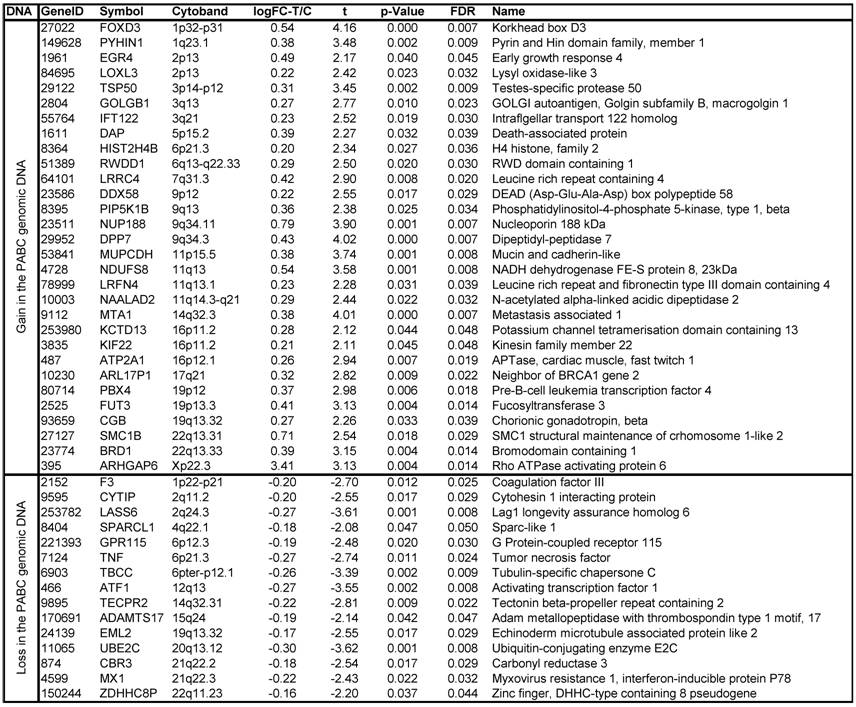
3. Significant gain or loss of DNA copy numbers in 45 chromosomal loci in all 4 PABC cases
Array-CGH detected significant DNA copy number changes in 45 chromosomal loci. Of these, 30 showed significant gain (≥1.2 fold), and 15 showed significant loss (≤0.8 fold) in all PABC samples, compared to non-PABC. Table 1 lists the chromosome band regions, statistic values, and harbored genes. The heat map and clusters in figure 4a-b visualize intuitive common differences of these 45 genes between the 4 paired DNA samples.
Significant gain or loss of DNA copy numbers in altered chromosomal loci of PABC
Log-FC-T/C: log 2 of fold changes (FC) between tumor (T) and control (C) samples. Moderated t-statistic, raw p-values, log2 fold changes in gene expression, and false discovery rate (FDR) on multiple statistical testing were calculated using the LIMMA package in software R/Bioconductor. The statistical significance was set at a p-value <0.05 and FDR ≤ 5%.
Significant gain or loss of DNA copy numbers in lobules with cytoplasmic p63 expression. Microdissected cells from 4 cases of PABC and 4 cases of morphologically similar non-PABC were subjected to array-CGH. Gain or loss of DNA copy numbers were identified arbitrarily by >1.2-fold change in the average of the background-subtracted mean intensity ratios between PABC and control samples. (A) Data normalization; (B) Heat map and clusters; (C) Control samples. The triplicate microarray experiments were performed for each sample.
4. Alterations in morphogenesis, angiogenesis, adhesion, and intra-cellular trafficking related genes
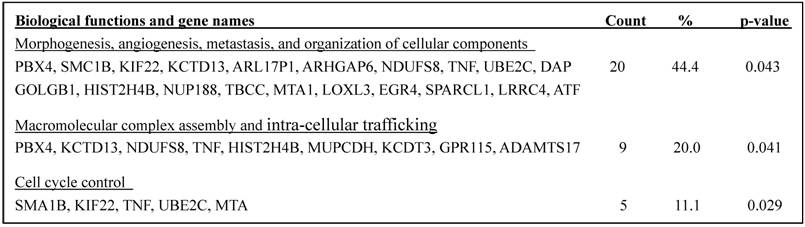
The known corresponding genes with the abnormal genomic DNA belong to three functionally distinct but correlated groups (Table 2). Twenty genes (44.4%), including PBX4, SMC1B, KIF22, KCTD13, ARL17P1, ARHGAP6, NDUFS8, TNF, UBE2C, GOLGB1, HIST2H4B, NUP188, TBCC, MTA1, LOXL3, EGR4, SPARCL1, LRRC4,.DAP, and ATF1, are known to be involved in morphogenesis, angiogenesis, and organization of cellular components and organelles Nine genes (20%), including PBX4, KCTD13, NDUFS8, TNF, HIST2H4B, MUPCDH, KCDT3, GPR115, and ADAMTS17, are known to involve in macromolecular complex assembly and intra-cellular trafficking. Five genes (11.1%), including SMC1B, KIF22, TNF, UBE2C, and MTA, are known to involve in controlling the cell cycle.
Biological functions and names of known genes affected by their genomic DNA gain or loss in PABC
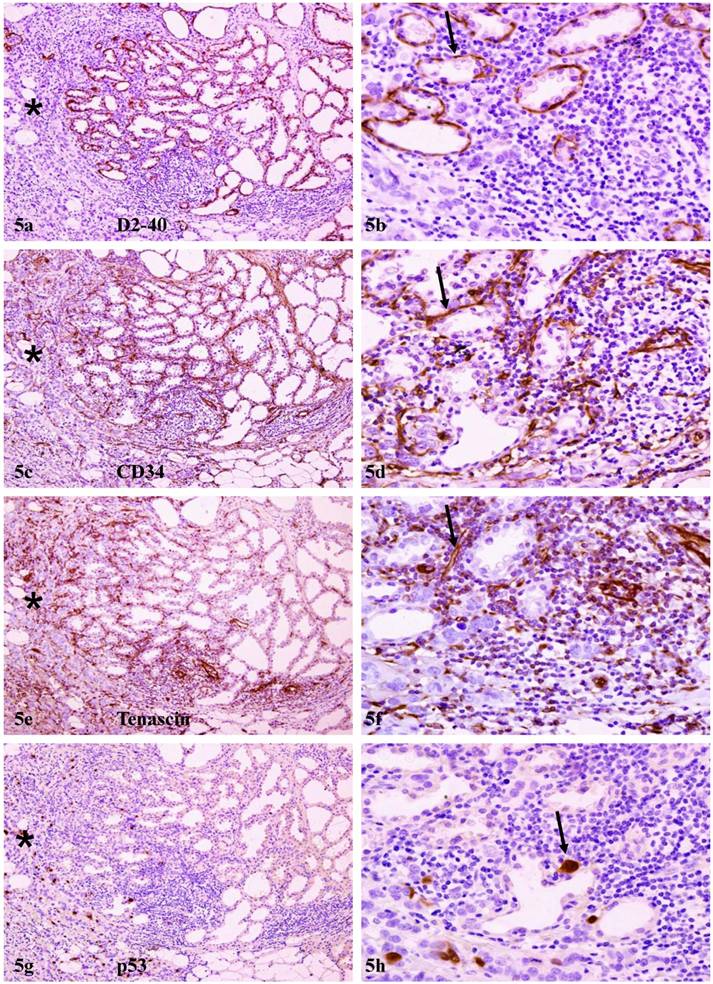
Array-CGH detected alterations in DNA copy numbers were in agreement with in vivo protein expression in adjacent tissue sections. In sections immunostained for angiogenesis-related marker D2-40 and CD34, strong and uniform immunoreactivities, in addition to those seen in lymphatic ducts and blood vessels, were also seen, in myoepithelial cell layers, which often formed a ring-like structure surrounding each of the acini (Fig 5). In adjacent sections immunostained for tenascin, a neonatal protein that facilitates early morphogenesis and tumor invasion [32], strong immunoreactivities were consistently detected within epithelial structures with focally disrupted myoepithelial cell layers, and also at the junctions between pre-invasive tissues and invasive lesions (Fig 5e-f), which consistently harbored detached individual cells or cell clusters that were strongly immunoreactive to p53 (Fig 5g-h).
Elevated expression of angiogenesis and growth-related genes. A set of four consecutive sections adjacent to sections used for array-CGH were immunostained for morphogenesis and angiogenesis-related biomarkers. Arrows identify aberrant expression of D2-40 and CD34 in myoepithelial cell layers, and aberrant expression of tenascin and p53 at the junctions between pre-invasive tissues and invasive lesions. Note that significantly higher expression of these markers were seen in normal appearing lobules adjacent to, compared to those distant from, invasive lesions (asterisks). a, c, e, and g: 80X; b, d, f, and h: a higher (200) magnification of a, c, e, and g, respectively.
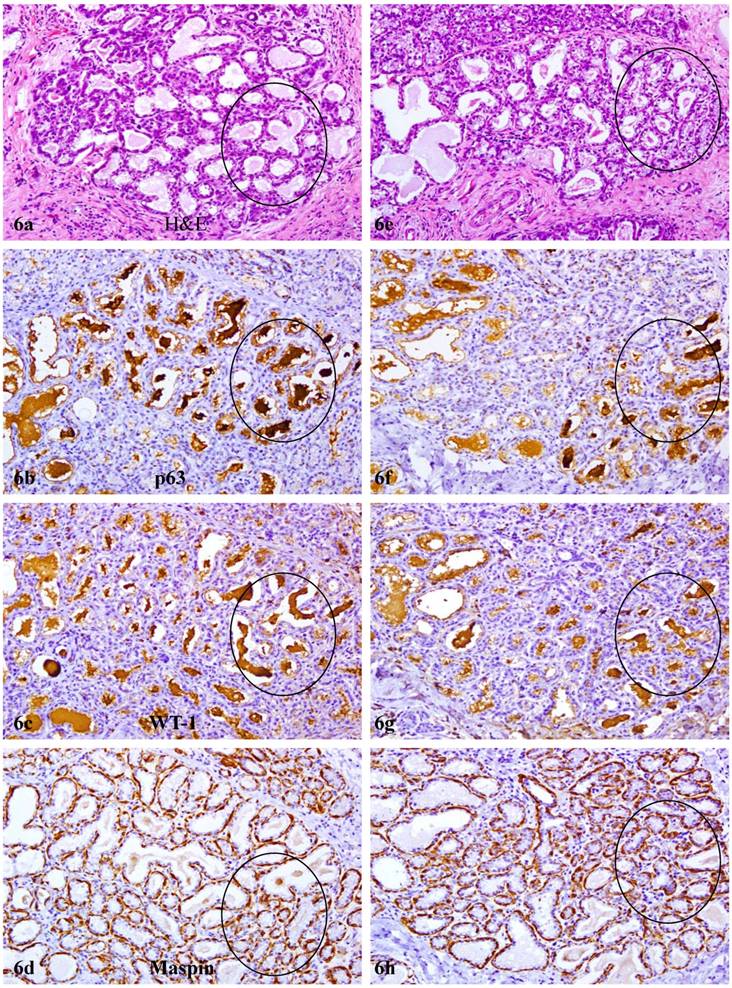
In contrast to reduction or loss of tumor suppressors in the myoepithelium, secretory products within the acinar lumen of most lobules in PABC were strongly immunoreactive to p63 and WT-1, but not to any of other antibodies used in the study. The pattern, intensity, and location of these immunoreactivities were similar with the peroxidase and alkaline phosphatase detection systems using DAB or AR-red chromogens (Fig 6). Pre-treatment with H2O2, lavamison, or biotin failed to eliminate or reduce immunoreactivities to WT-1 or p63. The substitution of the p63 and WT-1 primary antibodies with PBS or normal serum, however, resulted in total negativity (not shown). In sections immunostained for E-cadherin or beta-catenin, these acini consistently showed substantial reduction or absence of expression of these molecules, compared to their adjacent morphologically similar counterparts without cytoplasmic p63 expression (not shown).
Elevated expression of macromolecular complex assembly and intra-cellular trafficking related genes. Two sets of four consecutive sections adjacent to sections used for array-CGH were immunostained for p63, WT-1, and maspin. Circles identify acinal lumen with high level of secretory products. Note that strong p63 and WT-1 immunoreactivities were seen in these luminal secretory products, while maspin is only seen in the myoepithelium. 100X.
Discussion and Conclusions
The overall expression pattern of p63 expression in the myoepithelium and biological presentations in associated epithelium seen in our current study are in total agreement with those of our previous studies [9-11] and reports from other groups [5-8]. Our current study, however, has detected several unique alterations in normal or hyperplastic tissues of PABC that have not been previously reported, including: (1) immunohistochemical and morphological resemblance to adjacent invasive cancer, (2) significant gain of DNA copy number in chromosomal loci harbored morphogenesis, angiogenesis, and metastasis related genes, including early growth response 4, lysyl oxidase-like 3, and metastasis associated 1 [24-26, 29], and (3) significant loss of DNA copy number in chromosomal loci harbored tumor suppressor, cell adhesion, and macromolecular complex assembly or intra-cellular trafficking related genes, including Sparc-like 1, cytohesion 1 interacting protein, and G-protein coupled receptor115 [27,28,31]. Detected array-CGH alterations correlated well with in vivo expression of a number of corresponding proteins tested.
Together, our findings suggest that normal or hyperplastic appearing acinar cells with cytoplasmic p63 expression may represent a biologically more aggressive cell population, which could potentially progress to invasive lesions without undergoing through the stage of in situ cancer. Consistent with this possibility are a number of reports: (1) a subset of morphologically normal breast tissues share similar genetic and immunohistochemical profiles with malignant counterparts [33-35], (2) some “healthy” men between 19 and 29 years old had a spectrum of lesions, including hyperplasia and adenocarcinoma [36], (3) prostate tissues from some “healthy” men or morphologically normal prostate tissues harbored a DNA phenotype that is identical to that of prostate cancer [37,38], and (4) significant gene expression abnormalities were detected with microarrays using RNA extracted from microdissected histologically normal terminal ductal-lobular units (TDLU) of some breast tissues [39].
Normal or hyperplastic appearing acini with cytoplasmic p63 expression are likely to be formed from genetically altered progenitors for two main reasons: (1) TD and TDLU are the primary, if not the exclusive, site of progenitor cells, and (2) A vast majority of breast malignancies are derived from TDLU, which recapitulates the processes of early morphogenesis [40,41]. The formation of altered TD and associated acini is likely to result from two correlated pathways: (A) Genetic defects in myoepithelial cell self-renewal-related genes. As both p63 and WT-1 play significant roles in the early developmental of several organs and systems [42,43], it is possible that at the early stage of breast morphogenesis, the breasts of these patients may have exposed to certain external or internal insults that may have been causing permanent damages in DNA structures of some primitive stem cells. These DNA structural damages resulted in the inactivation of, or defects in, myoepithelial cell renewal-related genes, which impaired the myoepithelial cell replenishment process to replace the aged or injured myoepithelial cells, resulting in a “senesced” myoepithelial cell population with significantly reduced p63 and WT-1 expression. Consistent with this possibility is the fact that the expression of maspin, a tumor suppressor that is not involved in early development [6], is not affected in all PABC cases in this study. (B) Simultaneous activation and proliferation of multiple tumor stem cells. The DNA structural damages may have also caused the inactivation of, or defects in, cell cycle control- and apoptosis-related genes in the epithelial cell population, which allow these cells to proliferate to form clusters of ducts or acini with the same genetic defects. These acinar or duct clusters may progress rapidly after external or internal insults, leading to the early occurrence of breast cancer at young ages. On the other hand, these clusters may become maturation-arrested after a few cycles of cells divisions, but retain the potential for unlimited proliferation and multi-lineage differentiation [44]. During the last few weeks of pregnancy, these clusters are activated and simultaneously proliferating in responding to a sharp increase of estrogen, progesterone, and other hormones [1-3]. In agreement with this possibility is the fact that clinical morphological examinations have shown that a vast majority of the breast tumors are generally confined within a “sick” lobe [45].
Our immunohistochemical studies consistently showed a significant reduction or loss of nuclear p63 and WT-1 expression in a vast majority of the myoepithelial cells, while significantly increased cytoplasmic p63 expression in associated epithelial cells in PABC. Our array-CGH, however, failed to detect significant gain or loss in the genomic DNA that harbor genes coding for these two proteins. This disparity could potentially result from the following epigenetic alterations: (1) Aberrant intra-cellular transportation of the p63 protein. Similar to the p53 protein, p63 is synthesized in the cytoplasm and constantly transported through the nuclear pore complex into the nuclei [46,47]. A number of factors, including alterations of the elements and molecules involved in p63 nucleo-cytoplasmic transportation could significantly impact the p63 intra-cellular trafficking, and consequently alter its sub-cellular localization [46,47]. (2) Formation of protein-protein complexes. A previous study from our lab showed that only cells exhibiting secretory changes or secretory carcinomas had cytoplasmic p63 expression [48]. Thus, it is possible that secretory products in PABC may form protein-protein complexes with p63, which prevent the import of p63 protein into the nuclei. Consistent with this possibility is the fact that some proteins could form protein-protein complexes with p53, which results in accumulation of p53 in the cytoplasm [46]. (3) Interactions with WT-1 protein, which results in the secretion of both proteins into the acinar lumen. Consistent with this possibility is the fact that the secretory products within the acinar lumen of most lobules in PABC were strongly and uniformly immunoreactive to antibodies against p63 and WT-1, but not to any of other antibodies used in the study. In addition, our array-CGH analyses detected a number of factors that are closely involved in cellular organelle and molecular organization and transportation. Since previous studies have shown that cytoplasmic localization of p63 is associated with poor patient survival in lung cancer and is also associated with tumor grade in meningiomas [14,15], aberrant expression and sub-cellular localization of p63 and WT-1 may represent a novel mechanism for tumor aggressiveness and invasiveness. If confirmed, our findings could have significant implications: (1) changes of the tissue microenvironment may have direct impact on carcinogenesis, and (2) manipulation of the sub-cellular localization may have significant therapeutic value.
The specific cellular or molecular mechanism accounting for a correlation between tumor suppressor expression in the myoepithelium and biological behavior in the associated epithelium is unknown, but it is likely to result from aberrant tumor suppressor expression and focal disruptions in the myoepithelium. As the epithelium is normally devoid of both blood vessels and lymphatic ducts, and thus, totally relies on the stroma for its needed materials for normal functions and even survival, a focal myoepithelial cell layer disruption could have several consequences, including (1) an increase of permeability for oxygen, nutrients, and growth factors, which selectively triggers the exit of stem or progenitor cells from quiescence [49], (2) exposure of luminal cells to different cytokines, which facilitates tumor angiogenesis [50], (3) physical contact between luminal and stromal cells, which facilitates epithelial-mesenchymal transition (EMT) and cell motility [51], and (4) physical contact between luminal and immunoreactive cells, which directly cause genomic or cellular damages that trigger a cascade reaction of malignant transformation [52]. Consistent with these possibilities are the facts that our array-CGH revealed that all 4 PABC cases had a significant gain or loss in their genomic DNA harboring genes involved in morphogenesis, angiogenesis, and controlling the cell cycle.
No definite conclusions, however, could be drawn at present for two main reasons: (1) our study is the first one to assess the correlation between myoepithelia and associated epithelia in PABC with array-CGH and immunohistochemistry, and (2) our sample size is small and the clinical follow-up data are not available. On the other hand, if confirmed, our study will be the first one to provide in vivo evidence that suggests a correlation between reduced tumor suppressor expression in the myoepithelium and invasive behavior in the associated epithelium. Since degradation of the myoepithelium is an absolute pre-requisite for breast tumor invasion, our findings would have significant implications. Scientifically, these would suggest that the development of therapeutic agent to stimulate myoepithelial cell self-renewal or to prevent myoepithelial cell degradation may be more effective than the traditional approaches for breast cancer intervention and prevention. Clinically, as a vast majority of the breast malignancies are originated from TDLU, the development of more applicable methods to quantitatively assess the expression of p63 and WT-1 expression in this site may be more effective than other approaches for early detection of breast cancer. In addition, the elucidation of the mechanism of cytoplasmic p63 expression may lead to the development of new therapeutic agents to target cancer-related molecules in the cytoplasm or even in nuclei.
Acknowledgements
This study was supported in part by grant 2006CB910505 from the Ministry of Chinese Science and Technology Department, grant 30801176 from The National Natural Science Foundation of China, grants DAMD17-01-1-0129, DAMD17-01-1-0130, PC051308 from Congressionally Directed Medical Research Programs, grant BCTR0706983 from The Susan G. Komen Breast Cancer Foundation, and grant 2008-02 from US Military Cancer Institute and Henry M. Jackson Foundation.
The opinions and assertions contained herein represent the personal views of the authors and are not to be construed as official or as representing the views of the Department of the Army or the Department of Defense.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Mathelin C, Annane K, Treisser A. et al. Pregnancy and post-partum breast cancer: a prospective study. Anticancer Res. 2008;28(4C):2447-2452
2. Rodriguez AO, Chew H, Cress R. et al. Evidence of poorer survival in pregnancy-associated breast cancer. Obstet Gynecol. 2008;112(1):71-78
3. Polyak K. Pregnancy and breast cancer: the other side of the coin. Cancer Cell. 2006;9(3):151-153
4. Schedin P. Pregnancy-associated breast cancer and metastasis. Nat Rev Cancer. 2006;6(4):281-191
5. Sternlight MD, Barsky SH. The myoepithelial defense: a host defense against cancer. Med Hypotheses. 1997;48:37- 46
6. Zou Z, Anisowicz A, Hendrix MJ. et al. Maspin, a serpin with tumor-suppressing activity in human mammary epithelial cells. Science. 1994;263:526-529
7. Barbareschi M, Pecciarini L, Cangi MG. et al. p63, a p53 homologue, is a selective nuclear marker of myoepithelial cells of the human breast. Am J Surg Pathol. 2001;25:1954-1960
8. Yamamoto T, Oda K, Miyazaki K. et al. p73 is highly expressed in myoepithelial cells and in carcinomas with metaplasia. Int J Oncol. 2001;19:271-276
9. Man YG, Sang QXA. The significance of focal myoepitehlial cell layer disruptions in breast tumor invasion: a paradigm shift from the “protease-centered” hypothesis. Exp Cell Res. 2004;301:103-118
10. Li JH, Man YG. Dual usages of single Wilms' tumor 1 immunohistochemistry in evaluation of breast tumors: A preliminary study of 30 cases. Cancer Biomarkers. 2009;5(3):109-116
11. Xu ZL, Wang W, Deng CX, Man YG. Aberrant p63 and WT-1 expression in myoepithelial cells of pregnancy-associated breast cancer: implications for tumor aggressiveness and invasiveness. Int J Bio Sci. 2009;5:82-96
12. Tomkova K, Tomka M, Zajac V. Contribution of p53, 63, and p73 to the developmental diseases and cancer. Neoplasm. 2008;55(3):177-181
13. Westfall MD, Pietenpol JA. p63: Molecular complexity in development and cancer. Carcinogenesis. 2004;25(6):857-864
14. Narahashi T, Niki T, Wang T, Goto A, Matsubara D, Funata N. et al. Cytoplasmic localization of p63 is associated with poor patient survival in lung adenocarcinoma. Histopathol. 2006;49:349-357
15. Rushing EJ, Olsen C, Man YG. Correlation of p63 immunoreactivity with tumor grade in meningiomas. Int J Surg Pathol. 2008;16(1):38-42
16. Tavassoli FA, Man YG. Morphofunctional features of intraductal hyperplasia, atypical hyperplasia, and various grades of intraductal carcinoma. The Breast J. 1995;1(3):155-162
17. Man YG, Burgar A. An antigen unmasking protocol that satisfies both immunohistochemical and subsequent molecular biological assessments. Pathol Res Pract. 2003;199:815-825
18. Moinfar F, Man YG, Arnould L. et al. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: Implications for tumorigenesis. Cancer Res. 2000;60:2562-2566
19. Bai X, Wu J, Zhang Q. et al. Third-generation human mitochondria focused cDNA microarray and its bioinformatic tools for analysis of gene expression. Biotechniques. 2007;42:365-375
20. Su YA, Wu J, Zhang L. et al. Dysregulated mitochondrial genes and networks with drug targets in postmortem brain of patients with posttraumatic stress disorder (PTSD) revealed by human mitochondria-focused cDNA microarrays. Int J Biol Sci. 2008;4:223-235
21. Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185-193
22. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863-14868
23. Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - A Practical and Powerful Approach to Multiple Testing. J Royal Statistical Society Series B (Methodological). 1995;57:289-300
24. Wieland GD, Nehmann N, Muller D. et al. Early growth response proteins EGR-4 and EGR-3 interact with immunone inflammatory mediators NF-kappaB p50 and p65. J Cell Sci. 2005;118(pt 14):3203-3212
25. Payne SL, Fogelgren B, Hess AR. et al. Lysyl oxidase regulates breast cancer cell migration and adhesion through a hydrogen eroside-mediated mechanism. Cancer Res. 2005;65(4):11429-11436
26. Maddika S, Sy SM, Chen J. Functional interaction between Chfr and Kif22 controls genomic stability. J Biol Chem. 2009;284(19):12998-13003
27. Lau CP, Poon RT, Cheung ST. et al. SPARC and Hevin expression correlate with tumor aggiogenesis in hepatocellular carcinoma. J Pathol. 2006;210(4):459-468
28. Esposito I, Kayed H, Keleg S. et al. Tumor-suppressor function of SPARC-like protein 1/Hevin in pancreatic cancer. Neoplasia. 2007;9(1):8-17
29. Toh Y, Nicolson GL. The role of the MTA family and their encoded proteins in human cancer: molecular functions and clinical implications. Clin Exp Metastasis. 2009;26(3):215-227
30. Bjarnadóttir TK, Fredriksson R, Höglund PJ. et al. The human and mouse repertoire of the adhesion family of G-protein-coupled receptors. Genomics. 2004;84(1):23-33
31. Yona S, Lin HH, Siu WO. et al. Adhesion-GPRs: emerging roles for novel receptors. Trends Biochem Sci. 2008;33(10):491-500
32. Hancox RA, Allen MD, Holliday DL. et al. Tumor-associated tenascin-C isoforms promote breast cancer cell invasion and growth by matrix metalloproteinase dependent and independent mechanisms. Breast Cancer Res. 2009 [Epub ahead of print]
33. Moinfar F, Man YG, Bratthauer GL. et al. Genetic abnormalities in mammary ductal intraepithelial neoplasia-flat type (Clinging ductal carcinoma in situ)-A simulator of normal mammary epithelium. Cancer. 2000;88:2072-2081
34. Deng G, Liu Y, Ziotnikov G. et al. Loss of heterogosity in normal tissue adjacent to breast carcinomas. Science. 1996;274(5295):2057-2059
35. Man YG, Nieburgs HE. A subset of cell clusters with malignant features in morphologically normal and hyperplastic breast tissues. Cancer Detect Prev. 2006;30(3):239-247
36. Gardner WA, Culberson DE. Atrophy and proliferation in the young adult Prostate. J Urol. 1987;137(1):53-56
37. Malins DC, Anderson KM, Gilman NK. et al. Development of a cancer DNA phenotype prior to tumor formation. Proc Natl Acad Sci Sci USA. 2004;101:10721-10725
38. Malins DC, Gilman NK, Green VM. et al. A DNA phenotype in healthy prostates, conserved in tumors and adjacent normal cells, implies a relationship to carcinogenesis. Proc Natl Acad Sci USA. 2005;102:19093-19096
39. Tripathi A, King C, de la Morenas A. et al. Gene expression abnormalities in histologically normal breast epithelium of breast cancer patients. Int J Cancer. 2008;122(7):1557-1566
40. Smalley M, Ashworth A. Stem cells and breast cancer: A field in transit. Nat Rev Cancer. 2003;3(11):832-844
41. Polyak K. Breast cancer: origins and evolution. J Clin Invest. 2007;117(11):3155-3163
42. Barbieri CE, Pietenpol JA. p63 and epithelial biology. Exp Cell Res. 2006;312(6):695-706
43. Bourne TD, Bellizzi AM, Stelow EB. et al. p63 expression in olfactory neuroblastoma and other small cell tumors of the sinonasal tract. Am J Clin Pathol. 2008;130(2):213-218
44. Scharnhorst V, van der Eb AJ, Jochemsen AG. WT1proteins: functions in growth and differentiation. Gene. 2001;273(2):141-161
45. Tot T. The theory of the sick breast lobe and the possible consequences. Int J Surg Pathol. 2007;15(4):369-375
46. Liang SH, Clarke MF. Regulation of p53 localization. Eur J Biochem. 2001;268(10):2779-2783
47. O'Keefe K, Li H, Zhang Y. Nucleocytoplasmic shuttling of p53 is essential for MDM2-mediated cytoplasmic degradation but not ubiquitination. Mol Cell Biol. 2003;23(18):6396-6405
48. Bratthauer GL, Saenger JS, Strauss BL. Antibodies targeting p63 react specially in the cytoplasm of breast epithelial cells exhibiting secretory differentiation. Histopathology. 2005;47:611-616
49. Csete M, Walikonis J, Slawny N. et al. Oxygen-mediated regulation of skeletal muscle satellite cell proliferation and adipogrnesis in culture. J Cell Physiol. 2001;189:189-196
50. Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic mimicry and tumor-cell plasticity: lessons from melanoma. Nat Rev Cancer. 2003;3(6):411-421
51. Kang Y, Massague J. Epithelial-mesenchymal transition: twist in development and metastasis. Cell. 2004;118:277-299
52. Pawelek JM, Chakraborty AK. The cancer cell-leukocyte fusion theory of metastasis. Adv Cancer Res. 2008;101:397-444
Author contact
![]() Corresponding author: Yan-gao Man, MD., PhD., Director of Gynecologic and Breast Research Laboratory, Department of Gynecologic and Breast Pathology, Armed Forces Institute of Pathology and American Registry of Pathology. Tel: 202-782-1612; Fax: 202-782-3939; E-mail: manosd.mil. Or Ming-Chih Chou, MD., PhD., Professor, Department of Surgery, Member, Board of Trustees, Chung Shan Medical University, Taichung, Taiwan. Tel: 886-4-24730022; Fax: 886-4-24723229; E-mail: graduateedu.tw
Corresponding author: Yan-gao Man, MD., PhD., Director of Gynecologic and Breast Research Laboratory, Department of Gynecologic and Breast Pathology, Armed Forces Institute of Pathology and American Registry of Pathology. Tel: 202-782-1612; Fax: 202-782-3939; E-mail: manosd.mil. Or Ming-Chih Chou, MD., PhD., Professor, Department of Surgery, Member, Board of Trustees, Chung Shan Medical University, Taichung, Taiwan. Tel: 886-4-24730022; Fax: 886-4-24723229; E-mail: graduateedu.tw