Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2010; 6(6):569-583. doi:10.7150/ijbs.6.569 This issue Cite
Research Paper
Anterior Visceral Endoderm SMAD4 Signaling Specifies Anterior Embryonic Patterning and Head Induction in Mice
Cuiling Li1, Yi-Ping Li2, Xin-Yuan Fu3, Chu-Xia Deng1 ![]()
1. Mammalian Genetics Section, Genetics of Development and Disease Branch, National Institutes of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, 10/9N105, 10 Center Drive, Bethesda, MD 20892, USA
2. Department of Cytokine Biology, The Forsyth Institute, Boston, MA 02115, USA
3. Department of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, 8 Medical Drive, MD7 #02-03, Singapore 117597
Received 2010-9-16; Accepted 2010-9-27; Published 2010-9-27
Abstract
SMAD4 serves as a common mediator for signaling of TGF-β superfamily. Previous studies illustrated that SMAD4-null mice die at embryonic day 6.5 (E6.5) due to failure of mesoderm induction and extraembryonic defects; however, functions of SMAD4 in each germ layer remain elusive. To investigate this, we disrupted SMAD4 in the visceral endoderm and epiblast, respectively, using a Cre-loxP mediated approach. We showed that mutant embryos lack of SMAD4 in the visceral endoderm (Smad4Co/Co;TTR-Cre) died at E7.5-E9.5 without head-fold and anterior embryonic structures. We demonstrated that TGF-β regulates expression of several genes, such as Hex1, Cer1, and Lim1, in the anterior visceral endoderm (AVE), and the failure of anterior embryonic development in Smad4Co/Co;TTR-Cre embryos is accompanied by diminished expression of these genes. Consistent with this finding, SMAD4-deficient embryoid bodies showed impaired responsiveness to TGF-β-induced gene expression and morphological changes. On the other hand, embryos carrying Cre-loxP mediated disruption of SMAD4 in the epiblasts exhibited relatively normal mesoderm and head-fold induction although they all displayed profound patterning defects in the later stages of gastrulation. Cumulatively, our data indicate that SMAD4 signaling in the epiblasts is dispensable for mesoderm induction although it remains critical for head patterning, which is significantly different from SMAD4 signaling in the AVE, where it specifies anterior embryonic patterning and head induction.
Keywords: TGF-beta, SMAD4, AVE, epiblast, mesoderm patterning
Introduction
Gastrulation is a process in which embryos form three layered structures known as the mesoderm, ectoderm and endoderm [1-3]. Signaling from different layers interact with each other to ensure normal embryonic patterning and differentiation [1-3]. Within these three layers exist many molecules, including members of TGF-β superfamily, their receptors and intracellular mediators that are important for initiation of gastrulation, head induction and patterning [3-11]. For example, embryos carrying a mutation of TGF-β family members (such as BMP4 or Nodal), and their receptors (type I receptor ActRIB or the type II receptors ActRIIA/ActRIIB), either arrest at egg cylinder stages prior to mesoderm induction or die during gastrulation with severe patterning defects [12-15], highlighting an important function of TGF-β signaling in gastrulation and mesoderm induction. In light of these findings, it is important to point out that the precise role of each component of TGF-β signaling cascade in each of these distinct layers are often difficult to assess, as the conventional knockout approach disrupts genes in entire embryos.
SMAD proteins serve as a major intracellular component of TGF-β signaling pathway [7-9, 16]. There are eight members in the SMAD family where SMAD4 serve as a common mediator for TGF-β signaling. SMAD4 is a well-known tumor suppressor as its mutations have been detected in many human cancers, including pancreatic cancer, colon cancer, chlangieocarcinoma cancer, and gastric adenocarcinomas [17-19]. Mice carrying SMAD4 somatic mutations, generated by Cre-loxP approach to overcome embryonic lethality of germline mutation, develop cancers in the liver, pancreas, colon, skin, and many other organ/tissues [20-26]. To date, all SMADs have been knocked out in mice, and SMAD4‑deficient mice exhibited the most severe phenotype than any other mice carrying targeted mutations of other SMADs (reviewed in [10, 27], which is consistent with its function as a common mediator for TGF-β signaling.
Smad4-/- embryos died shortly after implantation at egg cylinder stages, exhibiting profound reduced cellular proliferation [28, 29]. Several lines of evidence indicated that the primary defect caused by SMAD4 deficiency is in the visceral endoderm. First, Smad4-/- embryos exhibited more severe abnormalities in the extraembryonic tissues than embryonic portion [29]. Second, Smad4-/- ES cells failed to make a normal visceral endoderm in embryoid body differentiated in vitro [28]. Third, embryos that carried a Cre-LoxP mediated deletion of SMAD4 in the epiblast but not in the visceral endoderm formed many mesodermal derivatives, including somites, heart, allantois and lateral plate mesoderm [30], suggesting the absence of SMAD4 in the epiblast does not block mesoderm formation.
Therefore, we hypothesized that SMAD4 signaling in visceral endoderm specify mesoderm formation. To test this hypothesis, we performed a tissue specific disruption of SMAD4 in the visceral endoderm. Our analysis indicated that SMAD4 mutant embryos failed to express a number of key molecules in the AVE and were arrested at E7.5-E8.5 lacking head and anterior embryonic structures. Our data reveals an essential role of SMAD4 in the visceral endoderm in anterior embryonic patterning and head induction during gastrulation.
Results
Expression of TTR-Cre
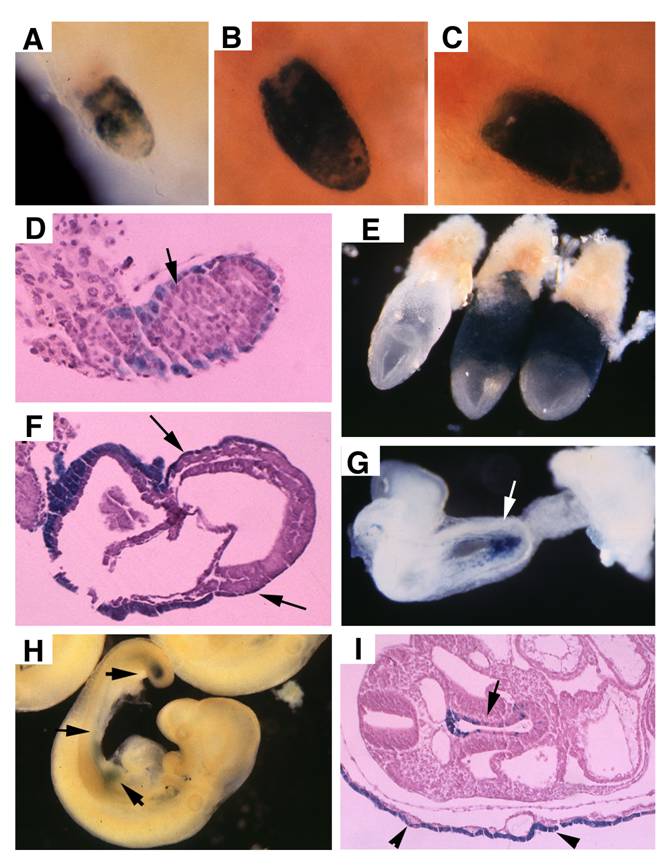
Transgenic mice carrying a Cre gene controlled by transthyretin (TTR) promoter was used to delete SMAD4. Using a Rosa-26 reporter strain (R26R) for Cre activity [31], TTR-Cre mediated recombination was detected in embryonic (E) 5.75 embryos at both embryonic and extraembryonic endoderm (Fig. 1A). The cells carrying TTR-Cre mediated recombination (β-gal positive) gradually spread into the entire endoderm of the embryos at E6-6.5 (Fig. 1B, C, D). As the mesoendoderm gradually replaces the distal endoderm, visceral endoderm gradually retreats toward extraembryonic portion. By E7.5, the β-gal positive cells were restricted to the visceral endoderm mainly at the extraembronic portion (Fig. 1E, F). At E8.5, Cre-mediated recombination is detected in the embryo starting from the primitive endoderm near the caudal neuropore (the tip of the hindgut) (Fig. 1G). At E9.5, the β-gal+ cells spreads to most part of the gut epithelium (arrows, Fig. 1H, I), and they also populated the endoderm-derived layer (the out layer) of the yolk sac (Fig. 1I).
TTR-Cre activity in early post-implantation embryos. (A-D), Images of Rosa-R26R;TTR-Cre embryos at E5.75-E6.5 revealed by X-Gal staining. (E,F) X-Gal staining in extraembryonic endoderm of two E7.5 Rosa-R26R;TTR-Cre embryos (right in E). Arrows point to weak X-Gal staining in visceral endoderm portion that covers the epiblast, which will development into the visceral yolk sac later (F). (G) At E8.5, TTR-Cre is expressed in the primitive endoderm near the caudal neuropore (arrows). (H,I) At E9.5, it expressed in tail bud and some tissues derived from midgut endoderm (arrows in H), and also in the visceral yolk sac (arrowhead in I, arrow points to gut epithelium). At least 10 embryos at each time point were analyzed for the staining.
Severe gastrulation defects in mice carrying a targeted deletion of SMAD4 in the visceral endoderm
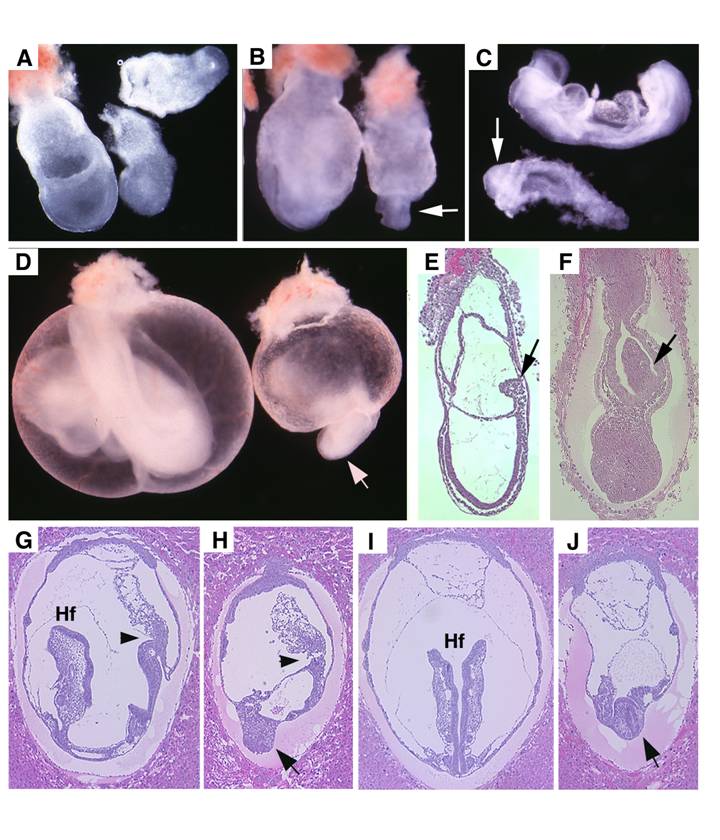
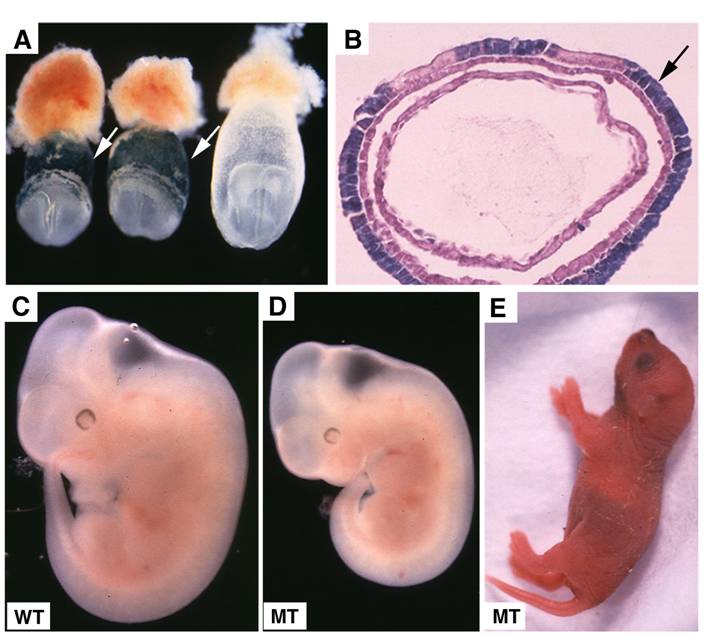
Visceral endoderm specific deletion of SMAD4 is achieved by crossing SMAD4 conditional mutant mice [32] with TTR-Cre transgenic mice. The Smad4+/Co;TTR-Cre mice were normal and were further crossed with Smad4Co/Co mice to generate Smad4Co/Co;TTR-Cre embryos. Our analysis on 41 offspring generated by this cross failed to obtain any Smad4Co/Co;TTR-Cre mice at post-neonatal stages, indicating that they were embryonic lethal (Table 1). Upon closer examination we determined that Smad4Co/Co;TTR-Cre embryos died before E9.5 (Table 1). Our data indicated that the mutant embryos were slightly smaller at E6.5 but became smaller at E7.5 (Fig. 2A) and at E8.5-9.5, the embryonic portion of mutant embryo was significantly smaller and maintained as a solid mass sticking out of the yolk sac (Fig. 2B-D). There was a correlation between the age of the embryos and their size. As they increased in age, their size significantly decreased. Upon histological examination, the Smad4Co/Co;TTR-Cre embryos were disorganized, lacking anterior and middle trunk structures (Fig. 2E-H). They had enlarged allantois and some visible, yet abnormal posterior structures (Fig. 2E-H). Cross section revealed the existence of neural epithelium although it was also quite abnormal (Fig. 2I,J). Together, the data indicate that the absence of SMAD4 does not block mesoderm induction; however, it causes severe defects during gastrulation.
Genotypes and phenotypes of offspring from crosses between Smad4Co/Co and Smad4Co/+;TTR-Cre micea
| Total | Co/Co | Co | Co/+;Cre | Co/Co;Cre b | resorption | |
|---|---|---|---|---|---|---|
| Neonates | 41 | 12 | 15 | 14 | 0 | 0 |
| E16.5 | 7 | 3 | 1 | 3 | 0 | 0 |
| E13.5 | 9 | 1 | 4 | 3 | 0 | 1 |
| E12.5-16.5 | 11 | 1 | 4 | 1 | 0 | 5 |
| E10.5 | 30 | 5 | 7 | 10 | 3 | 5 |
| E9.5 | 17 | 4 | 4 | 2 | 7 | 0 |
| E8.5 | 9 | 2 | 2 | 2 | 3 | 0 |
| E7.5 | 25 | 7 | 4 | 8 | 6 | 0 |
| E6.5 | 16 | 2 | 4 | 6 | 4 | 0 |
a. The cross between Smad4Co/Co and Smad4Co/+;TTR-Cre should yield equal number of offspring with each of four different genotypes if they are not lethal. However, there are no live Smad4Co/Co;TTR-Cre mice and Smad4Co/Co;TTR-Cre embryos were under represented starting from E10.5.
b. All Smad4Co/Co;TTR-Cre embryos are abnormal
Morphology and histology of Smad4Co/Co;TTR-Cre embryos. (A-D) Morphology of E7.5 (A) E8.5 (B,C) and E9.5 (D) embryos. Smad4Co/Co;TTR-Cre mutant embryos were pointed by arrows. Anterior region protrudes out from yolk sac in the Smad4Co/Co;TTR-Cre mutant embryos (arrows in B-D). (E-J) Histology of E7.5 (E,F) and E8.5 (G-J) control embryos (E,G,I) and Smad4Co/Co;TTR-Cre mutant embryos (F,H,J). Arrows in E,F, and arrowhead in G,H point allantois. Arrows in H,J point to anterior truncation in the mutant embryos presented at different angles. Hf: head-fold. At least 4 Smad4Co/Co;TTR-Cre mutant embryos at each time point were analyzed.
Loss of SMAD4 impairs AVE signaling and causes truncation of anterior and middle trunk of gastrulating embryos
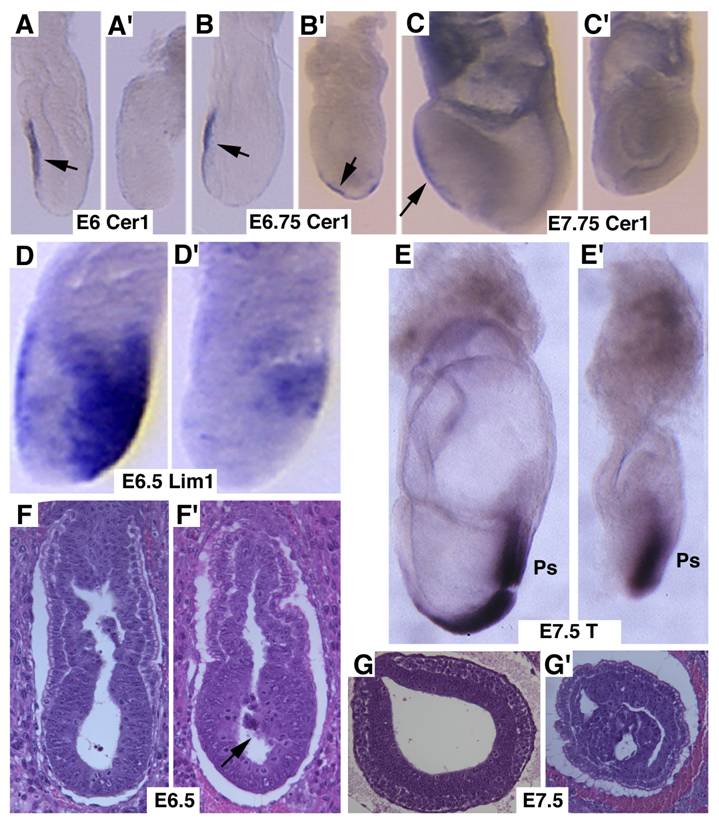
Next, we studied markers for AVE signaling as it plays a critical role in specifying head formation and anterior mesoderm patterning in mouse embryos [6, 33, 34]. At E6, Cer1, which marks the AVE and the earliest population of anterior definite endoderm [35] is detected in the AVE of wild-type embryos, and maintained distinct expression levels throughout E7.5 (Fig. 3A-C). However, Cer1 was significantly reduced or absent in SMAD4 mutant embryos at all the time points examined (Fig. 3A'-C'). Next, we examined Lim1, which is expressed in the AVE and also in a region of the epiblast where the future primitive streak forms prior to gastrulation [36]. At early gastrulation, Lim1 is present in the primitive streak, migrating mesodermal wings and the prechordal mesoderm that underlies the anterior portion of the future head-fold (Fig. 3D). In the SMAD4 mutant embryos, expression of Lim1 was significantly decreased in the posterior region and was diminished in the AVE (Fig. 3D'). We also observed similar reduced expression of Hnf3β (data not shown). Despite these changes, SMAD4 mutant embryos were able to initiate gastrulation as evidenced by expression of T gene in the primitive streak although the development is much delayed and the primitive streak is much shorter compared with the control embryos (Fig. 3E,E'). Histological sections prepared at different angles of embryos at E6.5 (Fig. 3F,F') and E7.5 (Fig. 3G,G') revealed abnormal accumulations of cells in the mutant embryos.
Marker analysis of Smad4Co/Co;TTR-Cre embryos at E6-7.5. (A-C) Cer1 expression is dramatically reduced in the Smad4Co/Co;TTR-Cre embryos (A', B', C') compared with controls (A-C). (D) Lim1 expression level is reduced in the posterior region and missing in the AVE of the mutant embryos (D'). (E) Expression of T, which marks the primitive streak (ps), is detected at both E7.5 control (E) and mutant (E') embryos, however the ps is much shorter in the mutant embryo. (F,G) Sagittal (F) and cross (G) sections of embryos showing accumulation of cells (arrow in F') and abnormally positioned cells (G') in the mutant embryos. At least 6 Smad4Co/Co;TTR-Cre embryos were analyzed for each marker at each time point.
Abnormal head-fold formation was further analyzed using molecular markers.
Otx2 is expressed in the epiblast prior to gastrulation in the wild-type embryos (Fig. 4A). No significant difference of Otx2 was observed in mutant embryos although they were smaller than controls at this stage (Fig. 4B). During gastrulation, Otx2 expressing cells progressively restricted to anterior portion of the embryo, and demarcated the anterior neuroectoderm in normal embryos (Fig. 4C,D). In the mutant embryos, the Otx2 expression domain was much smaller and presented in the poorly developed distal portion of the embryos (Fig. 4C,D). At E9.5 Otx2 expression marked the forebrain and midbrain (Fig. 4E), however, it disappeared in the mutant embryos (Fig. 4F).
Molecular marker analysis for Smad4Co/Co;TTR-Cre embryos. (A-E) Expression of Otx2 in control and mutant (Mt) embryos at different stages as indicated. Arrow in C points to the headfold of a control embryo. Arrows in D,E point to anterior of mutant embryos. (F-J) Expression of En1 (F), Krox20 (G), Mox1 (H,I) and Lim1 (J). Arrows in F,G, point to expression of Krox20 (F) and En1 (G). At least 4 Smad4Co/Co;TTR-Cre embryos were analyzed for each marker at each time point.
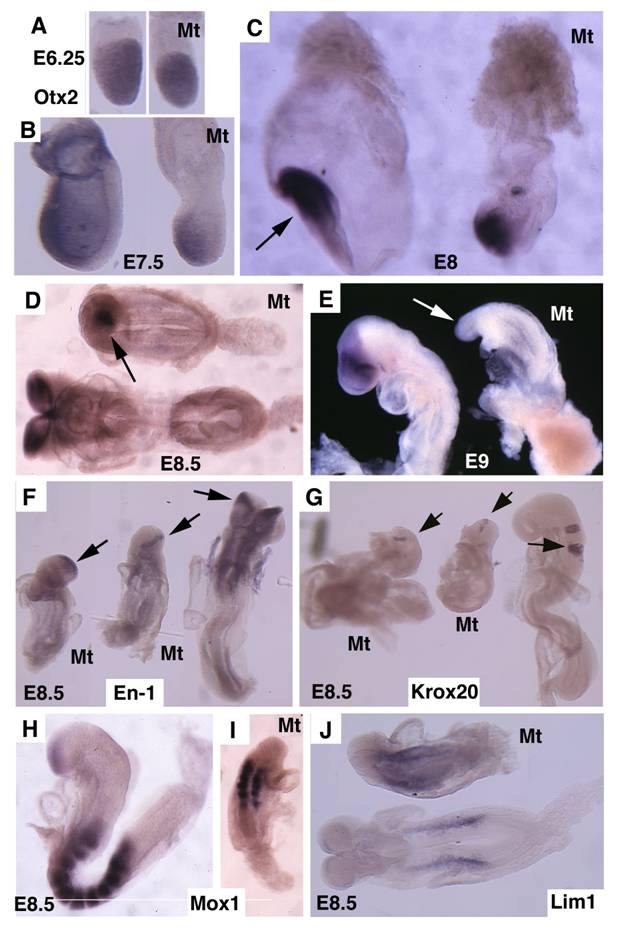
Next, we examined the development of the midbrain and hindbrain of SMAD4 mutant embryos. Staining of a midbrain marker, En-1, detected the existence of some residue cells in the midbrain (Fig. 4F). Mutant embryos also maintained a narrow stripe of rhombomere 5 revealed by using Krox20, which marks rhombomeres 3 and 5 (Fig. 4G). Staining of Mox1, a marker for somites (Fig. 4H,I), and Lim1, which marks areas in the urogenital precursor (Fig. 4J), revealed relative normal posterior structures. These data indicate that targeted disruption of SMAD4 in visceral endoderm resulted in a failure to establish the anterior-posterior (A-P) axis characterized by truncation of anterior and middle trunk of gastrulating embryos.
Wild-type tetraploid extraembryonic tissue rescues the lethality of Smad4Co/Co;TTR-Cre embryos
Tetraploid cells are known to preferentially contribute to extraembryonic tissues as demonstrated previously [37, 38] and in this study (Fig. 5A,B). To confirm that the lethality of Smad4Co/Co;TTR-Cre embryos is indeed caused by the deletion of SMAD4 in the extraembryonic endoderm, we aggregated Smad4Co/Co;TTR-Cre embryos with wild-type tetraploid embryos. In the three litters of embryos analyzed at E10.5-11.5, we found 5 morphologically normal Smad4Co/Co;TTR-Cre embryos (Fig. 5C,D) and one partially rescued SMAD4 mutant embryo (data not shown). Because we never observed any Smad4Co/Co;TTR-Cre embryos without fusing with tetraploid embryos that displayed normal or nearly normal morphology at these stages (Table 1), We concluded that wild-type tetraploid extraembryonic tissue repressed the lethality of Smad4Co/Co;TTR-Cre embryos and allowed them to develop through gastrulation. Some of them even advanced to birth (Fig. 5E). This observation provides strong evidence that extraembryonic tissue is critical for early post-implantation embryogenesis and normal patterning.
Embryonic lethality of Smad4Co/Co;TTR-Cre embryos was rescued by fusion with wild type tetraploid embryos. (A,B) Contribution of tetraploid cells to extraembryonic tissues analyzed at E7.5 embryos by wholemount view (A) and histological sections (B). Arrows point tetraploid beta-gal positive cells that are generated from embryos of Rosa-26 mice after fusion. (C,D) Tetraploid embryos fused wild type (C) and Smad4Co/Co;TTR-Cre (D) embryos at E11.5. (E) A rescued Smad4Co/Co;TTR-Cre mutant mouse at birth.
Deletion of SMAD4 in the epiblast yields less severe phenotypes than its deletion in the visceral endoderm
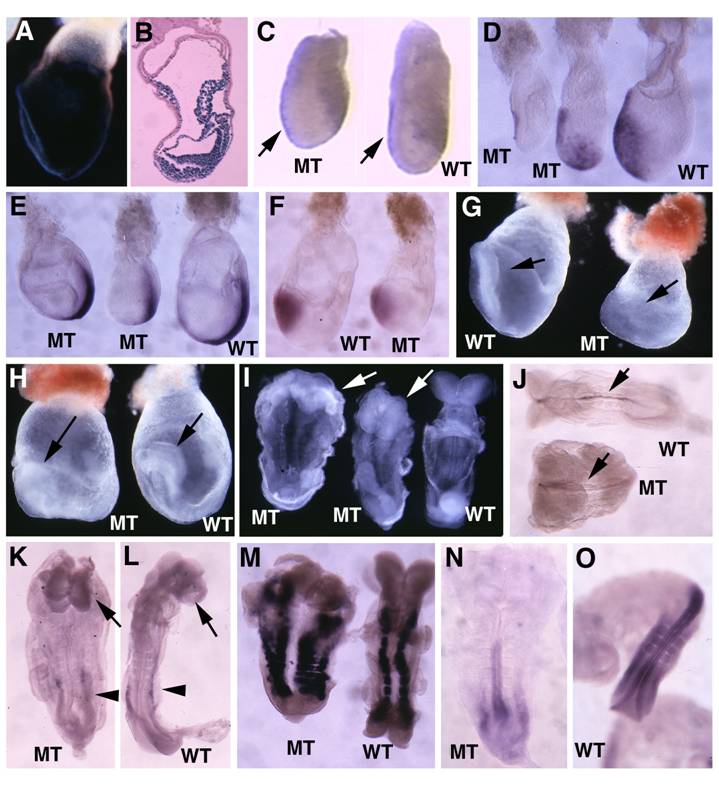
Previous investigations generated SMAD4 mutant embryos with wild-type endoderm either using wild-type tetraploid embryos to fuse with SMAD4-/- embryos [28] or knocking out SMAD4 in the epiblast specifically using Sox2-Cre [30]. Their data revealed that that wild-type visceral endoderm could rescue the gastrulation defect of Smad4-deficient embryos. To provide first hand comparative information regarding a role of SMAD4 in different germ layers during early embryonic development, we interbred the Smad4Co/Co mice with Mox2-Cre mice, which also specifically express Cre in the epiblast [39](Fig. 6A,B) but at a stage that slightly late than Sox2-Cre [40]. Our analysis indicated that the Smad4Co/Co;Mox2-Cre embryos were slightly smaller than control embryos in the early stages of gastrulation (Fig. 6C). Most Smad4Co/Co;Mox2-Cre embryos maintained relatively normal Lim1 (Fig. 6C) and Cer1 (Fig. 6D) expression. The mutant embryos were able to initiate gastrulation as revealed by the formation of the primitive streak although in most cases the mutant primitive streak is shorter (Fig. 6E). All mutant embryos advanced to the head-fold stage as revealed by Otx2 expression (Fig. 6F) and the morphological appearance at E7.75-E8.75 despite the fact that they were clearly abnormal at these stages (Fig. 6G-I). Further analysis using whole-mount in situ hybridization with lineage markers revealed that Smad4Co/Co;Mox2-Cre embryos formed notochord as revealed by sonic hedgehog (Shh, Fig. 6J), and forehead as revealed by brain factor (Bf1) (arrows, Fig. 6K,L). Of note, they also displayed expanded left-right (L-R) axis as revealed by paraxial mesoderm marker, Mox1, compared with controls (Fig. 6M). Hybridization with Lim1 for developing mesonephros in the middle trunk of the embryos (arrowheads, Fig. 6K,L) and HoxB9 for posterior neural ectoderm (Fig. 6N,L) detected slightly shortened posterior structures. Thus, despite the observation that Smad4Co/Co;Mox2-Cre embryos exhibited many patterning and morphogenesis defects, the absence of SMAD4 in the epiblast does not block anterior mesoderm formation and head-fold induction. The Smad4Co/Co;Mox2-Cre embryos also survived, on the average, one day longer than Smad4Co/Co;TTR-Cre embryos. These data suggest that SMAD4 plays a more critical role in the visceral endoderm than it does in the epiblast.
Analysis for Smad4Co/Co;Mox2-Cre embryos. (A,B) Images of Rosa-R26R;Mox2-Cre embryos at E7.5 revealed by X-Gal staining. (C). Wholemount in situ hybridization with Lim1 in E6.5 mutant (MT) and control (WT) embryos. (D-F) Expression of Cer1 (D), T (E) and Otx2 (F) in E7.5 embryos. (G,H) Morphological of E7.75 (G) and E8.5 (H) embryos before dissecting out from yolk sac. Arrow point to head-fold. (I) E8.5 embryos after dissecting out from yolk sac. (J-O) Expression of SSH (J), BF1 (arrows in K,L), Lim 1 (arrowheads, in K,L), and Mox 1 (M) in E8.75 embryos, as well as HoxB9 (N,O) in E9 embryos. At least 4-10 Smad4Co/Co;Mox2-Cre embryos were analyzed for each marker at each time point.
Absence of SMAD4 lost responsiveness to TGF-β induced morphogenetic changes and visceral endoderm gene expression in embryoid bodies
Embryoid bodies serve as an excellent model for studying developmental signaling and early embryonic development. Therefore, we studied the response of embryoid body differentiation to TGF-β signaling. Upon TGF-β treatment, embryoid body underwent faster differentiation (Fig. 7A) compared with untreated controls (Fig. 7A'). We also detected up-regulation of VE marker genes, such as Hex1 (Fig. 7B), Cer1 (Fig. 7C), and Lim1 (Fig. 7D) in the TGF-β treated samples compared with controls (Fig. 7B', C',D'). We next isolated RNA from both TGF-β treated and untreated embryoid bodies at different time points and analyzed gene expression (Fig. 7E). We found that Lim1 level increased 24 hours upon TGF-β treatment. EndoA, also called mouse keratin 8 that serves as an endodermal marker for early embryos [41], was initially expressed at relatively higher levels and its expression became weaker during later days of differentiation in the untreated embryos. Treatment of TGF-β maintained higher expression levels at all time points examined, consistent with a role of TGF-β signaling in induction and maintaining endoderm cell fate. Expression of other two later endoderm marker genes, Gata4 [42] and Hnf1 [43] was induced 2 days after treatment. Meanwhile, expression of T gene, a marker for mesoderm, was induced 6 days after treatment. Collectively, the data indicate that morphogenetic change induced by TGF-β treatment is associated with distinct expression changes of genes involved in early embryonic development and germ layer formation.
Embryoid body culture and expression analysis of molecular markers. (A) Morphology of embryoid bodies that have been treated (A) or untreated (A') with TGF-β1 (10ng/ml) for 4 days. Arrows in A point to cavities induced by TGF-β1. (B-D) Gene expression of in TGF-β1 treated (B-D) or untreated (B'-D') embryoid bodies revealed by wholemount in situ hybridization. (E). RT-PCR analysis of gene expression. RNA was isolated at different time points from both TGF-β1 treated (T) and untreated (C) embryoid bodies. TGF-β1 was added 48 hours after starting the culture, so the third day counts as treatment day 1 (T1). The treatment was lasted for 4 days. (F) RNA was isolated from both wild type and SMAD4-/- embryoid bodies at different time points, from day 0 (ES), 3 (EB3), 6 (EB6), 9 (EB9) and 12 (EB12).
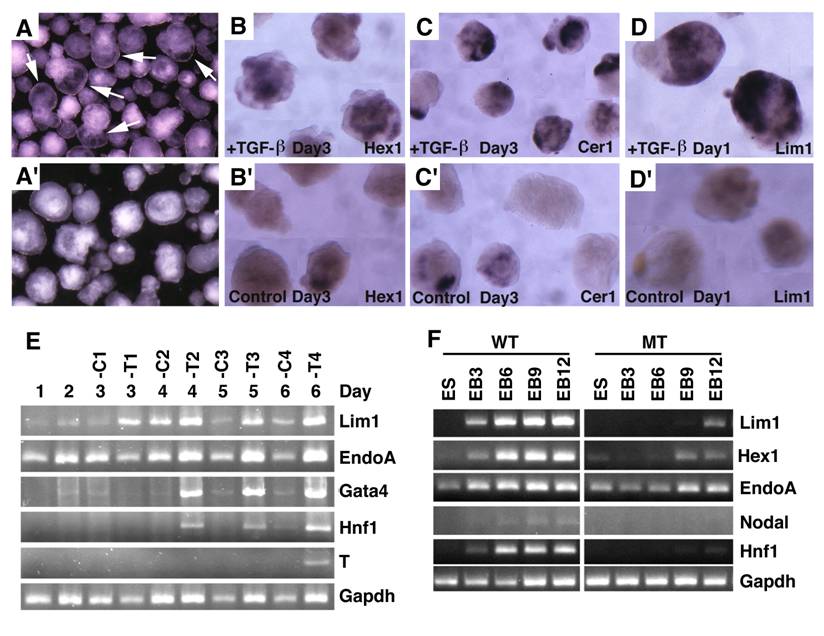
To investigate whether this action of TGF-β is mediated by SMAD4, we treated embryoid bodies formed by SMAD4-/- and wild-type ES cells with TGF-β. Our data indicated while TGF-β treatment readily up-regulated expression of genes involved in egg cylinder development (Lim1 and Hex1) and endoderm formation (Endo A and Hnf1), SMAD4 mutant embryoid bodies showed impaired response or responsiveness to the treatment (Fig. 7F). Expression of Nodal is first detected in primitive streak-stage embryos at about the time of mesoderm formation, and then becomes highly localized in the node at the anterior of the primitive streak [15]. Consistent with late appearance of T expression (Fig. 7E), we detected weak expression of Nodal in E6 embryoid bodies, but its expression was not detected in SMAD4-/- ES cells (Fig. 7F). These data provide strong evidence that SMAD4 mediates TGF-β signaling which in turn regulates expression of genes during early embryonic development and endoderm differentiation.
Discussion
In the present study, we disrupted TGF-β/SMAD4 signaling in the visceral endoderm by crossing SMAD4 conditional mutant mice with mice carrying a TTR-Cre transgene. We illustrated that the Smad4Co/Co;TTR-Cre embryos displayed severe abnormalities in the development of anterior structures; however, they had relatively normal posterior structures. This is unlikely caused by a lack of Cre activity as Cre-mediated deletion of lac-Z reporter gene is detected in the entire visceral endoderm at early post-implantation stages. Rather, this finding suggests that TGF-β/SMAD4 signaling has a more critical role in the AVE than it does in posterior visceral endoderm.
We also confirmed that the lethality of Smad4Co/Co;TTR-Cre embryos is indeed due to SMAD4 deficiency in the extraembryonic endoderm by aggregating Smad4Co/Co;TTR-Cre embryos with wild-type tetraploid embryos. It has been shown that tetraploid wild-type cells preferentially incorporate into visceral endoderm and extraembryonic endoderm but they cannot participate in embryonic development [37, 38], and therefore entire embryos are formed by Smad4Co/Co;TTR-Cre embryos. The fact that Smad4Co/Co;TTR-Cre tetraploid embryos survive through the gastrulation process provides strong evidence that TGF-β/SMAD4 signaling in the visceral endoderm and extraembryonic tissue is critical for early post-implantation embryogenesis and normal patterning. Due to the fact that the expression of TTR-Cre is in the entire visceral endoderm (Fig. 1), the TTR-Cre transgenic mice should serve as an important tool for testing functions of genes in this tissue for their ability in regulating gene expression and embryonic patterning prior to and during gastrulation. Of note, a recent study also generated a TTR-Cre transgenic mouse line, which showed similar Cre activity by using R26R reporter line [44]. The TTR promoter used to drive Cre is known to have a similar expression pattern compared with the endogenous TTR gene in some adult organs such as liver, pancreas, gut, and gallbladder [44, 45]. TTR-Cre transgenic mice can also be used for studying gene function in these tissues.
The AVE provides nutrition and physical support to embryos, and also plays a critical role for mesoderm formation, and head induction and patterning [4-6, 33, 34, 46]. Many molecules, including members of TGF-β superfamily (Nodal, Lefty1), receptors (ActRIB, ActRIIA, ActRIIB), and intracellular mediators (Smad2, and Smad4) and those that are not directly related to TGF-β, such as Hex1, Lim1, HNF3β, Cer1, Otx2, mDkk1, and Goosecoid, are expressed in the AVE [4, 6, 33, 34]. Embryos deficient for some of these genes exhibit phenotypes including failure to initiate gastrulation, mesoderm induction (ActRIB [12], ActRIIA and ActRIIB double mutants [13], Smad2 [47] and Smad4 [28, 29]), mesoderm patterning defects (Nodal [15], Hnf3-β [48]), L-R axis position defect (Lefty1 [49]), and anterior truncation (Otx2 [50], Lim1 [36]. However, the precise role of these molecules in embryonic patterning and their relationship in signaling cascades remain elusive.
We show here that the abnormal development of the Smad4Co/Co;TTR-Cre embryos is associated with significantly reduced or absence of expression of many genes important for early embryonic development. The absence of these genes could either serve as a causative reason or simply as a victim to the abnormal development in mutant embryos. To distinguish this, we studied early post-implantation embryos, and found that expression of Lim1, Cer1 and Hnf3β is already diminished in Smad4Co/Co;TTR-Cre embryos at stages prior to the onset of morphological abnormalities. This data suggests that the absence of expression of these genes is not secondary to the SMAD4 deficiency, rather it argues that their expression is positively regulated by SMAD4 signaling. This notion is confirmed by our in vitro study where the treatment of TGF-β induces expression of these genes in embryoid bodies formed by wild type ES cells, but not by SMAD4-/- ES cells. Thus, our study reveals a cascade of TGF-β/SMAD4 signaling in the AVE through regulating expression of some important genes, including Cer1, Hnf3β, and Lim1 that determine anterior embryonic patterning and head induction in mouse embryos.
In addition to the AVE, many members of TGF-β superfamily are expressed in the underlying epiblasts [4-7, 11, 16]. As a common mediator for TGF-β superfamily [8, 9], SMAD4 should mediate TGF-β signaling from both the AVE and epiblast. To investigate this, we have also deleted SMAD4 in the epiblast using Mox2-Cre. We found that many of the Smad4Co/Co;Mox2-Cre embryos developed into head-fold stages and exhibited relatively normal head process. Indeed, the SMAD4-deficient epiblast seems to be able to form most, if not all, mesoderm derivatives. Loss of SMAD4 in the epiblast also does not affect expression of genes that is severely affected in Smad4Co/Co;TTR-Cre embryos. A previous investigation also deleted SMAD4 in the epiblast using Sox2-Cre [30]. The Sox2Cre;Smad4CA/N embryos exhibited profound failure to pattern derivatives of the anterior primitive streak [30], which is more severe than the Smad4Co/Co;Mox2-Cre embryos generated here perhaps due to slightly earlier expression of Sox2-Cre [40] than Mox2-Cre [39]. Despite this, the Sox2Cre;Smad4CA/N embryos and the SMAD4-/- embryos fused with wild-type tetraploid embryos [28] do not display defects in many well-characterized TGF-β regulated processes involved in mesoderm formation and patterning. These studies, together with our analysis of Smad4Co/Co;Mox2-Cre embryos (Fig. 6) indicate that SMAD4 signaling in the epiblasts is dispensable for mesoderm induction although it is critical for head patterning.
In summary, we have studied functions of SMAD4 in the visceral endoderm and epiblast using a Cre-LoxP mediated tissue specific gene knockout approach. In contrast to our previous finding that loss of SMAD4 in both germ layers blocked egg cylinder elongation and mesoderm induction [29], we showed that SMAD4 deficiency in the epiblast has no major impact in mesoderm induction although it results in abnormal mesoderm patterning during gastrulation. This data is consistent with a similar finding reported previously [28, 30]. Importantly, analyzing a novel mutant strain generated by deleting SMAD4 in the visceral endoderm, we uncovered an essential role of SMAD4 in the visceral endoderm that mediates TGF-β signaling and regulates expression of genes in the AVE, where they specify anterior embryonic patterning and head induction. These data indicate that TGF-β/SMAD4 signaling has a distinct role in the visceral endoderm and underlying epiblast during gastrulation in mesoderm patterning and head induction, while joined signaling from both layers is critical for egg cylinder development and mesoderm induction, leading to the initiation of gastrulation.
Materials and Methods
Mice and genotyping analysis
The Smad4Co/Co;TTR-Cre mice and Smad4Co/Co;Mox2-Cre mice were generated by crossing the Smad4Co/Co mice [32] with TTR-Cre transgenic mice (X.Y. Fu, manuscript in preparation), and Mox2-Cre transgenic mice [39], respectively. Smad4 conditional mutant mice are genotyped by PCR using a pair of primers flanking one of the loxP sites: forward primer (5'-GACCCAAACGTCACCTTCAC-3') and reverse primer-1 (5'-GGGCAGCGTAGCATATAAGA-3'). This pair of primer amplifies a fragment of about 450 pb from wild type allele and a fragment of about 510 pb from Smad4 conditional allele. After the Cre mediated recombination, the mutant Smad4 allele is genotyped by using the forward primer and reverse primer-2 (5'-AAGAGCCACAGGTCAAGCAG-3'). The PCR product is about 500 bp. We used a pair of common Cre primers, Cre-1 (5'-CCT GTT TTG CAC GTT CAC CG-3') and Cre-2 (5'-ATG CTT CTG TCC GTT TGC CG-3') to genotype TTR-Cre and Mox2-Cre. Animals were handled in accordance with guidelines of the NIDDK Animal Care and Users Committee.
Generation of tetraploid embryos and rescue of SMAD4 mutant mice
Generation of tetraploid embryos was performed as described [37, 38]. Briefly, CD-1 females (Charles River) were mated with stud males and sacrificed at E1.5. Two-cell stage embryos were flushed out of oviducts and electrofused using cell fusion machine (CF-150 Hungary) to form one-cell stage tetraploids. Smad4Co/Co female mice were mated with Smad4+/Co;TTR-Cre males to produce diploid embryos with various genotypes, including Smad4Co/Co;TTR-Cre, Smad4Co/Co, and Smad4+/Co;TTR-Cre. Two tetraploids were co-cultured with one diploid embryo to aggregate overnight at 37°C. The blastocysts were then transferred to CD-1 foster mothers that were sacrificed at day 10.5-11.5 of pregnancy. Genotyping of embryos was performed as described above. As Smad4Co/Co;TTR-Cre embryos without fusion with tetraploid cells do not survive at these stages, any Smad4Co/Co;TTR-Cre embryos with normal appearance were considered as embryos that are completely rescued by tetraploid cells.
Whole-mount in situ hybridization
Whole-mount in situ hybridization was carried out as described [51]. Anti-sense RNA probes were synthesized using the DIG RNA Labeling Kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's recommendations.
Histology
Tissue was fixed in 10% neutral-buffered formalin (Sigma) at 4oC overnight, dehydrated through a graded alcohol series, xylene and paraffin, and then embedded in paraffin. Sections of 5 mm were prepared for H&E and antibody staining using regular procedures.
Embryoid body differentiation and TGF-β treatment
Embryoid body differentiation was performed as described [52]. Briefly, 6X106 cells were plated in 10-cm gelatin-coated dishes in the absence of feeder cells in ES cell medium. After 2 days in culture, small ES cell clumps were lifted off the plates by gentle trypsinization and transferred into suspension culture in ES cell medium without LIF in 10-cm bacteria plate. Cells were treated with 10ng/ml TGF-β1 (R&D Systems) after 48 hours, harvest samples at different time points as indicated in figure 7 for analysis.
X-gal staining
Embryos were stained in X-gal overnight at 37º C as described [53]. Embryos were washed in PBS twice after staining, then fixed in 4% paraformaldehyde for one hour, dehydrated through a graded alcohol series, treated with xylene, and embedded in paraffin. Five μm sections were prepared and counterstained with Harris hematoxylin according to standard procedures.
RT-PCR
Total RNA was extracted from cells using RNA STAT-60 (Tel-Test Inc.). RT was carried out using the Cell to cDNA kit from Ambion. RT-PCR was performed using the following primers for the genes of Hnf1, Gata4, Nodal, EndoA, Hex, T, and Lim1: Hnf1-F: 5'-GAA AGC AAC GGG AGA TCC TCC GAC-3', Hnf1-R: 5'-CCT CCT CCA CTA AGG CCT CCC TCT CTT CC-3'; Gata4-F: 5'-CAG CCC CTA CCC AGC CTA CAT-3', Gata4-R: 5'-GTG CCC CAG CCT TTT ACT TTG-3'; Nodal-F: 5'-CCC CAC AGG GTT AGG ACA CTC G-3', Nodal-R: 5'-TGC TGA AAG TGC TGT CTG TCT GCT C-3'; EndoA-F: 5'-TTC AGC AGC CGC TCG TTC AC-3', EndoA-R: 5'-TTC TCC TGA GTG CGC ACA GC-3'; Hex-F: 5'-GTT CTC CAA CGA CCA GAC CG-3',Hex-R: 5'-GGA GGG TGA ACA CTG CGA AC-3'; T-F: 5'-AGA AAG AAA CGA CCA CAA AGA TG-3', T-R: 5'-ATT TAT TTA TTT TTC CCT TGT CC-3'; Lim1-2F: 5'-CCA AGC GAT CTG GTT CGC AG -3', Lim1-3R: 5'-GAT AAC ACG CAT GTT GAG GC - 3'; Gapdh-1: 5′-ACAGCCGCATCTTCTTGTGC-3′; Gapdh-2: 5′-TTTGATGTTAGTGGGGTCTCGC-3′.
Acknowledgements
We thank Dr. T Mak for providing Smad4-/- ES cells, and members of Dr. Deng lab for their critical discussion of this work. This work was supported by the Intramural Research Program of the National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, USA.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Nakaya Y, Sheng G. Epithelial to mesenchymal transition during gastrulation: an embryological view. Dev Growth Differ. 2008;50:755-766
2. Chenoweth JG, McKay RD, Tesar PJ. Epiblast stem cells contribute new insight into pluripotency and gastrulation. Dev Growth Differ. 2010;52:293-301
3. Arnold SJ, Robertson EJ. Making a commitment: cell lineage allocation and axis patterning in the early mouse embryo. Nat Rev Mol Cell Biol. 2009;10:91-103
4. Tam PP, Loebel DA, Tanaka SS. Building the mouse gastrula: signals, asymmetry and lineages. Curr Opin Genet Dev. 2006;16:419-425
5. Chea HK, Wright CV, Swalla BJ. Nodal signaling and the evolution of deuterostome gastrulation. Dev Dyn. 2005;234:269-278
6. Perea-Gomez A, Rhinn M, Ang SL. Role of the anterior visceral endoderm in restricting posterior signals in the mouse embryo. Int J Dev Biol. 2001;45:311-320
7. Weinstein M, Yang X, Deng C. Functions of mammalian smad genes as revealed by targeted gene disruption in mice [In Process Citation]. Cytokine Growth Factor Rev. 2000;11:49-58
8. Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465-471
9. Massague J. TGF-beta signal transduction. Ann Rev Biochem. 1998;67:753-791
10. Weinstein M, Deng CX. Genetic disruptions within the murine genome reveal the numerous roles of the Smad gene family in development, disease, and cancer. In: (ed.) Dijke PT, Heldin CH. Smad signal Transduction: Smads in proliferation, differentiation and disease. US: Springer. 2006:151-171
11. Wu MY, Hill CS. Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev Cell. 2009;16:329-343
12. Gu Z, Nomura M, Simpson BB. et al. The type I activin receptor ActRIB is required for egg cylinder organization and gastrulation in the mouse. Genes Dev. 1998;12:844-857
13. Song J, Oh SP, Schrewe H. et al. The type II activin receptors are essential for egg cylinder growth, gastrulation, and rostral head development in mice. Dev Biol. 1999;213:157-169
14. Winnier G, Blessing M, Labosky PA. et al. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995;9:2105-2116
15. Zhou X, Sasaki H, Lowe L. et al. Nodal is a novel TGF-beta-like gene expressed in the mouse node during gastrulation. Nature. 1993;361:543-547
16. Moustakas A, Heldin CH. The regulation of TGFbeta signal transduction. Development. 2009;136:3699-3714
17. Howe JR, Roth S, Ringold JC. et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis [see comments]. Science. 1998;280:1086-1088
18. Hahn SA, Hoque AT, Moskaluk CA. et al. Homozygous deletion map at 18q21.1 in pancreatic cancer. Cancer Res. 1996;56:490-494
19. Friedl W, Kruse R, Uhlhaas S. et al. Frequent 4-bp deletion in exon 9 of the SMAD4/MADH4 gene in familial juvenile polyposis patients. Genes Chromosomes Cancer. 1999;25:403-406
20. Li W, Qiao W, Chen L. et al. Squamous cell carcinoma and mammary abscess formation through squamous metaplasia in Smad4/Dpc4 conditional knockout mice. Development. 2003;130:6143-6153
21. Bornstein S, White R, Malkoski S. et al. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. J Clin Invest. 2009;119:3408-3419
22. Yang G, Yang X. Smad4-mediated TGF-beta signaling in tumorigenesis. Int J Biol Sci. 2010;6:1-8
23. Xu X, Ehdaie B, Ohara N. et al. Synergistic action of Smad4 and Pten in suppressing pancreatic ductal adenocarcinoma formation in mice. Oncogene. 2010;29:674-686
24. Xu X, Kobayashi S, Qiao W. et al. Induction of intrahepatic cholangiocellular carcinoma by liver specific disruption of Smad4 and Pten in mice. Journal of Clinic Investigation. 2006 in press
25. Yang L, Mao C, Teng Y. et al. Targeted disruption of Smad4 in mouse epidermis results in failure of hair follicle cycling and formation of skin tumors. Cancer Res. 2005;65:8671-8678
26. Qiao W, Li AG, Owens P. et al. Hair follicle defects and squamous cell carcinoma formation in Smad4 conditional knockout mouse skin. Oncogene. 2006;25:207-217
27. Weinstein M, Monga SP, Liu Y. et al. Smad proteins and hepatocyte growth factor control parallel regulatory pathways that converge on beta1-integrin to promote normal liver development. Mol Cell Biol. 2001;21:5122-5131
28. Sirard C, de la Pompa JL, Elia A. et al. The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo. Genes Dev. 1998;12:107-119
29. Yang X, Li C, Xu X. et al. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc Natl Acad Sci U S A. 1998;95:3667-3672
30. Chu GC, Dunn NR, Anderson DC. et al. Differential requirements for Smad4 in TGFbeta-dependent patterning of the early mouse embryo. Development. 2004;131:3501-3512
31. Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70-71
32. Yang X, Li C, Herrera PL. et al. Generation of Smad4/Dpc4 conditional knockout mice. Genesis. 2002;32:80-81
33. de Souza FS, Niehrs C. Anterior endoderm and head induction in early vertebrate embryos. Cell Tissue Res. 2000;300:207-217
34. Beddington RS, Robertson EJ. Axis development and early asymmetry in mammals. Cell. 1999;96:195-209
35. Shawlot W, Deng JM, Behringer RR. Expression of the mouse cerberus-related gene, Cerr1, suggests a role in anterior neural induction and somitogenesis. Proc Natl Acad Sci U S A. 1998;95:6198-6203
36. Shawlot W, Behringer RR. Requirement for Lim1 in head-organizer function. Nature. 1995;374:425-430
37. Nagy A, Gocza E, Diaz EM. et al. Embryonic stem cells alone are able to support fetal development in the mouse. Development. 1990;110:815-821
38. Li C, Guo H, Xu X. et al. Fibroblast growth factor receptor 2 (Fgfr2) plays an important role in eyelid and skin formation and patterning. Dev Dyn. 2001;222:471-483
39. Tallquist MD, Soriano P. Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis. 2000;26:113-115
40. Hayashi S, Lewis P, Pevny L. et al. Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Gene Expr Patterns. 2002;2:93-97
41. Thorey IS, Meneses JJ, Neznanov N. et al. Embryonic expression of human keratin 18 and K18-beta-galactosidase fusion genes in transgenic mice. Dev Biol. 1993;160:519-534
42. Soudais C, Bielinska M, Heikinheimo M. et al. Targeted mutagenesis of the transcription factor GATA-4 gene in mouse embryonic stem cells disrupts visceral endoderm differentiation in vitro. Development. 1995;121:3877-3888
43. Abe K, Niwa H, Iwase K. et al. Endoderm-specific gene expression in embryonic stem cells differentiated to embryoid bodies. Exp Cell Res. 1996;229:27-34
44. Kwon GS, Hadjantonakis AK. Transthyretin mouse transgenes direct RFP expression or Cre-mediated recombination throughout the visceral endoderm. Genesis. 2009;47:447-455
45. Costa RH, Van Dyke TA, Yan C. et al. Similarities in transthyretin gene expression and differences in transcription factors: liver and yolk sac compared to choroid plexus. Proc Natl Acad Sci U S A. 1990;87:6589-6593
46. Srinivas S. The anterior visceral endoderm-turning heads. Genesis. 2006;44:565-572
47. Weinstein M, Yang X, Li C. et al. Failure of egg cylinder elongation and mesoderm induction in mouse embryos lacking the tumor suppressor smad2. Proc Natl Acad Sci U S A. 1998;95:9378-9383
48. Ang SL, Rossant J. HNF-3 beta is essential for node and notochord formation in mouse development. Cell. 1994;78:561-574
49. Meno C, Shimono A, Saijoh Y. et al. lefty-1 is required for left-right determination as a regulator of lefty-2 and nodal. Cell. 1998;94:287-297
50. Acampora D, Mazan S, Lallemand Y. et al. Forebrain and midbrain regions are deleted in Otx2-/- mutants due to a defective anterior neuroectoderm specification during gastrulation. Development. 1995;121:3279-3290
51. Riddle RD, Johnson RL, Laufer E. et al. Sonic-hedgehog mediates the polarizing activity of the zpa. Cell. 1993;75:1401-1416
52. Xiong JW, Battaglino R, Leahy A. et al. Large-scale screening for developmental genes in embryonic stem cells and embryoid bodies using retroviral entrapment vectors. Dev Dyn. 1998;212:181-197
53. Mansour SL, Goddard JM, Capecchi MR. Mice homozygous for a targeted disruption of the proto-oncogene int-2 have developmental defects in the tail and inner ear. Development. 1993;117:13-28
Author contact
![]() Corresponding author: Chu-Xia Deng, Phone: 301-402-7225; Fax: 301-480-1135; Email: chuxiadniddk.nih.gov
Corresponding author: Chu-Xia Deng, Phone: 301-402-7225; Fax: 301-480-1135; Email: chuxiadniddk.nih.gov