Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
INTRODUCTION
MATERIALS AND METHODS
RESULTS
DISCUSSION
Acknowledgements
CONFLICTS OF INTEREST
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2010; 6(6):599-612. doi:10.7150/ijbs.6.599 This issue Cite
Research Paper
Aberrant Cytoplasm Localization and Protein Stability of SIRT1 is Regulated by PI3K/IGF-1R Signaling in Human Cancer Cells
Vanessa Byles, Laura K. Chmilewski, Joyce Wang, Lijia Zhu, Lora W. Forman, Douglas V. Faller, Yan Dai ![]()
Cancer Research Center and Department of Medicine, Boston University School of Medicine, Boston, Massachusetts 02118, USA.
Received 2010-8-25; Accepted 2010-10-5; Published 2010-10-7
Abstract
SIRT1, an NAD-dependent histone/protein deacetylase, has classically been thought of as a nuclear protein. In this study, we demonstrate that SIRT1 is mainly localized in the nucleus of normal cells, but is predominantly localized in the cytoplasm of the cancer / transformed cells we tested. We found this predominant cytoplasmic localization of SIRT1 is regulated by elevated mitotic activity and PI3K/IGF-1R signaling in cancer cells. We show that aberrant cytoplasmic localization of SIRT1 is due to increased protein stability and is regulated by PI3K/IGF-1R signaling. In addition, we determined that SIRT1 is required for PI3K-mediated cancer cell growth. Our study represents the first identification that aberrant cytoplasm localization is one of the specific alternations to SIRT1 that occur in cancer cells, and PI3K/IGF-1R signaling plays an important role in the regulation of cytoplasmic SIRT1 stability. Our findings suggest that the over-expressed cytoplasmic SIRT1 in cancer cells may greatly contribute to its cancer-specific function by working downstream of the PI3K/IGF-1R signaling pathway.
Keywords: SIRT1, cytoplasm localization, protein stability, cancer cells, PI3K/IGF-1R
INTRODUCTION
Silent information regulator 1 (SIRT1) is a nicotinamide adenine dinucleotide (NAD)-dependent histone deacetylase (class III HDAC), which has been reported to be a crucial regulator in the pathophysiology of aging, metabolic disease and neurodegenerative disorders [1-6]. In addition, SIRT1 has been shown to be a cancer cell-specific growth factor [7-10]. The knockdown of SIRT1 expression halts the growth of various tumor cell lines, but does not impact normal human epithelial cell growth [7-9].
SIRT1 has classically been thought of as a nuclear protein that functions to regulate histones and non-histone substrates with its deacetylase activity [4-6, 11-16]. Many factors in the nucleus have been identified as SIRT1 substrates, such as p53 [17], FOXO [18, 19], E2F [20], HIC1 [21], Rb [22], AR [23] and WRN [24, 25]. These SIRT1 nuclear substrates are involved in various biological processes including stress-induced apoptosis, cell senescence, cell growth and DNA repair. Recent evidence suggests, however, that SIRT1 can also localize to the cytoplasm such as in murine pancreatic beta cells and rat cardiomyocytes [26-29]. In cancer cells, SIRT1 cytoplasmic localization remains largely unidentified, and it is unclear if SIRT1 localization changes during carcinogenesis.
In this study, we report for the first time that while SIRT1 is mainly localized in the nucleus of normal / primary cells, it is predominantly localized in the cytoplasm of cancer / transformed cells. Not only have we observed this localization pattern in prostate cancer cell lines, but we have also seen this in lung and breast cancer cells, transformed cell lines and prostate carcinoma tissues. We found that inhibition of PI3K/IGF-1R signaling or reduction of mitotic activity in cancer cells reduces the abundance and protein stability of cytoplasmic SIRT1. Moreover, we show that SIRT1 is required for PI3K induced growth in prostate cancer cells. These results indicate that aberrant cytoplasmic localization of SIRT1 is a cancer specific alteration; PI3K/IGF-1R signaling increases the stability of cytoplasmic SIRT1 and ultimately results in its aberrant cytoplasmic localization. Because SIRT1 is an important protein deacetylase with both histone and non-histone substrates, changes in its localization will greatly affect its targets and will likely translate further into important functional consequences for cell growth, survival and proliferation. Our findings suggest that the over-expressed cytoplasmic SIRT1 in cancer cells may greatly contribute to its cancer-specific function by working downstream of the PI3K/IGF-1R signaling pathway.
MATERIALS AND METHODS
Antibodies, plasmids and cell lines
SIRT1 antibody (sc-74504, sc-19857) and Lamin A/C (sc-20681) were purchased from Santa Cruz Biotechnology (Santa Cruz, California). α-Tubulin (T6074) and β-Actin (A-1978) were purchased from Sigma Aldrich (St. Louis, Missouri). Anti-BrdU (NMM1645458) and anti-SIRT1 (07-131, 05-707) were purchased from Upstate-Millipore (Billerica, MA). SIRT1 antibody (C05187) was purchased from Epitomics (Burlingame, CA). And anti-hSir2 antibody (856) [17] is a gift from Dr. Roy A. Frye (Pittsburgh Veterans Administration Medical Center, Pittsburgh, PA 15240). The SIRT1 RNAi vector (pSUPER.retro.puro-SIRT1) was generously provided by Dr. F. Picard (Laval University, Quebec, Canada).
DU145, PC3, MCF-7, H460 were maintained in 1x DMEM with 10% FBS. LNCaP, MRC5, BR3 and BR3neo cells were maintained in 1640 RPMI with 10% FBS. PZ-HPV-7 cells were maintained in Keratinocyte Serum-Free Medium (Invitrogen, catalog no. 10724) + Supplements (Invitrogen, catalog no. 37000-015). All cell lines were obtained from ATCC except the BR3 cell line, which was a gift from Penny A. Jeggo (University of Sussex, East Sussex, BN1 9RQ, UK). Prostate cancer tissues and normal prostate tissues were purchased from Cybrdi, Inc (Rockville, Maryland)
Isolation of nuclear and cytoplasmic protein
Nuclear and cytoplasmic proteins were isolated using NE-PER Nuclear and Cytoplasmic Extraction Reagents kit and following the detailed instructions provided (Pierce Biotechnology). Protease inhibitor tablets (Roche Diagnostics) and the phosphatase inhibitors, 10mM sodium pyrophosphate (Fisher Scientific) and 100 μM sodium orthovanadate (Fisher Scientific), were added to the CERI and NER extraction reagents prior to use.
Immunoblot
Cells were lysed with 1% Nonidet P-40 lysis buffer (1% NP40, 150mm NaCl, 50mM Tris-HCl) with protease inhibitor (Roche Diagnostics). The concentration of cell extract was measured by Bradford assay. The protein samples were subsequently separated on an 8% SDS gel and analyzed with antibodies anti-SIRT1, Lamin A/C, β-Actin and α-Tubulin.
Immunofluorescence Assay
Cells (103-104) were plated in a Chamber Slide and incubated for 24 hours. Cells were fixed with 4% paraformaldehyde, treated with 0.1% Sodium Citrate + 0.1%Triton 100 and with Image-iT FX Signal Enhancer (Invitrogen). The cells were blocked with 3% BSA, treated with anti-SIRT1 antibodies, then incubated with Alexa Fluor secondary antibodies (Invitrogen) and stained with DAPI.
BrdU immunostainning was performed with mouse anti-BrdU antibody. After being labeled with 20uM BrdU for 12 hours, standard indirect immunofluorescence was performed as described above, except for one hour DNase I treatment before the blocking step. Images were collected using a deconvolution microscope (Nikon deconvolution wide-field Epifluorescence system).
Establishment of stable SIRT1-knockdown cell lines
The pSUPER.retro.puro-SIRT1 or the pSUPER.retro vectors was used [30]. The Phoenix packaging cell line was transfected with the pSUPER.retro.puro-SIRT1 or the pSUPER.retro vectors separately, using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). After 48 hr, the medium containing retrovirus was collected, filtered, treated with polybrene, and transferred to DU145, PC3, LNCaP, H460 and PZ-HPV-7 cell cultures. Infected cells were selected with puromycin for isolation of stably infected colonies.
Cell viability assay
Cells were plated at a density of 5x103 cells per well on 96-well plates and cultured for 24hr, followed by treatment with Ly294002 (440202, CALBIOCHEM ) for 72hr. Then 20 µl MTS (G3580, Promega) was added to the medium for 1 h, and the absorbance was read at 490 nm in an Elisa plate reader.
BrdU based cell proliferation assay
2x104cells/well was seeded in a 96 well plate and BrdU (5mM) was added to each well for 24hr. The cells were fixed and stained with anti-BrdU antibody for 20hr at RT. Anti-mouse IgG HRP was added for 30 min and 100ul/well TMB peroxidase substrate was added for 30min. The integration of BrdU into the cell was determined at 450mM in the spectrophotometer microplate reader. The wells that did not receive cells and wells that contain cells but did not receive any BrdU reagent were included as controls. The detailed procedure can be found in the BrdU Cell Proliferation Assay (Cat# 2750) from Milipore (Billerica, MA)
Real-Time RT-PCR
Total cellular RNA was isolated using Trizol reagent (Invitrogen Life Technologies, Carlsbad, CA) according to the manufacturer's instructions. A two-step RT-PCR method was employed to synthesize single-stranded cDNA (SuperScript TM III First Strand kit, Invitrogen, 18080-051). Target genes were analyzed by real-time PCR using Applied Biosystems 7500 Fast Real-Time PCR System with SYBR Green I dye (Applied Biosystems, 4309155). The primers used were: SIRT1 forward: TAGTGACTGGACTCCAAGGCC, SIRT1 reverse: CAGTGTCATATCATCCAACTCAGGT; β-actin forward: GAGAAAATCTGGCACCACACC; β-actin reverse: ATACCCCTCGTAGATGGGCAC. PCR reactions were performed in triplicate.
RESULTS
SIRT1 is predominantly localized in the cytoplasm in cancer / transformed cells
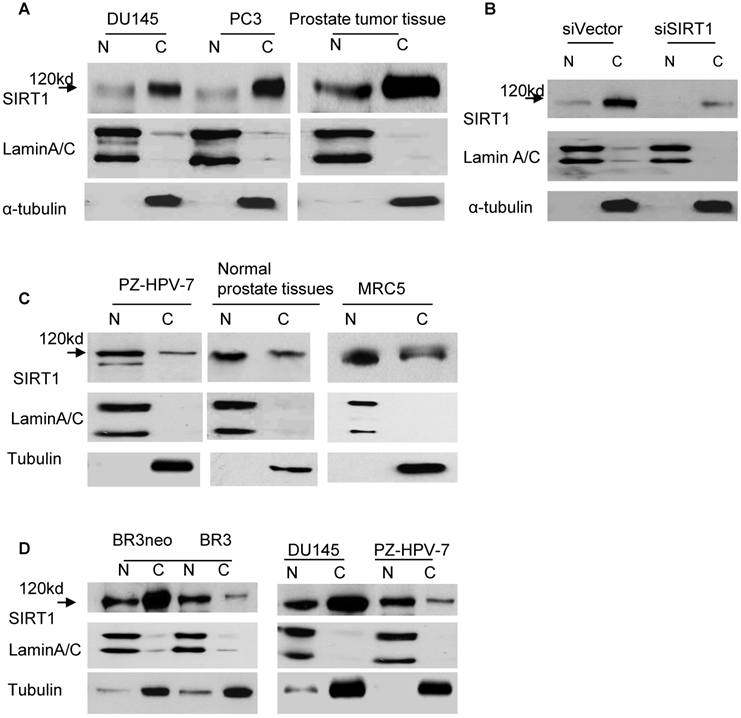
In order to clearly understand the localization pattern of SIRT1 in cancer cells, we performed immunoblots combined with cell fractionation to analyze the localization of SIRT1. Lung, breast and prostate cell lines, as well as tissues, were analyzed. The whole cell or tissue lysates were separated into nuclear and cytoplasmic fractions, and immunoblots were performed with anti-SIRT1 antibodies. Unexpectedly, we found that SIRT1 (MW=120kd) was predominantly localized in the cytoplasm, rather than in the nucleus of cancer cells lines including DU145, PC3, LNCaP and LNCaP-C4-2B prostate cancer cell lines, H460 lung cancer cells, MCF-7 breast cancer cell lines, and prostate cancer tissues (Figure 1A and data not shown). Fractionation efficiency was confirmed by immunoblot with the α-tubulin antibody that recognizes the protein in the cytoplasm, and the lamin A/C that binds the protein in the nucleus (Figure 1A). To ensure that this cytoplasmic localization of SIRT1 was not occurring as a result of antibody epitope specificity, we subsequently performed immunoblots with several other SIRT1 antibodies and similar localization patterns of SIRT1 were observed (Figure S1). In order to further confirm the specificity of localized cytoplasmic SIRT1, we sought to determine if cytoplasmic SIRT1 could be specifically knocked down via RNA interference. Our results demonstrated that both nuclear and cytoplasmic SIRT1 (MW=120kd) levels are greatly reduced by RNA interference of SIRT1 (siSIRT1) as compared to vector-transfected cells (siVector) in DU145, PC3, LNCaP and H460 cell lines (Figure 1B and data not shown).
Localization patterns of SIRT1 in cancer and normal cells. A. Cytoplasmic and nuclear fractions of DU145 and PC3 cells and prostate cancer tissues were isolated, the concentration of nuclear and cytoplasmic protein was measured by Bradford assay, the same amount of nuclear and cytoplasmic protein was subjected to SDS gel, and immunoblots were performed with anti-SIRT1, α-tubulin and LaminA/C antibodies. B. Cytoplasmic and nuclear proteins were isolated from DU145 cells transfected with RNA interference of SIRT1 (siSIRT1) or vector control (siVector). Equal amounts of nuclear and cytoplasmic protein were loaded, and immunoblots were performed with the same antibodies used in Fig.1A. C. The nuclear and cytoplasmic fractions of normal prostate cell PZ-HPV-7, normal lung cell MRC5 and normal prostate tissues were prepared. The same amount of nuclear and cytoplasmic protein was loaded, and immunoblots were performed with the same antibodies as Fig.1A. D. The nuclear and cytoplasmic fractions of primary and transformed human fibroblast cells BR3 and BR3neo, normal prostate cell PZ-HPV-7 and prostate cancer cell line DU145 were prepared. Equal amounts of nuclear and cytoplasmic protein were loaded and immunoblots were performed with the same antibodies as Fig.1A.
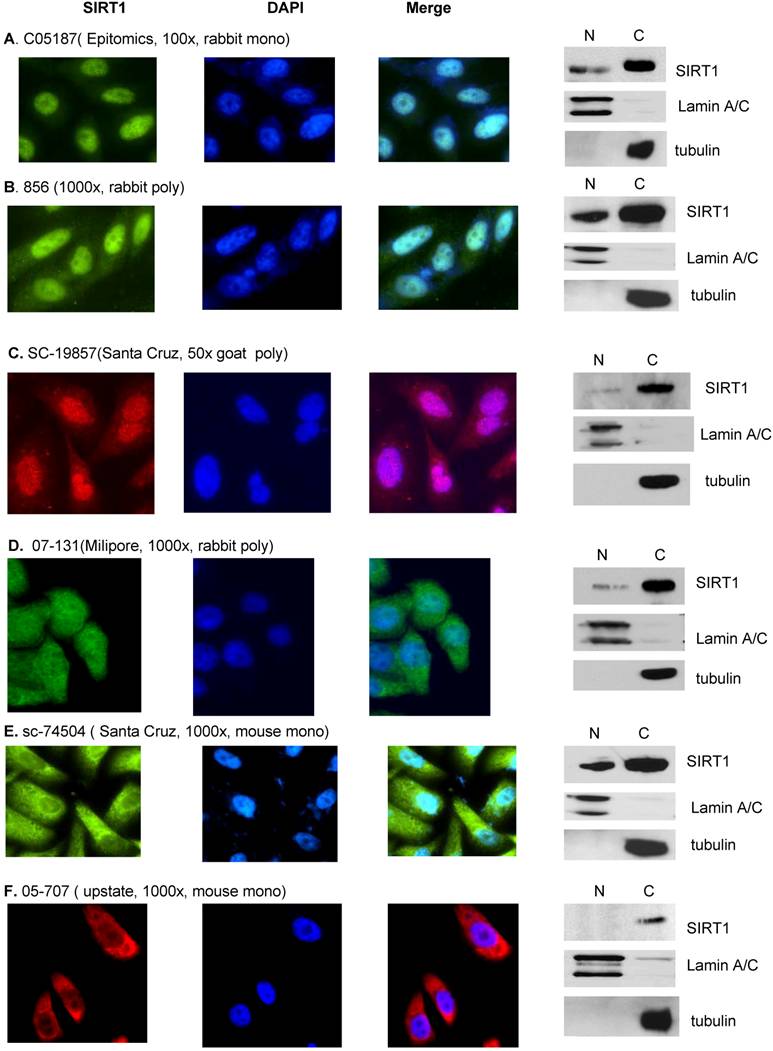
SIRT1 localization determined by both Immunofluorescence and immunobolt analysis. DU145 cells were cultured in the chamber slide with DMEM+10% FBS for the immunoflurorescence analysis (left panel). A. Anti-SIRT1 (C05187, Epitomics, 1:100, RT 1hr) and Alexa Fluor 488 goat anti-rabbit (Invitrogen, 200x, RT 1hr). B. Anti-hSir2 (856, 1:500, RT 1hr) and Alexa Fluor 488 goat anti-rabbit (Invitrogen, 200x, RT 1hr). C. Anti-SIRT1 (SC-19857, Santa Cruz, 1:50. RT 1hr) and Alexa Fluor 546 donkey anti-goat (Invitrogen, 100x, RT 1hr). D. Anti-SIRT1 (07-131, Milipore, 1:50, RT 1hr) and Alexa Fluor 488 goat anti-rabbit (Invitrogen, 200x, RT 1hr). E. Anti-SIRT1 (sc-74504, Santa Cruz, 1:1000, RT 1hr) and Alexa Fluor 488 goat anti-mouse (Invitrogen, 200x, RT 1hr). F. Anti-SIRT1 (05-707, upstate, 1:1000, RT 1hr) and Alexa Fluor 594 goat anti-mouse (Invitrogen, 100x, RT 1hr). Cytoplasmic and nuclear fractions of DU145 cells were isolated and same amount of nuclear and cytoplasmic protein were subjected to SDS gels, and immunoblots were performed (right panel). A. Anti-SIRT1 (C05187, Epitomics, 1:500, RT 1hr). B. Anti-hSir2 (856, 1:1000, RT 1hr). C. Anti-SIRT1 (SC-19857, Santa Cruz, 1:200, 4oC O/N). D. Anti-SIRT1 (07-131, Milipore, 1:500, 4oC O/N). E. Anti-SIRT1 (sc-74504, Santa Cruz, 1:1000, RT 1hr). F. Anti-SIRT1 (05-707, upstate, 1:1000, RT 1hr).
We subsequently analyzed the localization patterns of SIRT1 in normal cell lines and tissues. In contrast to cancer cell lines and tissues, we found SIRT1 predominantly localized in the nucleus of normal cells, including the prostate cell line, PZ-HPV-7, normal prostate tissues, the normal lung cell line, MRC5 (Figure 1C), and the normal human fibroblast cell line, BR3 (Figure 1D). These results indicate that the SIRT1 localization pattern differs between cancer and normal cells. SIRT1 is predominantly localized in the cytoplasm of cancer cells and in the nucleus of normal cells.
In addition, we compared the localization patterns of SIRT1 in transformed and primary cells. We established a transformed and immortalized fibroblast cell line, BR3neo, through transfection of SV40 Large T to a normal primary fibroblast cell line, 1BR3 [31]. As shown in Fig.1D, SIRT1 is predominantly localized in the nucleus in BR3, the primary cell line, whereas it is predominantly localized in the cytoplasm in the BR3neo transformed cells. These results indicate that the predominant cytoplasmic localization of SIRT1 is also associated with cell transformation.
The over-expression of SIRT1 in cancer cells is mainly attributed to the elevation of cytoplasmic SIRT1
After finding SIRT1 predominantly localized to the cytoplasm of cancer cells, we subsequently asked whether the previously documented overexpression of SIRT1 in cancer cells and tissues [32-36] is mainly attributed to increased cytoplasmic SIRT1. The levels of cytoplasmic and nuclear SIRT1 in cancer and normal cells were compared. The results demonstrated that the cytoplasmic SIRT1 level is robustly elevated in cancer cells compared to normal cells, whereas the nuclear SIRT1 is only marginally increased in cancer cells, such as prostate cancer cell line DU145 versus normal prostate cell PZ-HPV-7 and the transformed cell line BR3neo versus its parental normal cell BR3 (Figure 1D). Thus, the over-expression of SIRT1 in cancer cells is mainly attributed to the elevation of cytoplasmic SIRT1.
Increased mitotic activity of cancer cells promotes cytoplasmic localization of SIRT1
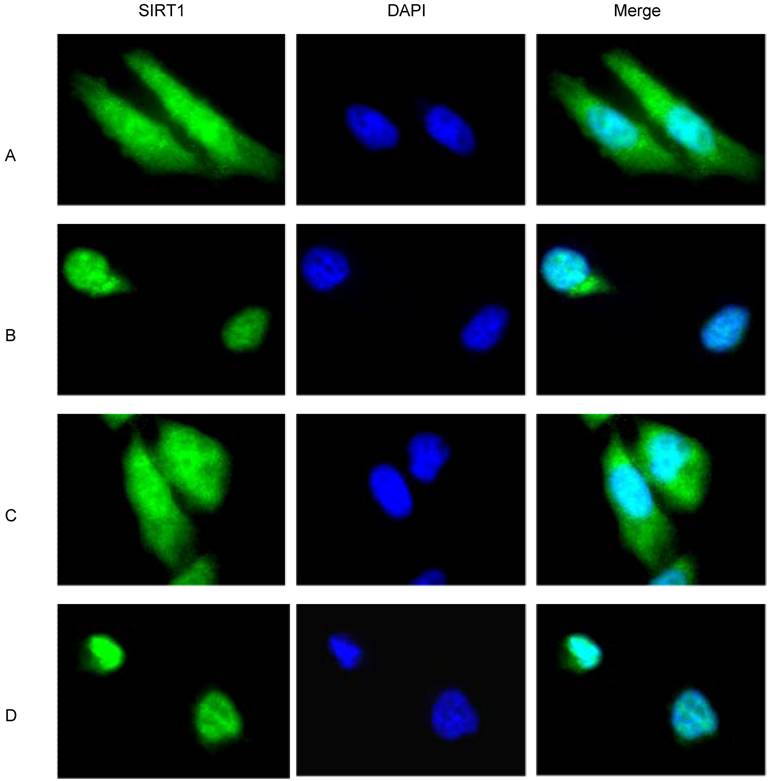
Because cancer cells present a higher level of mitotic activity compared to normal cells, the finding that cytoplasmic SIRT1 is robustly increased in cancer cells seems to suggest a correlation between the SIRT1 localization and the mitotic activity of cancer cells. Indeed, the doubling time of all of our analyzed cancer cells with predominant cytoplasmic localization of SIRT1 including DU145, PC3, H460 and BR3neo cells is less than 8 hours, which contrasts with the doubling time of 4 to 8 days for all the normal cells with predominant nuclear localization of SIRT1 including PZ-HPV-7, MRC5 and BR3 cells (data not shown). In order to understand whether the increased mitotic activity of cancer cells regulates the predominant cytoplasmic localization of SIRT1, we treated the cells with and without serum or hydroxyurea, which blocks the cell cycle, to study if reducing / blocking cancer cell proliferation / division would affect SIRT1 localization. Immunofluorescence assays showed that SIRT1 is localized in both the nucleus and cytoplasm under normal culture conditions (10% FBS) (Figure 2A). However, after serum depletion, we observed SIRT1 was largely diminished from the cytoplasm (Figure 2B). When the serum was replaced in the starved cells for 24hr, the depleted cytoplasmic SIRT1 exhibited restoration (Figure 2C). Hydroxyurea treatment, specifically the S phase and cell division, had an effect similar to that of the serum depletion treatment (Figure 2D). These results suggest that increased mitotic activity or growth signaling promotes the cytoplasmic localization of SIRT1, and conversely, the reduced mitotic activity or reduced growth signaling decreased cytoplasmic SIRT1.
Mitotic signaling regulates cytoplasmic localization of SIRT1. DU145 cells were cultured in the chamber slide with DMEM+10% FBS. The immunofluorescence was performed with anti-SIRT1 antibody (sc 74504, Santa Cruz 1:1000, RT 1hr) and Alexa Fluor 488 anti-mouse (Invitrogen, 1:200, RT 1hr). Nuclear protein was stained with DAPI (1:10,000). A. DU145 cells were cultured with DMEM+10% FBS. B. The DU145 cells that were cultured in the DMEM+10% FBS were shifted to serum-free DMEM for 48 hr. C. After culturing the DU145 cells in serum-free medium for 48hr, the medium was replaced with DMEM+10% FBS for 24hr. D. DU145 cells that were cultured in DMEM+10%FBS were treated with hydroxyurea for 24 hr.
Protein abundance rather than the translocation plays a dominant role in SIRT1 cytoplasm localization
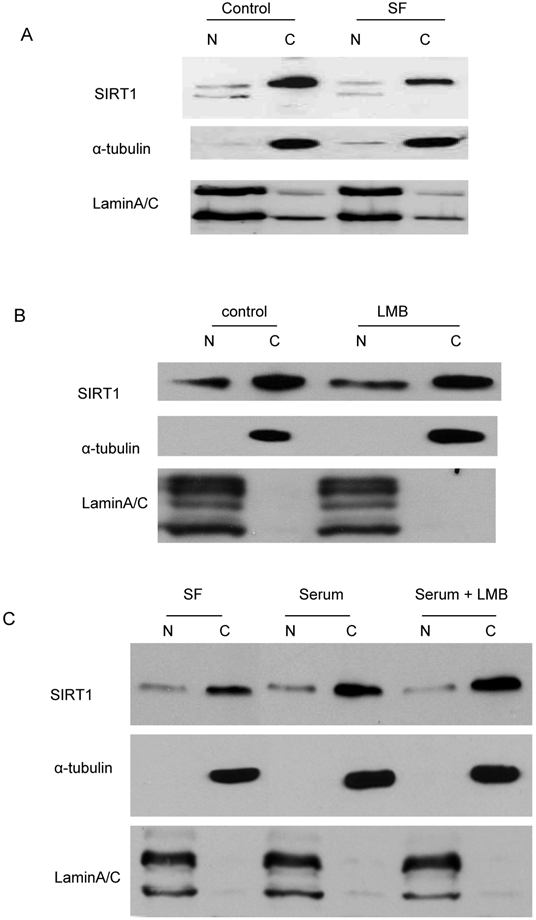
Since murine SIRT1, which has 84% homology with human SIRT1, has been reported to be a nucleo-cytoplasmic shuttling protein mediated by a NLS and NES-dependent mechanism [37], we asked whether the decreased cytoplasmic SIRT1 induced by serum depletion and hydroxyurea treatment in cancer cells had translocated to the nucleus. Immunoblot analysis was performed with the fractionated nuclear and cytoplasmic proteins, which were treated with serum depletion or hydroxyurea. The results showed no increase in nuclear SIRT1 when cytoplasmic SIRT1 is diminished by starvation (Figure 3A) or hydroxyurea treatment (data not shown). In addition, we treated the cells with leptomycin B (LMB), an inhibitor of the NES-dependent nuclear exporter, and found SIRT1 in cancer cells neither accumulated in the nucleus nor diminished from the cytoplasm in the presence of LMB (Figure 3B). Additionally, we treated the starved cells with serum with or without LMB and found that LMB treatment did not affect the cytoplasmic SIRT1 restoration by serum replacement (Figure 3C). These results suggest that predominant cytoplasmic localization induced by mitotic activity or growth signals is not due to SIRT1 translocation, but rather to the change in the abundance of cytoplasmic SIRT1.
Translocation does not play a dominant role in SIRT1 cytoplasm localization. A. The DU145 cells that were cultured in the DMEM+10% FBS were shifted to serum-free DMEM for 24 hr. The nuclear and cytoplasmic proteins were fractionated and equal amounts of nuclear and cytoplasmic protein were loaded, and immunoblots were performed with anti-SIRT1, Lamin A/C and tubulin antibodies. B. The DU145 cells were treated with either vehicle or 10nM LMB for two hours. The nuclear and cytoplasmic proteins were fractionated and equal amounts of nuclear and cytoplasmic protein were loaded and immunoblots were performed with anti-SIRT1, Lamin A/C and tubulin antibodies. C. The DU145 cells that were cultured in the serum-free DMEM were shifted to DMEM+10% FBS with or without LMB for 2hr. The nuclear and cytoplasmic proteins were fractionated and the same amounts of nuclear and cytoplasmic protein were loaded and immunoblots were performed with anti-SIRT1, Lamin A/C and tubulin antibodies.
PI3K/IGF-1R signaling regulates protein stability of cytoplasmic SIRT1
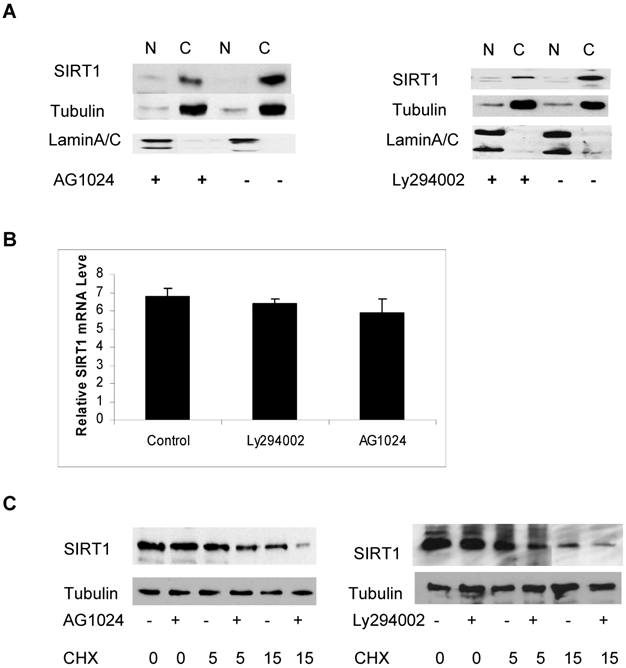
In order to identify which signaling pathway may regulate the abundance of cytoplasmic SIRT1, we treated the cells with various kinase inhibitors including inhibitors Ly294002 and AG1024 for PI3K and IGF-1R respectively, as well as Rottlerin, a PKC inhibitor and determined their effect on the abundance of cytoplasmic SIRT1. We found that treatment with Ly294002 and AG1024 reduced levels of cytoplasmic SIRT1, as determined by immunoblot (Figure 4A). The quantitative RT-PCR analysis shows no obvious change in SIRT1 mRNA levels in response to PI3K and IGF-1R inhibition (Figure 4B). We subsequently wanted to determine whether the reduced abundance of cytoplasmic SIRT1 was due to a protein stability change. The cytoplasmic and nuclear SIRT1 were isolated from the cancer cells, and either treated with Ly294002 or AG1024 or left untreated and then combined with cycloheximide (CHX) to inhibit protein synthesis, The SIRT1 protein level was then determined by immunoblotting. The results showed that inhibition of either IGF-1R or PI3K kinase activity reduced cytoplasmic SIRT1 stability (Figure 4C) but had no effect on the stability of nuclear SIRT1 (data not shown), as the half-life of cytoplasmic SIRT1 changed from 11hr to 6hr for IGF-1R inhibition, and 11hr to 4hr for PI3K inhibition (Figure 4C). These results suggest that PI3K/IGF-1R signaling regulates the protein stability of cytoplasmic SIRT1.
PI3K and IGF-1R regulates the protein stability of cytoplasmic SIRT1. A. DU145 cells were treated with or without AG1024 (4µM) (left panel) or Ly294002 (20µM) (right panel) for 12hr. The nuclear and cytoplasmic proteins were prepared and equal amounts of nuclear and cytoplasmic protein were loaded to SDS gel. The immunoblot was performed with anti-SIRT1, tubulin and LaminA/C antibodies. B. Transcript levels of SIRT1 was measured by quantitative RT-PCR analysis of RNA extracted from the cells treated with or without AG1024 (4µM) or Ly294002 (20µM) for 12hr. Transcript levels are expressed relative to β-actin transcripts. C. DU145 cells were pretreated with or without AG1024 (4µM) (left panel) or Ly294002 (20µM) (right panel) for 30minutes, then treated with cycloheximide (10μg/ml). The nuclear and cytoplasmic proteins were isolated after treatment with cycloheximide for 0, 5 and 15hr and the same amounts of nuclear and cytoplasmic proteins were subjected to SDS gel. The immunoblot was performed with anti-SIRT1 and anti-tubulin antibodies.
SIRT1 functions downstream of PI3K and regulates prostate cancer cell growth
PI3K and IGF-1R have been reported to play an important role in prostate cancer cell growth and survival [38, 39]. We found that PI3K can regulate cytoplasmic SIRT1 stability, suggesting that SIRT1 may play a role in PI3K-mediated cancer cell growth. We therefore firstly performed an MTS assay to study the effect of SIRT1 knockdown on PI3K inhibitor induced cell growth arrest. The results showed that Ly294002 treatment induced great inhibition in SIRT1-overexpressed prostate cancer cells, induced 50% growth inhibition in PC3 cells and 45% in LNCaP cells with wt SIRT1, while inducing only a 20% inhibition in SIRT1 knockdown cells (Figure 5A & 5B).
SIRT1 silencing attenuates PI3K inhibition-mediated prostate cancer cell growth arrest. Prostate cancer cell lines PC3 (5A) and LNCaP (5B) in which SIRT1 had been knocked down by siRNA expression (siSIRT1) or empty-vector-transfected cells (siVector) were exposed to the Ly292002 (20µM).Viable cells were quantitated at 72hr by MTS assay. The error bars represent the SEM. The SIRT1 knockdown efficiency was shown in 5A and 5B lower panel. C. The SIRT1 knockdown (siSIRT1) and empty-vector control (siVector) PC3 cells were treated with or without Ly292002 (20µM) for 24hr, then exposed to the BrdU (20uM) for another 24hr. The immunostaining was performed with anti-BrdU antibody (NMM1645458) and anti-mouse 594 secondary antibody. The images were taken with Nikon deconvolution wide-field Epifluorescence system. 100x. D. The SIRT1 knockdown (siSIRT1) and empty-vector control (siVector) PC3 cells were treated with or without Ly292002 (20µM) for 24hr, then exposed to the BrdU (4uM) for 24hr. The cells were fixed, immunostainned with anti-BrdU antibody and peroxidase conjugate secondary antibody; the peroxidase substrate was added and plates were read at 450nM. The error bars represent the SEM. E. MTS analysis for normal prostate cells PZ-HPV-7 cells. The experiment conditions are the same as 5A and 5B. The SIRT1 knockdown efficiency was shown in 5E lower panel.
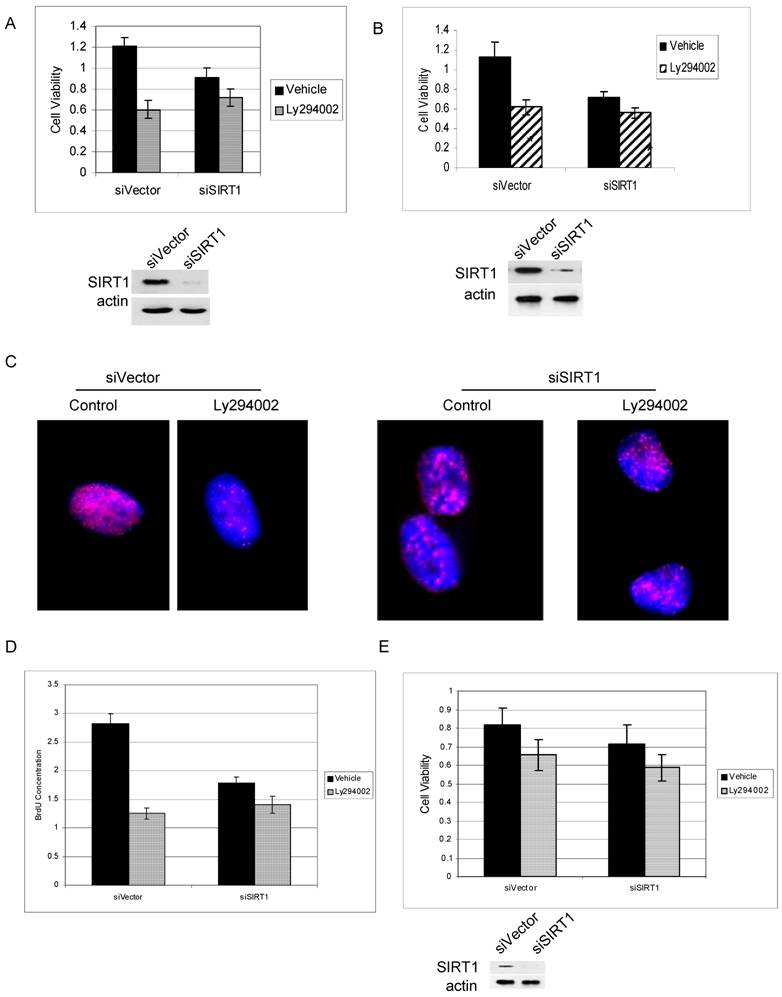
Similar results were seen using Brdu immunostaining (Figure 5C) and BrdU-based proliferation assay (Figure 5D). The results show that PI3K inhibition result in much more reduction of Brdu integration to the cells in SIRT1 wild type cells than in SIRT1 knockdown cells (Figure 5C & 5D). However, in normal prostate PZ-HPV-7 cells in which SIRT1 expression is lower and is predominantly in the nucleus, PI3K inhibition induce little growth arrest for both SIRT1 wt and SIRT1 knockdown cells (Figure 5E). These results suggest that SIRT1 is an important downstream target of PI3K in the regulation of cancer cell growth and survival, and overexpressed cytoplasmic SIRT1 in cancer cells may play an important role in this regulation.
DISCUSSION
In this study we demonstrated that SIRT1 is predominantly localized in the cytoplasm of cancer cells, while in normal cells, SIRT1 is predominantly localized in the nucleus. SIRT1 cytoplasmic localization in cancer cells is largely unidentified and it is unclear if SIRT1 localization has any changes during carcinogenesis. Our study represents the first identification that aberrant cytoplasmic localization is one of the cancer-specific alternations to SIRT1. This finding may suggest a new mechanism for SIRT1 function as a cancer-specific survival factor by targeting cytoplasmic proteins.
In contrast to its well-described role in the nucleus, the function of cytoplasmic SIRT1 is largely unknown. However, recent findings that some cytoplasmic proteins can be deacetylated by SIRT1 provide important insights into the function of cytoplasmic SIRT1. SIRT1 was found to enhance IGF-1 signaling by deacetylating ISH-2, which is a cytoplasmic protein [27]. SIRT1 also deacetylates cytoplasmic cortactin and promotes cancer cell migration [40]. In addition, SIRT1 was found to promote the activation of cytoplasmic kinases, including AMPK, Ras-MAPK, Erk and S6K1 [8, 41, 42]. Furthermore, SIRT1 was shown to regulate nucleo-cytoplasmic shuttling of transcription factors, including FOXO3a, FOXO1, and PGC-1 [43-47]. Because many known SIRT1 substrates, including p53 and NF-κB, also shuttle between the nucleus and cytoplasm, SIRT1 could regulate their function via cytoplasmic deacetylase activity. Recent study show that cytoplasmic SIRT1 can promote nerve growth factor (NGF)-induced neurite outgrowth [48]. In addition, we found that SIRT1 can form a complex with E-cadherin in the cytoplasm and regulate cell-cell adhesion (unpublished data). These results provide support for the association of cytoplasmic SIRT1 with kinase signaling, nucleo-cytoplasmic shuttling of the transcription factors, and with the proteins involved in cell adhesion and migration. SIRT1 may now prove to be more multifaceted in function due to its newly discovered predominant cytoplasmic localization, which may play a critical role in cancer cell development and progression by targeting cytoplasmic substrates.
In this paper we also elucidated the regulation of SIRT1 predominant cytoplasmic localization in cancer cells. Ectopically expressed murine SIRT1 has been reported as a nucleocytoplasmic shuttle protein mediated by a NLS and NES-dependent mechanism [37]. However in human cancer cells, our results show that the nucleocytoplasmic shuttle does not seem to play a significant role in the regulation of increased cytoplasmic localization of SIRT1. Although we cannot exclude that there may exist translocation for small amount of SIRT1 in the cancer cells, the nuclear-cytoplasm shuttle is not a major mechanism in the regulation of cytoplasmic localization of SIRT1 in cancer cells, as there is no obvious change in the nuclear SIRT1 level while cytoplasmic SIRT1 is altered. Instead, we found that PI3K/IGF-1R signaling regulates cytoplasmic SIRT1 stability and abundance, which suggests the PI3K/IGF-1R-mediated cytoplasmic SIRT1 stability plays a dominant role in the regulation of aberrant cytoplasm localization. The elevated SIRT1 protein levels in cancer cells may reflect abnormal stabilization induced by overactive PI3K/IGF-1R signaling.
Activated phosphoinositide 3-kinase (PI3K) is an important signaling molecule and acts as a key survival factor involved in the control of cell proliferation, apoptosis and oncogenesis [49]. Our results show that SIRT1 is required for PI3K mediated cancer cell growth. The tumor suppressor PTEN is an inhibitor of PI3K and its loss or mutation has been associated with many different types of cancer; in the absence of PTEN, PI3K is constitutively activated and strongly inhibits apoptosis and promotes cell survival and proliferation [39, 50]. Interestingly, SIRT1 has been shown to deactivate PTEN by deacetylation [51] and our results show that SIRT1 can be regulated by PI3K. It seems that there could be a positive feedback loop in the cell whereby SIRT1 is a downstream target of PI3K signaling, which may sequester SIRT1 in the cytoplasm to deacetylate PTEN and act on other non-histone substrates regulating cell growth and survival. Indeed we found SIRT1 is required for PI3K mediated cell growth in prostate cancer cells in which cytoplasmic SIRT1 is overexpressed, but inhibiting PI3K in the SIRT1 deficient cells, or normal prostate cells in which SIRT1 expression is lower and is predominantly in the nucleus, produces a much lower percentage of growth inhibition (Figure 5). These results suggest that overexpressed cytoplasmic SIRT1 in cancer cells may be an important downstream target of PI3K in the regulation of prostate cancer cell growth and survival. However, further investigation is needed to elucidate the detailed mechanism by which cytoplasmic SIRT1 is involved in P13K-mediated cell growth.
Finally, in this study we have clarified the cause of the controversial report for SIRT1 localization in cancer cells [13, 26, 28, 34]. Due to the fact that published data for SIRT1 localization is mainly generated from immunofluorescence analysis, we performed an immunofluorescence assay with the most available SIRT1 antibodies to study whether antibody specificity played a role in the controversial results. When we stained the same cells with a different anti-SIRT1 antibody, the immunofluorescence results show that while some antibodies only stain nuclear SIRT1 (C05187 Epitomics) [36], others were either specific for cytoplasmic SIRT1 (05-707 upstate) [26] or recognized both nuclear and cytoplasmic SIRT1 forms ( 856 [17, 52], sc-74504, and SC-19857 (Santa Cruz) and 07-131(milipore) (Figure S1). This data suggests that antibody specific epitopes play a dominant role in determining if SIRT1 is localized in the nucleus or cytoplasm while using the immunofluorescence assay, and may explain the controversial reports about SIRT1 localization. In order to eliminate the antibody specificity issues produced in the immunofluorescence experiments, we performed an immunoblot combined with nuclear-cytoplasmic SIRT1 fractionation to determine SIRT1 localization. All tested antibodies in the immunoblot assays have shown that SIRT1 is predominantly localized in the cytoplasm regardless of whether or not the antibody in the immunofluorescence assay recognized only nuclear SIRT1, only the cytoplasmic SIRT1, or both the nuclear and cytoplasmic SIRT1 (Figure S1). Thus, the immunoblot method can overcome the antibody specificity issues that arise in immunofluorescence assays. It is unclear why the SIRT1 antibody has such a dramatic effect in recognizing nuclear or cytoplasmic SIRT1 in immufluorescence. SIRT1 may form some structure or conformation with other proteins or organelles, thereby hiding the isotope and making recognition more difficult by some antibodies. It is also possible that some SIRT1 antibodies may have non-specific recognition for the nuclear proteins. Therefore, extra caution should be used when interpreting the immunoflurescence or IHC results regarding SIRT1 localization or expression. The nuclear and cytoplasmic fractionation combined with immunoblot with SIRT1 antibody is a good way to study SIRT1 localization.
Acknowledgements
We sincerely thank Dr. F. Picard (Laval University, Canada) and Dr. R.A. Frye (VA Medical Center, Pittsburgh, PA) for reagents. Thanks to Jenna Lovaas and Katie Enzer (Cancer Center, Boston University) for the proofreading of this manuscript. This work was supported by grants from the National Cancer Institute (1R21CA141036) (Y.D.), the Clinical and Translational Science Institute award of NIH (UL1RR025771)(Y.D.), the American Cancer Society (IRG-72-001-27-IRG) (Y.D.), the National Cancer Institute (CA101992) (D.V.F.) and Karin Grunebaum Cancer Research Foundation (Y.D.)
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest
References
1. Saunders L.R, Verdin E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene. 2007;26(37):5489-504
2. Yamamoto H, Schoonjans K, Auwerx J. Sirtuin functions in health and disease. Mol Endocrinol. 2007;21(8):1745-55
3. Zeng L, Chen R, Liang F. et al. Silent information regulator, Sirtuin 1, and age-related diseases. Geriatr Gerontol Int. 2009;9(1):7-15
4. Donmez G, Guarente L. Aging and disease: connections to sirtuins. Aging Cell. 2010;9(2):285-90
5. Haigis M.C, Sinclair D.A. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253-95
6. Deng CX. SIRT1, is it a tumor promoter or tumor suppressor? Int J Biol Sci. 2009;5(2):147-52
7. Ford J, Jiang M, Milner J. Cancer-specific functions of SIRT1 enable human epithelial cancer cell growth and survival. Cancer Res. 2005;65(22):10457-63
8. Ota H, Tokunaga E, Chang K. et al. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene. 2006;25(2):176-85
9. Liu T, Liu P.Y, Marshall G.M. The critical role of the class III histone deacetylase SIRT1 in cancer. Cancer Res. 2009;69(5):1702-5
10. Kojima K, Ohhashi R, Fujita Y. et al. A role for SIRT1 in cell growth and chemoresistance in prostate cancer PC3 and DU145 cells. Biochem Biophys Res Commun. 2008;373(3):423-8
11. Haigis M.C, Guarente L.P. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20(21):2913-21
12. Dai Y, Faller DF. Transcription Regulation by Class III Histone Deacetylases (HDACs)—Sirtuins. Translational Oncogenomics. 2008;3:137-149
13. Michishita E, Park J.Y, Burneskis J.M, Barrett J.C, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 2005;16(10):4623-35
14. Longo V.D, Kennedy B.K. Sirtuins in aging and age-related disease. Cell. 2006;126(2):257-68
15. Gasser S.M, Cockell M.M. The molecular biology of the SIR proteins. Gene. 2001;279(1):1-16
16. Pruitt K, Zinn R.L, Ohm J.E. et al. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet. 2006;2(3):e40
17. Vaziri H, Dessain S.K, Eaton EN. et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107(2):149-59
18. Huang H, Tindall D.J. Dynamic FoxO transcription factors. J Cell Sci. 2007;120(Pt 15):2479-87
19. Giannakou M.E, Partridge L. The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol. 2004;14(8):408-12
20. Wang C, Chen L, Hou X. et al. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat Cell Biol. 2006;8(9):1025-31
21. Chen W.Y, Wang D.H, Yen R.C. et al. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123(3):437-48
22. Wong S, Weber J.D. Deacetylation of the retinoblastoma tumour suppressor protein by SIRT1. Biochem J. 2007;407(3):451-60
23. Fu M, Liu M, Sauve A.A. et al. Hormonal control of androgen receptor function through SIRT1. Mol Cell Biol. 2006;26(21):8122-35
24. Li K, Wang R, Lozada E. et al. Acetylation of WRN protein regulates its stability by inhibiting ubiquitination. PLoS One. 2000;5(4):e10341
25. Uhl M, Csernok A, Aydin S. et al. Role of SIRT1 in homologous recombination. DNA Repair (Amst). 2010;9(4):383-93
26. Stunkel W, Peh B.K, Tan Y.C. et al. Function of the SIRT1 protein deacetylase in cancer. Biotechnol J. 2007;2(11):1360-8
27. Zhang J. The direct involvement of SirT1 in insulin-induced insulin receptor substrate-2 tyrosine phosphorylation. J Biol Chem. 2007;282(47):34356-64
28. Moynihan K.A, Grimm A.A, Plueger M.M. et al. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005;2(2):105-17
29. Chen I.Y, Lypowy J, Pain J. et al. Histone H2A.z is essential for cardiac myocyte hypertrophy but opposed by silent information regulator 2alpha. J Biol Chem. 2006;281(28):19369-77
30. Dai Y, Ngo D, Forman L.W. et al. Sirtuin 1 is required for antagonist-induced transcriptional repression of androgen-responsive genes by the androgen receptor. Mol Endocrinol. 2007;21(8):1807-21
31. Dai Y, Kysela B, Hanakahi L.A. et al. Nonhomologous end joining and V(D)J recombination require an additional factor. Proc Natl Acad Sci U S A. 2003;100(5):2462-7
32. Kwon H.S, Ott M. The ups and downs of SIRT1. Trends Biochem Sci. 2008;33(11):517-25
33. Hida Y, Kubo Y, Murao K, Arase S. Strong expression of a longevity-related protein, SIRT1, in Bowen's disease. Arch Dermatol Res. 2007;299(2):103-6
34. Kuzmichev A, Margueron R, Vaquero A. et al. Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc Natl Acad Sci U S A. 2005;102(6):1859-64
35. Bradbury C.A, Khanim F.L, Hayden R. et al. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia. 2005;19(10):1751-9
36. Huffman D.M, Grizzle W.E, Bamman M.M. et al. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007;67(14):6612-8
37. Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J Biol Chem. 2007;282(9):6823-32
38. Saikali Z, Setya H, Singh G, Persad S. Role of IGF-1/IGF-1R in regulation of invasion in DU145 prostate cancer cells. Cancer Cell Int. 2008;8:10
39. Lin J, Adam R.M, Santiestevan E, Freeman M.R. The phosphatidylinositol 3'-kinase pathway is a dominant growth factor-activated cell survival pathway in LNCaP human prostate carcinoma cells. Cancer Res. 1999;59(12):2891-7
40. Zhang Y, Zhang M, Dong H. et al. Deacetylation of cortactin by SIRT1 promotes cell migration. Oncogene. 2009;28(3):445-60
41. Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci U S A. 2007;104(17):7217-22
42. Huang J, Gan Q, Han L. et al. SIRT1 overexpression antagonizes cellular senescence with activated ERK/S6k1 signaling in human diploid fibroblasts. PLoS ONE. 2008;3(3):e1710
43. Anderson R.M, Barger J.L, Edwards M.G. et al. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell. 2008;7(1):101-11
44. Lan F, Cacicedo J.M, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008;283(41):27628-35
45. Han M.K, Song E.K, Guo Y. et al. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell. 2008;2(3):241-51
46. Subauste A.R, Burant C.F. Role of FoxO1 in FFA-induced oxidative stress in adipocytes. Am J Physiol Endocrinol Metab. 2007;293(1):E159-64
47. Smith J.S, Avalos J, Celic I. et al. SIR2 family of NAD(+)-dependent protein deacetylases. Methods Enzymol. 2002;353:282-300
48. Sugino T, Maruyama M, Tanno M. et al. Protein deacetylase SIRT1 in the cytoplasm promotes nerve growth factor-induced neurite outgrowth in PC12 cells. FEBS Lett. 2010;584(13):2821-6
49. Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal J.F. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets. 2008;8(3):187-98
50. Deocampo N.D, Huang H, Tindall D.J. The role of PTEN in the progression and survival of prostate cancer. Minerva Endocrinol. 2003;28(2):145-53
51. Ikenoue T, Inoki K, Zhao B, Guan K.L. PTEN acetylation modulates its interaction with PDZ domain. Cancer Res. 2008;68(17):6908-12
52. Langley E, Pearson M, Faretta M. et al. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. Embo J. 2002;21(10):2383-96
Author contact
![]() Corresponding author: Yan Dai, Ph.D, Cancer Research Center, Boston University School of Medicine, 72 E. Concord St, L913, Boston, MA 02118. Tel: (617) 638-5650; Fax: (617) 638-5609 ; E-mail: yandaiedu.
Corresponding author: Yan Dai, Ph.D, Cancer Research Center, Boston University School of Medicine, 72 E. Concord St, L913, Boston, MA 02118. Tel: (617) 638-5650; Fax: (617) 638-5609 ; E-mail: yandaiedu.