Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2010; 6(7):700-715. doi:10.7150/ijbs.6.700 This issue Cite
Research Paper
Bone morphogenetic protein 4 (BMP4) signaling in retinoblastoma cells
Maike Haubold1, Andreas Weise1, Harald Stephan2, Nicole Dünker1 ![]()
1. Institute for Anatomy, Department of Neuroanatomy, University of Duisburg-Essen, Medical Faculty, 45122 Essen, Germany
2. Division of Haematology and Oncology, Children's Hospital, University of Duisburg-Essen, 45122 Essen, Germany
Received 2010-10-5; Accepted 2010-11-22; Published 2010-11-24
Abstract
Bone morphogenetic proteins (BMPs) - expressed in the developing retina - are known to be involved in the regulation of cell proliferation and apoptosis in several tumor entities. The objective of this study was to determine the role of the BMP4 pathway in retinoblastoma cells, which are absent in a functional retinoblastoma (RB1) gene. BMP receptors were detected in all retinoblastoma cell lines investigated. A correct transmission of BMP signaling via the Smad1/5/8 pathway could be demonstrated in WERI-Rb1 retinoblastoma cells and application of recombinant human BMP4 resulted in an increase in apoptosis, which to a large extend is caspase independent. Cell proliferation was not affected by BMP4 signaling, although the pRb-related proteins p107 and p130, contributing to the regulation of the same genes, are still expressed. WERI-Rb1 cells exhibit elevated endogenous levels of p21CIP1 and p53, but we did not detect any increase in p53, p21CIP1or p27KIP1 expression levels. Id proteins became, however, strongly up-regulated upon exogenous BMP4 treatment. Thus, RB1 loss in WERI-Rb1 cells is obviously not compensated for by pRb-independent (e.g. p53-dependent) cell cycle control mechanisms, preventing an anti-proliferative response to BMP4, which normally induces cell cycle arrest.
Keywords: retinoblastoma, apoptosis, proliferation, cell culture, bone morphogenetic protein
Introduction
The expanding transforming growth factor-ß (TGF-ß) superfamily includes three main members: the TGF-ßs, the activins, and the bone morphogenetic proteins (BMPs) [1-3]. Members of the TGF-ß superfamily bind to two different types of serine/threonine kinase receptors termed type I and II receptors [1,2] and are known to play multiple roles during nervous system development including stimulation or inhibition of cell proliferation, triggering programmed cell death (PCD), and promoting neuronal survival [4-10]. BMPs are widely expressed in the embryonic nervous system including the developing retina [11-14].
BMP activity has been associated with developmental PCD in general [15,16] and with apoptosis in the prospective neural crest in particular [17]. Trousse et al. demonstrated that BMP4 mediates apoptotic cell death in early chick retinal development [11] and recently we showed that BMP4 triggers ganglion cell death in the developing murine retina [18]. BMPs including BMP4 have been shown to induce apoptosis i.e. in human myeloma cells [19-21]. Overexpression of BMP4 has been reported for melanoma, hepatocellular carcinoma, adenocarcinoma and colorectal cancer cell lines and is frequently accompanied by an enhanced migratory and invasive potential of the tumors [22-25]. Loss of TGF-ß receptors (TßRs) which normally induce growth inhibition and hypophosphorylation of the retinoblastoma gene product Rb represent one mechanism through which retinal progenitor cells escape from control and form retinoblastomas [26-28]. The inactivation of BMP signaling via mutation of the BMPR-IA causes familial juvenile polyposis, an inherited gastrointestinal cancer predisposition syndrome [29] Some human myeloma cell lines likewise lack BMPR-IA and BMPR-IB [30]. On the other hand, a significant increase in BMPR-IB levels was found in malignant human glioma cells [31].
Key proteins in the BMP signaling cascade are Smad and Id (inhibitor of differentiation) proteins. BMPRI activates the R-Smads Smad1, Smad5, and Smad8, which bind to Co-Smads and translocate into the nucleus [32,33]. The inhibitory I-Smads Smad6 and Smad7 block the activation of R- and Co-Smads by TGF-ß and BMPs [34]. In some cell lines, which turned out to be resistent to TGF-ß signaling, an overexpression of Smad 7 has been detected [35].
Via the Smad pathway BMP normally activates p21, a CDK (cyklin dependent kinase) inhibitor protein. P21 is able to hypophosphorylate and therefore activate the Rb1 tumor suppressor protein, which causes cell cycle arrest [36,37]. In retinoblastoma cell lines, however, the Rb gene is either deleted or non-functional. Thus, these cells might differ in their response to BMP4.
Id proteins are induced in response to the Smads [38]. These proteins inhibit bHLH (basic helix-loop-helix) transcription factors like E2A by forming heterodimeric complexes and consequently promote cell cycle progress [39] Furthermore, Id proteins induce apoptosis or function as survival factors, depending on the cell context [40]. The effect of BMP4 on expression of Id proteins in retinoblastoma cells, however, has not yet been investigated.
Despite the tremendous progress in delineating functional derangements of BMP pathways in carcinogenesis during the last decade, the biological significance of BMPs in human retinoblastoma has received little if any attention.
In the present study, we investigated the expression profiles of endogenous BMP4 and BMP receptors in different retinoblastoma cell lines compared to the healthy human retina. Changes in endogenous BMP4 expression itself, in the expression of BMP4 pathway molecules and/or loss or reduced levels of BMPRs might provide an additional mechanism through which retinoblastoma cells escape from apoptosis and cell cycle control. On the other hand, if expression levels of all signalling components are normal and BMPRs are still expressed in retinoblastoma cells, differences in response to BMP4 compared to other cancer cell lines investigated so far could reflect the role of the Smad pathway in the absence of functional pRb.
Materials and Methods
Collection of tissue and preparation of human retina
All investigations followed the tenets of the Declaration of Helsinki and were approved by the institutional human experimentation committee. Human retinas were obtained post-mortem from cornea donors. Consent for donation and research was given by the family and confidentiality was maintained. After removal of the cornea, the remainder of the eye was stored in a moist sterile container at 4°C until further use. Donor tissues were harvested within 12 - 48 hrs. Donors ranged in age from 20 years old to 85, with most being over 50 years old. A circumferential incision was made in the sclera of the globes approximately 6 mm behind the limbus. The posterior segment, including the vitreous, retina, choroid, and posterior sclera, was placed in a Petri dish. The vitreous was excised under a dissecting microscope. The retina was carefully separated from the retinal pigment epithelium and excised from the posterior segment and washed in 0.1 M phosphate buffer (pH 7.4). Freshly excised retinas were either immediately frozen in liquid nitrogen or fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) overnight at 4°C for immunocytochemistry.
Cell culture
We investigated 8 retinoblastoma cell lines: WERI-Rb1 [41], Y-79 [42], RBL-13, RBL-15, RBL-30, RB 247C3, RB 355 and RB 383 [43]. Cell lines were cultivated in Dulbecco`s modified Eagles medium (DMEM; Sigma) with 10% fetal calf serum (FCS; Cave), 100 U/ml penicillin (Invitrogen), 100 µg/ml streptomycin (Invitrogen), 4mM L-glutamine (Sigma), 50 µM ethandiol (Sigma) and 10 µg/ml insulin (Sigma) at 37°C, 10% CO2 and 80% humidity.
Cells were treated with different concentrations of recombinant human BMP4 (R&D Systems, Wiesbaden, Germany) diluted from a 100ng/µl stock solved in 0.1% BSA in 4mM HCl for different time spans as indicated in the figures and figure legends. If WERI-Rb1 cells were treated with BMP4 for more than 24 hrs, the medium - containing 10% FCS - was not changed. For detection of cell proliferation, 5 µM BrDU (Sigma) was added 6h before end of the culture. DNA and protein were as extracted from these cell lines using the protocols described below. For immunocytochemistry, cells were seeded on Poly-D-lysine coated (Sigma) coverslips and fixed with ice cold acetone for 10 min or cold 4% paraformaldehyde for 1h.
Semiquantitative RT-PCR assay
Retinas and cells were homogenized in 1 ml Trizol (Invitrogen) by manual disruption with an autoclaved plastic pestle and passing the tissue or cells, respectively through a 25G needle with a 1ml insulin syringe several times. The RNA was extracted according to the manufacturer's instructions (Invitrogen). Equal amounts of RNA were subjected to an RT-reaction using oligo(dt)20 primers (Invitrogen), employing the SuperScript III reverse transcriptase system (Invitrogen) and following the manufacturer`s instructions. An additional DNase I (Invitrogen) digestion was performed in advance to avoid cross contamination with genomic DNA. 1.5 µl of the RT-reaction product were taken for PCR with BMPR-IA, BMPR-IB, BMPR-II and GAPDH (Glyceraldehyde-3-phosphate dehydrogenase) specific primers already published [37] BMPR-IA_for: 5`-TAAGGTGACAGTACACAGGAACA-3`, BMPR-IA_rev: 5`-TCTATGATGGCAAAGCAATGTCC-3` (amplicon: 298 bp; Tm. 55°C); BMPR-IB_for: 5`-GTTGTAAATGCCACCACCATT-3`; BMPR-IB_rev 5`-GTCTGGTTTCTTGTCTTTTAT-3` (amplicon: 378 bp; Tm : 58°C); BMPR-II_for: 5`-TCCTCTCATCAGCCATTTGTCCTTC-3`; BMPR-II_rev: 5`-AGTTACTACACATTCTTCATAG-3` (amplicon: 457 bp; Tm 55°C); Id1_for 5´- CGGATCTGAGGGAGAACAAG - 3´; Id1_rev 5´- TGAGAAGCACCAAACGTGAC - 3´ (amplicon: 211bp; Tm 59°C); Id2_for 5´- CGTGAGGTCCGTTAGGAAAA - 3´; Id2_rev 5´- ATAGTGGGATGCGAGTCCAG - 3´ (amplicon: 237 bp; Tm 60°C); Smad7_for 5´- AGCAGGCCACACTTCAAACT - 3´; Smad7 _rev 5´- GTGTCCTGCCGATCATACCT - 3´ (amplicon: 219 bp; Tm 59°C); GAPDHfor 5`-CATCACCATCTTGGAGC-3`; GAPDHrev 5`-ATGACCTTGCCCACAGCCTT-3` (amplicon: 384 bp Tm 20°C; Invitrogen). PCR products were amplified using an expand high fidelity Taq polymerase (Roche) and employing the following conditions: (i) 5 min at 94°C; (ii) 35 to 37 amplification cycles (for GAPDH: 27 cycles); 1 min at 94°C, 1 min at 55°C (or 58°C), 30 sec at 72°C; (ii) 5 min at 72°C. The PCR products were separated on a 2% agarose gel and stained with ethidium bromide.
Quantitative Real-time PCR
Quantitative real time PCR analyses were performed using a 7300 Real-Time PCR System (Applied Biosystems). The following Taqman Gene Expression Assays (Applied Biosystems) were used: Id1: ID Hs00357821_g1; Id2: ID Hs00747379_m1; Id3: ID Hs00171409_m1; p21 (CDKN1A): ID Hs99999142_m1; p27: ID Hs00153277_m1; p53: ID Hs00153340_m1; p130: ID Hs00180562_m1; p107: ID Hs00765707_m1; BMP4: ID Hs00181626_m1. GAPDH (ID Hs99999905_m1) was used as an endogenous control. Real time PCR reactions were performed in duplicate and a total volume of 20 µl was applied to the following program: 2 minutes 50°C, 10 minutes 95°C followed by 40 cycles of 15 seconds 95°C and 60 seconds 60°C. Relative quantification was calculated by the 7300 Real-Time PCR System software (Applied Biosystems). Results represent mean values of at least three independent experiments.
Antibodies
For detection of BMPR-IA and BMPR-IB we used rabbit polyclonal BMPR-IA (H-60; Santa Cruz, sc-20736) and BMPR-IB (H-44; Santa Cruz, sc-25455) antibodies raised against amino acids 24-83 of BMPR-IA and 15-58 of BMPR-IB of human origin, respectively. BMPR-II was detected by a polyclonal BMPR-II (R&D, AF811) antibody produced in goats immunized with purified, NS0-derived, recombinant human BMPR-II extracellular domain. PC-3 cell lysate (Santa Cruz, sc-2220) was used as positive controls. For Smad translocation studies, a mouse monoclonal smad1 antibody (A-4; Santa Cruz; sc-7965) was used. For pathway studies, the following antibodies were used: a mouse monoclonal p21Waf1/Cip1 (DCS60) antibody (Cell Signaling #2946),, a rabbit polyclonal Bax antibody (Cell Signaling #2772), a rabbit polyclonal P-Smad1/5/8 antibody (Cell Signaling #9511). For BrdU immunocytochemistry a rat anti-BrDU antibody (Abcam; BU1/75 (ICR1); ab6326) was used.
Western blot analysis
For Western blot analysis, retinas and cells were homogenized in modified Ripa buffer (0.5% sodium deoxycholate / 0.1% SDS in PBS) or IPN150 buffer (50 mM TrisHcl / 150 mM NaCl / 5 mM MgCl2 / 0.1% NP40 / 1mM DTT / 0.1M NaVan / 0.1 M PMSF / 1M NaF / complete protease inhibitor cocktail), respectively. The extracts were fractionated in a 7.5 - 12% SDS-PAGE and transferred to a nitrocellulose membrane by tank blotting with 25 mM Tris/HCl, pH 8.7, 192 mM glycine, 10% methanol as transfer buffer. Membranes were blocked in phosphate buffer saline (PBS; pH 7.4) with 0.1% Tween-20 (PBS-T) and 5% bovine serum albumin (BSA) at 4°C overnight or for 30 min at room temperature. For the detection of BMPRs the membranes were incubated with the rabbit polyclonal BMPR-IA (1:100), the goat polyclonal BMPR-IB (1:500) and the goat polyclonal BMPR-II (1:200) primary antibodies in 3% milk powder/PBS/0.05% Tween-20 (BMPR-IA and B) or 3% BSA/PBS/0.05% Tween-20 (BMPR-II) for 2 hours at room temperature. For the detection of p21, Bax, P-Smad1/5/8, and Smad1 membranes were incubated with the primary antibodies (all 1:1000) in 5% milk powder/TBS/0.1% Tween-20 or TBS-T (Tris / NaCl / Tween-20) / 0.02% NaN3 / 0.025% BSA at 4°C with gentle shaking overnight. After three washes with TBS, the membranes were incubated for 1h at room temperature with the respective secondary antibody (horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-goat IgG (DAKO, Germany) at a dilution of 1:1500 (anti-rabbit HRP) - 1:10,000 (anti-goat HRP) in 2% milk powder/PBS/0.05% Tween-20. Signals were developed by the SuperSignal West Pico Chemiluminescent Substrate (Pierce) and visualized in a chemiluminescence imager (Chemi Genius, Bio Imaging Systems, Syngene). Densitometric measurements were performed by quantifying the density of the bands vs the local background using Bio Imaging System software (Syngene). Depending on the origin of primary antibody (rabbit vs goat), blots were re-stained with a goat polyclonal anti-actin antibody (I-19; Santa Cruz, sc-1616, 1:500) or a mouse monoclonal antibody to GAPDH (6C5; Abcam, ab8245, 1:1000) to verify equal protein loading in all lanes.
Immunocytochemistry
Immunostaining was performed using the same antibodies used for immunoblots. After a 1h blocking step with 10 % normal goat serum (NGS; for BMPR-IA and B) or 10% bovine serum albumin in phosphate buffer saline (PBS; pH 7.4; for BMPR-II), ten-micrometer cryo-sections of human retinas or retinoblastoma cells seeded and fixed on coverslips were incubated with the primary antibodies at a dilution of 1:200 (BMPR-IA and B) or 1:100 (BMPR-II), respectively. For detection of ganglion cells the sections were double stained with the ganglion cell-specific, nuclear marker Brn3a (Chemicon; MAB1585; 1:100). Following 1h permeabilisation with 3 mg/ml BSA/100 mM glycine/0.25% triton X-100, endogenous biotin was blocked using a biotin blocking kit (DAKO). After overnight incubation at 4°C with the respective makers, the reaction was visualized using the respective biotinylated IgGs (1:200) and streptavidin-conjugated Cy3 or FITC secondary antibodies (MoBiTec) at a dilution of 1:800. Sections were analyzed with a NIKON Eclipse E600 microscope equipped with epifluorescence, a NIKON CCD camera and NIKON Eclipsenet software. As controls, in all cases PBS was substituted for the primary antisera in order to test for nonspecific labeling. No specific cellular staining was observed when the primary antiserum was omitted.
For BrdU immunocytochemistry, cells were permeabilised in 1% triton X-100 for 30 min. To denature the DNA, cells were incubated in 2N HCl at 37°C for 60 min. The HCl was neutralized with sodium borate and unspecific staining was blocked by 1h incubation in PBS / 0.3% triton X-100 / 4% BSA / 5% NGS. Cells were incubated with the BrDU antibody diluted 1:1000 in PBS / 0.1% triton / 4% BSA / 1% NGS at 4°C overnight and the reaction was visualized with an goat anti rat FITC (1:1000) antibody. For immunolocalisation of Smad 1, cells were permeabilised in 100% cold MeOH for 5 min on ice, washed 3 times in PBS, blocked in PBS / 0.3% triton X-100 / 4% BSA / 5% normal goat serum (NGS) for 1h at room temperature and incubated with the SMad1 antibody diluted 1:200 in PBS /0.1 triton X-100 / 4% BSA / 1% NGS at 4°C overnight.
Cell cycle analysis
For cell cycle analysis by FACS, cells suspended in Deitch buffer (10 mM Tris-hydrochloride (pH 7.5) / 5 mM MgCl2) and stained with 100 µg/ml propidium iodide44 were analyzed in a Coulter EPICS XL flow cytometer using SYSTEM II Version 3.0 software (Beckman-Coulter, Krefeld, Germany). The percentage of cells present in the sub-G1 peak, representing apoptotic cells, was calculated after exclusion of cell doublets. The sum of cells in G2 and S phase was defined as the percentage of proliferating cells. Alternatively, proliferating cells and pyknotic nuclei were counted manually from BrdU- and 4',6-Diamidino-2-phenylindole (DAPI)-stained cells on coverslips, respectively. For this purpose, in a survey, at least 10 different sections of one coverslip and at least 1000 cells were counted and the number of apoptotic, clearly pyknotic nuclei (at least 10) or clearly BrDU-positive stained cells was determined.
Inhibition of endogenous caspase activity
In order to block endogenous caspase activity, Boc-D-fmk (Merck, Germany), a broad spectrum caspase inhibitor was used. WERI-Rb1 cells were seeded on Poly-D-lysine coated coverslips and pre-incubated in 38µM Boc-D-fmk for 30 min. Afterwards cells were incubated for 24h in DMEM supplemented with 40nM recombinant human BMP4 or BMP4 together with the caspase inhibitor. The number of pyknotic nuclei was assed by DAPI cell counts.
Results
Expression of BMPR subtypes in the human retina
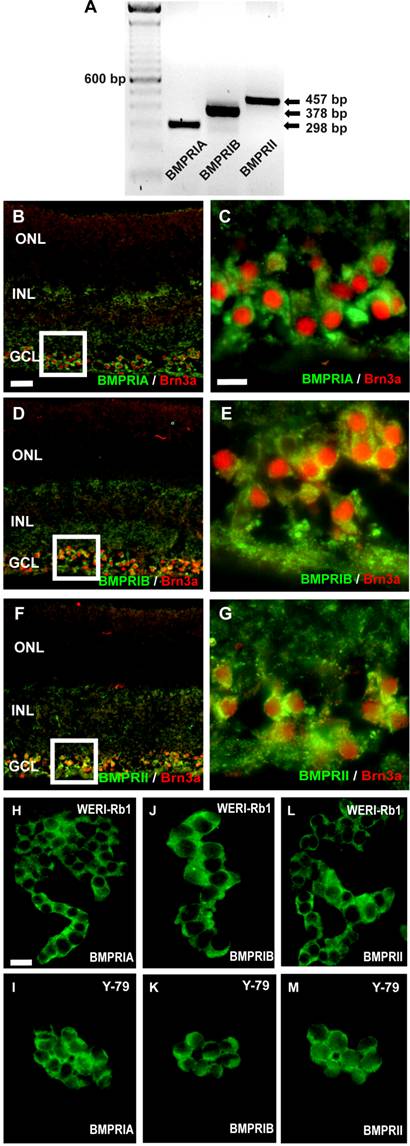
RNA was isolated from pooled human retinas of 5 cornea donors and cDNA was amplified by the use of specific primers, testing for BMPR-IA, BMPR-IB, or BMPR-II transcripts (Fig. 1A). Amplification products for all three BMPR subtypes were clearly visible after separation in an agarose gel. The cellular localization of BMPRs in the human retina was revealed in cryosections, stained with specific antibodies for the different receptor subtypes. Double labeling studies with the established ganglion cell marker Brn3 revealed that all BMPR subtypes are expressed on the surface of ganglion cells (Fig. 1C-G). No specific labeling for BMPRIA, BMPRIB, or BMPRII was detected in the photoreceptor layer or the inner nuclear layer.
Detection of BMPRs in the human retina (A-G) and retinoblastoma cell lines (H-M) by reverse transcription-polymerase chain reaction (RT-PCR; A) and immunohistochemistry (B-M). (A) PCR amplification products of the expected sizes were detected for each BMPR subtype. For orientation, the size of the products is indicated to the right and the prominent 600 bp line of a 100-bp DNA marker is indicated to the left. (B-G) Immunohistochemical detection of BMPR-IA, BMPR-IB, and BMPR-II in cryosections of the human retina. Immunohistochemical double labeling studies with the established ganglion cell marker Brn3a revealed that BMPR-IA is localized on the surface of ganglion cells (B,C). The same holds true for the BMPR-IB (D,E) and the BMPRII (F,G). The white insets in B,D,F are shown in higher magnification in C,E,G, respectively. Abbreviations: GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer. Scale bar in (B) = 50 µm also applies for D,F; Scale bar in (C) = 20 µm also applies to E,G. (H-M) Immunocytochemical labelling of BMPR subtypes in the retinoblastoma cell line WERI-Rb-1 (H,J,L) and Y-79 (I,K,M). Scale bar in (H) = 20 µm also applies to I-M.
Immunocytochemical detection of BMPRs on retinoblastoma cells
Immunocytochemical labeling of WERI-Rb1 and Y-79 cells with specific antibodies for the three BMPR subtypes BMPR-IA (Fig. 1H,I), BMPR-IB (Fig. 1J,K) and BMPR-II (Fig. 1L,M) revealed the expression of all BMPRs on the surface of these well characterized and frequently used retinoblastoma cell lines.
Transcripts for BMPRs in retinoblastoma cell lines
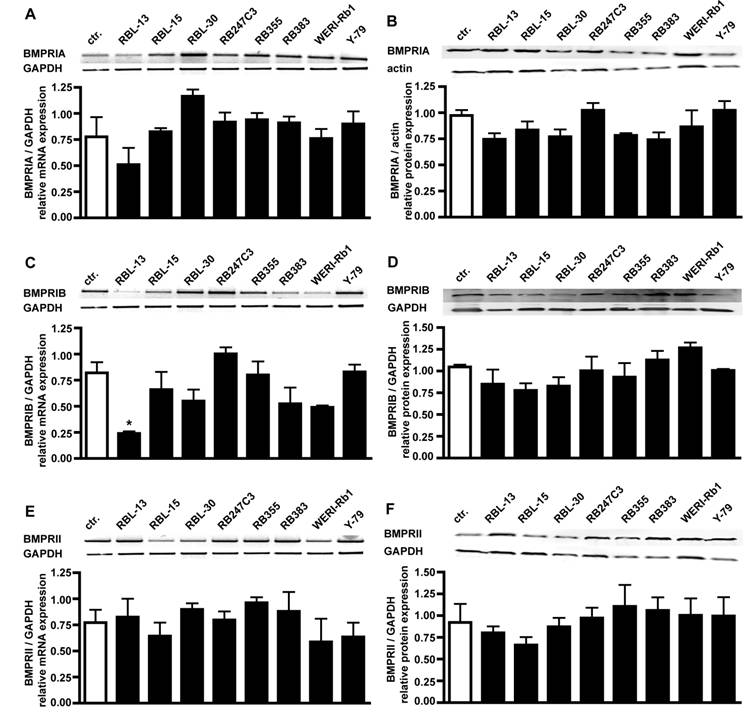
RT-PCR analysis for BMPR transcripts in 8 retinoblastoma cell lines confirmed that all BMPR subtypes are expressed in these cells (Fig. 2). Semi-quantification of the RT-PCR data showed that the levels of BMPR-IA (Fig. 2A), BMPR-IB (Fig. 2C) and BMPR-II (Fig. 2E) transcripts are not significantly reduced in retinoblastoma cell lines compared to the healthy human retina controls, except for the level of BMPR-IB in RBL-13 cells.
Comparison of BMPR-IA, BMPR-IB, and BMPR-II mRNA (A,C,E) and protein (B,D,F) expression levels in healthy human retina (ctr.) and eight retinoblastoma cells lines using reverse transcription-polymerase chain reaction (RT-PCR) and Western Blot analysis. The integrity of the cDNA was tested by amplification of the GAPDH transcript. Depending on the origin of the primary antibodies (α-rabbit BMPRIA; α-goat BMPRIB and BMPRII), either α-goat actin or α-mouse GAPDH were used as housekeeping enzymes to normalize equal protein loading. Quantification of the RT-PCR data revealed that the level of BMPR-IA (A), BMPR-IB (C) and BMPR-II (E) transcripts in retinoblastoma cell lines resembles the mRNA levels found in the healthy human retina control (ctr.), except for the level of BMPR-IB in RBL-13 cells. Quantification of the Western Blot data confirmed the presence of BMPR-IA (B), BMPR-IB (D), and BMPR-II (F) protein in the healthy human retina and all retinoblastoma cells investigated. Values are means of three independent experiments (n=3) ± s.e.m. * P<0.05; statistical differences compared to basal group, calculated by one way Anova test and Newman-Keuls Post test comparing all experimental groups.
Expression of BMPRIA, BMPRIB, and BMPRII in retinoblastoma cells
Western Blot analysis of BMPR expression in retinoblastoma cell lines confirmed their presence on protein level, revealing equal expression of BMPR-IA (Fig. 2B), BMPR-IB (Fig. 2D), and BMPRII (Fig. 2F) protein in the healthy human retina control and all retinoblastoma cell lines (Fig. 2).
Expression level of endogenous BMP4 in retinoblastoma cells
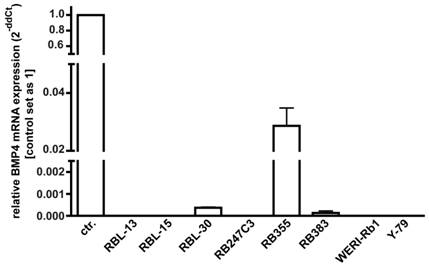
As overexpression of BMP4 has been reported for different cancer cell lines, changes in BMP4 levels might provide a mechanism through which retinoblastoma cells escape from apoptosis and cell cycle control, we investigated endogenous BMP4 expression by real-time PCR. Figure 3 summarizes the results of three independent PCRs, revealing that no endogenous BMP4 expression was detectable in 5 out of 8 retinoblastoma cell lines investigated. Only RB 355, RB 30, and RB 383 cells express endogenous BMP4 at detectable levels, which are, however, significantly lower compared to the normal human retina, used as a positive control.
Endogenous expression of BMP4 in retinoblastoma cell lines. The expression of BMP4 mRNA was analyzed by real-time PCR in the retinoblastoma cell lines RBL-13, RBL-15, RBL-30, RB 247C3, RB 355, RB 383, WERI-Rb1, and Y-79. Only RBL-30, RB 355 and RB 383 expressed BMP4 at detectable levels. Normal human retina mRNA was used as a positive control (ctr.), and its expression level was set as 1. Values are means of three independent experiments (n=3).
Effects of exogenous BMP4 application on apoptosis and cell proliferation of WERI-Rb1 cells
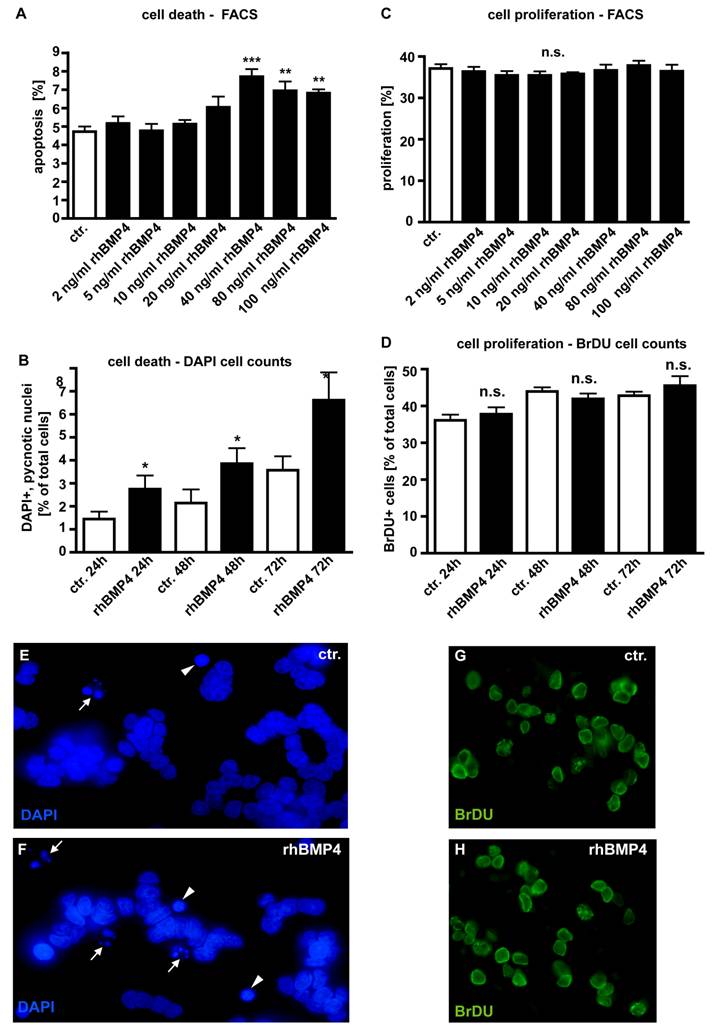
In dose-response experiments, WERI-Rb1 cells were treated with different concentrations of recombinant human BMP4 (Fig. 4) to test its effect on retinoblastoma cell apoptosis and proliferation. Apoptosis and cell proliferation levels were quantified by flow-cytometric analysis (Fig. 4A,C) and manual counts of DAPI and BrDU stained cells (Fig. 4B,D). No pro-apoptotic effect could be detected for exogenously applied BMP4 at concentrations lower than 40ng/ml (2, 5, 10, 20ng/ml). At a concentration of 40ng/ml, BMP4 treatment results in a significant increase in the number of pyknotic cells. Higher concentrations (80 and 100ng/ml) of recombinant BMP4 could not further increase this pro-apoptotic effect (Fig. 4A). Application of 40ng/ml recombinant human BMP4 resulted in a doubling in the number of DAPI-stained, apoptotic cells after 24hrs, 48hrs and 72hrs (Fig. 4B). No effects of BMP4 administration upon WERI-Rb1 cell proliferation could be detected irrespective of the concentration (Fig. 4C) and the culture time (Fig. 4D).
Effect of recombinant human BMP4 on the number of apoptotic (A,B;E,F) and proliferating (C,D;G,H) WERI-Rb1 cells. Data gained from FACS analyses after 24 hrs (A,C), manual DAPI (B) and BrDU cell counts (D) after 24, 48 and 72hrs, as indicated and DAPI stains on coverslips after 24h (E,F). Cells that were only treated with 0.1% BSA in 4mM HCl, the solvent for recombinant human BMP4, were taken as a control (ctr.) group. In dose-response experiments, retinoblastoma cells were treated with different concentrations of recombinant human BMP4. For FACS analysis cells were stained with propidium iodide and the percentage of cells present in the sub-G1 peak, representing apoptotic cells, was calculated. The sum of cells in G2 and S phase was defined as the percentage of proliferating cells. Administration of 40ng/ml recombinant BMP4 showed a significant pro-apoptotic effect in FACS analyses (A), inducing a doubling in the number of DAPI positive, pyknotic nuclei after 24, 48 and 72hrs (B), also clearly visible in images from DAPI stains of control (E) vs. BMP4 treated cells (F). White arrowheads (E,F) demarcate condensed nuclei of apoptic cells; small white arrows indicate apoptotic bodies in pyknotic cells. (C,D). FACS and BrDU cell counts revealed no effects of BMP4 administration on cell proliferation, neither after 24hrs nor after 48 or 72hrs. Values are means of three independent experiments (n=3) ± s.e.m. **P<0.01; *** P<0.001 calculated by one way Annova test and Newman-Keuls Post test comparing all experimental groups. (G,H) BrDU stains of cells, 24 hrs after BMP4 treatment, resemble those of control (ctr.) groups.
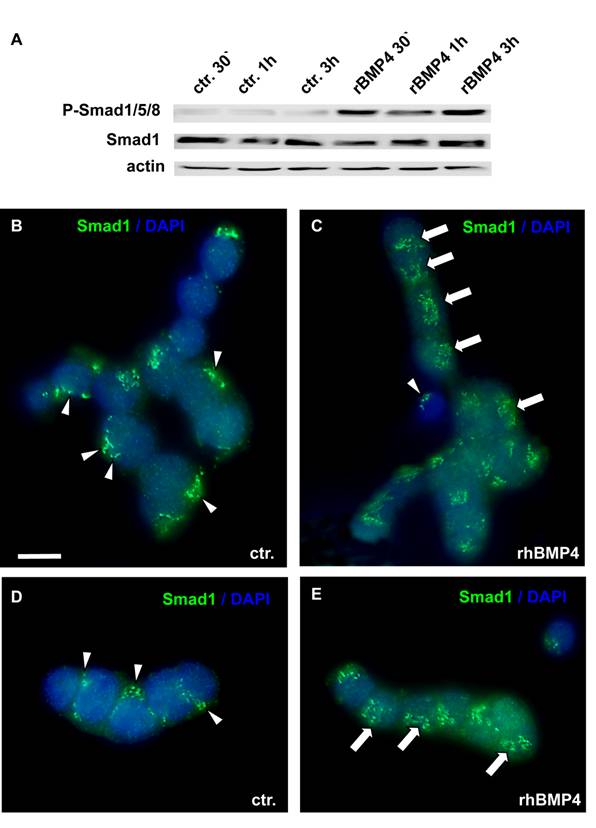
Transmission of BMP signaling
BMP actions on cell proliferation and apoptosis are normally mediated by Smad proteins. As WERI-Rb1 cells lack functional pRb, which is normally activated by the Smad pathway, we checked for a correct transmission of BMP4 signals in these cells. BMP4 treatment induced a significant up-regulation of phosphorylated Smad1, Smad5 and Smad8 levels, as determined by Western blots with a specific antibody, detecting all three Smads (Figure 5A). Phosphorylation of Smad1/5/8 was already detected 30min after BMP4 administration. Besides, an evident translocation of Smad1 into the nucleus was observed upon 1h BMP4 treatment, as visualized by immunocytochemical staining (Figure 5B-E). Thus, BMP4 signals are still transmitted correctly to the nucleus in WERI-Rb-1 cells. The dotted Smad1 staining in the control groups (Figure 5 B,D) appears to show discrete localization in the cytoplasm. If there, however, was a correspondence to organelles of the cytoplasm, the staining pattern should disappear when Smad1 translocates to the nucleus, but Smad1 staining remains punctate in BMP4 treated cells (Figure 5C,E).
Smad induction and nuclear translocation in WERI-Rb-1 cells after BMP4 treatment. (A) Western blot with a specific P-Smad1/5/8 antibody demonstrating a significant up-regulation of activated, phoshorylated Smad1/5/8 already 30min after addition of recombinant BMP4. Total Smad1 levels served as an internal control and remained constant. (B-E) Immunocytochemical staining of cells cultured for 1h in the presence of 40ng/ml recombinant BMP4 revealed that BMP4 treatment results in a significant translocation of Smad 1 into the nucleus (C,E), whereas the Smad staining is mainly cytoplasmic in the control (ctr.) group, only treated with 0.1% BSA in 4mM HCl, the solvent for recombinant human BMP4 (B,D). Arrowheads point to cytoplasmic Smad staining. Arrows demarcate Smad translocation to the nucleus. Scale bar in (B) = 50 µm also applies for C-E
Involvement of caspases and Bax in BMP4 mediated apoptosis
We studied the involvement of caspases, the major cell death executioners, in the execution of cell death in WERI-RB-1 cells. We determined that in WERI-RB-1 cells endogenous apoptosis is caspase independent since the number of pycnotic nuclei was not significantly reduced in cells treated for 24 hrs with Boc-D-fmk, a broad spectrum caspase inhibitor (Fig. 6A). BMP4 mediated cell death is at least partially caspase-dependent, as the number of pycnotic nuclei is marginally, but significantly lower after BMP4/BOC-D-fmk-inhibitor double treatment compared to BMP4 treatment alone (Fig. 6A). The predominant part of BMP4 mediated apoptosis in WERI-RB1 cells is, however, caspase independent and accordingly, levels of activated caspases appeared to be too low to be detected by Western Blot analyses, where we were not able to detect cleaved caspase-3, cleaved caspase-9, or cleaved caspase-8 although WERI-RB-1 cells express high levels of the respective pro-caspases (data not shown).
(A) Involvement of caspases in endogenous and BMP4 meditated cell death of WERI-RB-1 cells. Treatment of WERI-RB1 cells with Boc-D-fmk, a broad spectrum caspase inhibitor, revealed that endogenous apoptosis is caspase independent, while BMP4 mediated apoptosis in these retinoblastoma cells is at least partially caspase-dependent. Values are means of three independent experiments (n=3) ± s.e.m. *** P<0.001 calculated by one way Annova test and Newman-Keuls Post test. (B) Western Blot analyses of changes in Bax protein levels following 24hr BMP4 treatment of WERI-Rb1 cells. Levels of the pro-apoptotic protein Bax remain unchanged (B).
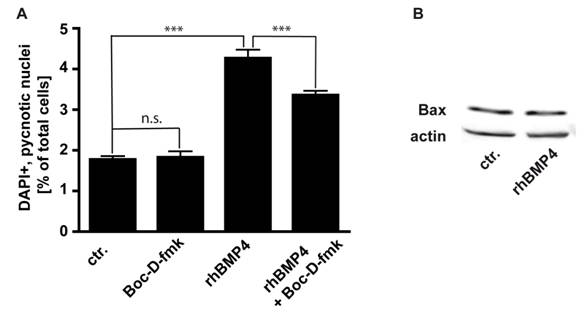
Pro-apoptotic Bcl-2 family members have been shown to be involved in the signaling of BMPs. Levels of the pro-apoptotic protein Bax remain, however, unchanged after BMP4 treatment (Fig. 6B),
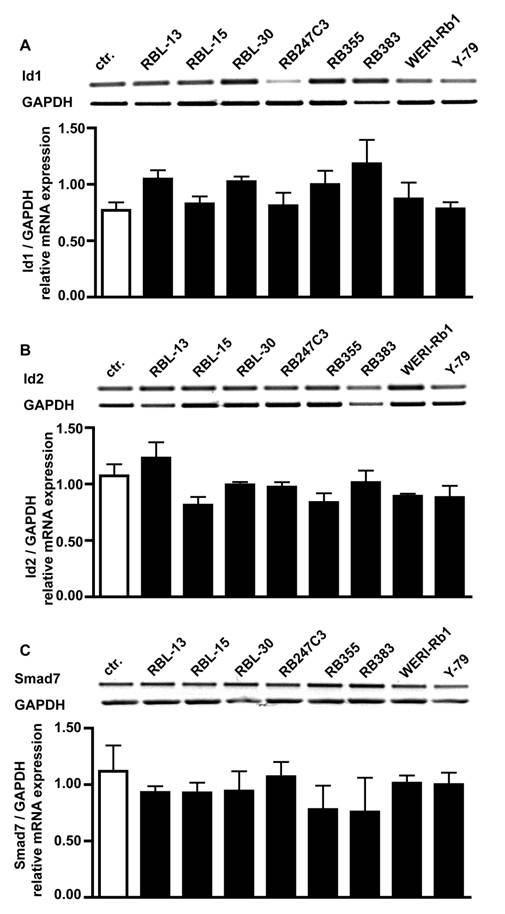
Expression levels of Id1, Id2 and Smad7 in retinoblastoma cells
One reason for the missing effect of BMP4 treatment on cell proliferation could be that Id proteins or the inhibitory Smad7 protein are constantly up-regulated in retinoblastoma cells. RT-PCR analysis for BMPR transcripts revealed, however, that compared to healthy human retina controls mRNA levels for Id1 (Fig. 7A), Id2 (Fig. 7B) and Smad7 (Fig. 7C) are not altered in all 8 retinoblastoma cell lines investigated.
Comparison of Id1, Id2 and Smad7 mRNA expression levels in healthy human retina (ctr.) and 8 retinoblastoma cells lines (RBL-13, RBL-15, RBL-30, RB 247C3, RB 355, RB 383, WERI-Rb1, Y-79) by RT-PCR. Quantification of RT-PCR data from three (smad7) to five (Id1, Id2) independent experiments revealed that the level of Id1 (A), Id2 (B) and smad7 (C) transcripts in retinoblastoma cell lines resembles the mRNA levels found in the healthy human retina control.
Induction of Id proteins and influence of BMP4 administration upon p53, p21, p27, p107 and p130 expression levels
It has been reported that Id proteins become concomitantly up-regulated upon BMP4 signaling in various cell lines, preventing the suppression of cell proliferation [34, 36]. In the present study, we likewise found mRNA expression levels of Id1, Id2 and Id3 to be significantly up-regulated 24hrs after administration of 40ng/ml recombinant BMP4 (Fig. 8A).
Induction of Id1-3 (A), endogenous expression of p21 and p53 (B), and expression of p53 (C), p21 (D), p27 (E), p107 (F) and p130 (G) upon recombinant BMP4 administration to cell cultures 24hrs after administration of 40ng/ml recombinant BMP4. Messenger RNA expression levels of Id1, Id2 and Id3 are significantly up-regulated in WERI-Rb-1 (A) upon a BMP4 administration, whereas no changes in p53, p21, p27, p107 and p130 expression level were detectable (C-G). Endogeneous levels of p21 and p53 are, however, elevated in WERI-Rb1 cells (B). Data were gained from real-time PCR analyses of three independent experiments. Cells that were only treated with 0.1% BSA in 4mM HCl, the solvent for recombinant human BMP4, were taken as a control (ctr.) group for A and C-G, healthy human retina was used as a reference in B and the expression level was set as 1, respectively.
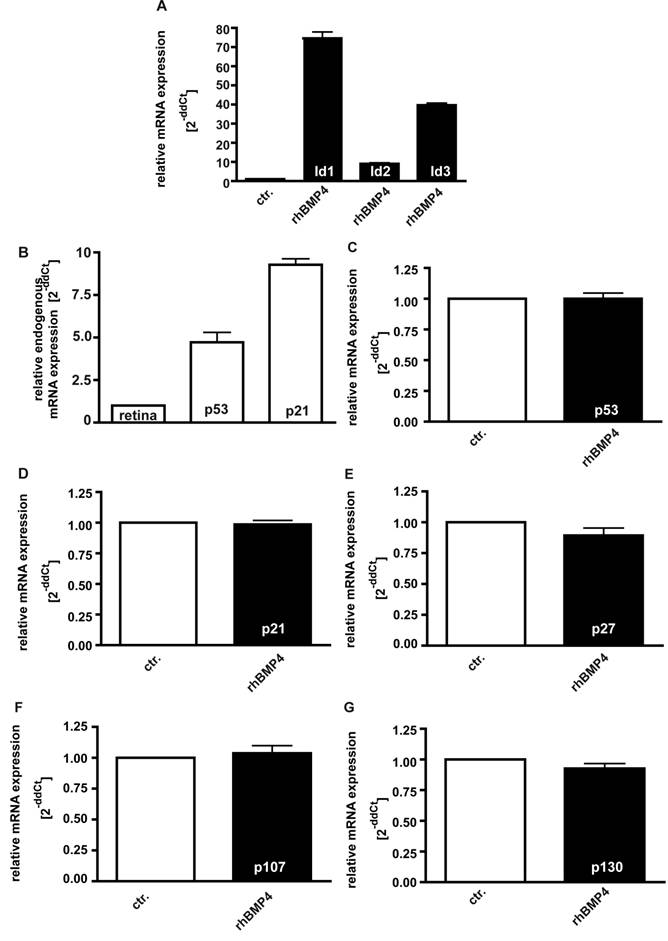
On the other hand, it has been observed that pRb-deficient hepatocytes exhibit elevated levels of p53 and p21CIP1and BMPs have been reported to cause an up-regulation of the CDK inhibitors p21 and p27 in human cancer cell lines [37]. Endogenous levels of the cell cycle regulator p53 and p21 are likewise significantly elevated in WERI-Rb1 cells compared to normal human retinas (Fig. 8B), but in the present study, we did not detect any increase in p53, p21, and p27 expression levels upon exogenous BMP4 treatment (Fig. 8C-E). The pRb-related proteins p107 and p130, which are still expressed in RB1 deficient WERI-Rb-1 cells (data not shown), are likewise not induced upon administration of recombinant BMP4 (Fig.8F,G).
Discussion
The expression of BMPRs has been described for the chick and murine retina by our group and others [12,14,18,45,46]. We showed recently that BMPRs are localized on retinal ganglion cells of the developing murine retina [18]. The particular expression of BMPR on human retinal ganglion cells observed in the study presented here can probably be explained in terms of BMP activity being associated with PCD and its involvement in retinal patterning and formation of retinotectal projections. We and others demonstrated that BMP4 mediates apoptotic cell death in the developing chick and murine retina, where BMP4 triggers ganglion cell death [18,45,46]. PCD is a physiological phenomenon and represents the most common mechanism to regulate the size of cell populations during development as well as in adult life. As the retina develops from a single layer of undifferentiated neuroectoderm extraneous cells unable to make functional neural connections are eliminated by apoptosis. As ganglion cells are the neurons that finally transmit the signal to the brain, it is reasonable to assume that their number requires an especially tight regulation, which is obviously mediated by BMP4 signaling. This might explain why especially ganglion cells express the required receptors. Besides, BMPs have been shown to play a key role in dorso-ventral patterning of the retina and formation of retinotectal projections (for review see: [46]). In this context, it has been reported that targeted deletion of BMPRIB in mice resulted in failure of ventral ganglion cells to enter the optic nerve (for review see: [46]).
BMP7 expression had been examined in the adult human retina [46], but until now, BMP receptor expression in the human eye was only systematically studied in the cornea, conjunctiva and the trabecular meshwork and optic nerve head in the context of glaucoma [47-50]. Yamada et al. investigated BMPR expression in malignant glioma tumors [31], but to our knowledge, the present study is the first investigating BMPR expression in retinoblastoma cell lines and comparing expression levels of healthy retinal neurons with those in pathologically altered retinoblastoma cells.
It has been shown that retinoblastoma cells (i.e. Y-79 and WERI-Rb1) are resistant to TGF-ß activity due to the absence of TGF-ß binding [27]. Horie et al. demonstrated that Y-79 and WERI-Rb1 retinoblastoma cells do not express TßRII mRNA, whereas TßRI mRNA was detected [28]. Loss of TßRII represents one mechanism through which retinoblastoma cells escape from growth control [26]. It has been shown that some human myeloma cell lines lack BMPRIA and BMPRIB [19]. Thus, one might have speculated that retinoblastoma cells likewise lack BMPRs. In the present study we, however, demonstrated that all BMPR subtypes are expressed on all 8 retinoblastoma cell lines investigated, including Y-79 and WERI-Rb1. All retinoblastomas develop from the immature retina and have mutations of both RB alleles, but it is reasonable to assume that mutations in additional genes contribute to promotion and progression of this tumor. A mutational loss or reduced levels of BMPRs, found in other tumor cells, would provide an additional mechanism through which developing retinal cells escape from apoptosis control and form retinoblastomas. If the fact that retinoblastoma cells show no BMPR loss but exhibit full BMPR equipment and are thus receptive to BMP4 stimuli provides a mechanism by which BMPs excert inhibitory or stimulatory effects on invasive tumor growth needs to be determined. Along this line a positive correlation between the malignancy grade of glioma and BMPR expression has, however, been found as BMPRIB is progressively expressed in malignant glioma tumors compared with low-grade astrocytomas and gliosis [31], suggesting that tumor cells may be more responsive to the action of BMPs.
It has been reported that BMP4 is overexpressed in melanoma cell lines [22]. BMP4 overexpression followed by enhanced invasiveness was likewise reported for hepatocellular carcinoma, adenocarcinoma and colorectal cancer [23-25]. BMP2 expression, by contrast, is not prominent in melanomas and was only detected in two out of nine cell lines analyzed [22]. Comparable to latter results, we could only detect BMP4 expression in three out of eight retinoblastoma cell lines in the present study. As we and others demonstrated that BMP4 induces apoptosis in normal retinal cells [11,18] and inhibits cell proliferation in cancer cells [20,21], intrinsic down-regulation of endogenous BMP4 might provide an additional mechanisms by which retinoblastoma cells escape from apoptosis and cell cycle control.
It is not known if the impairment of pRb would alter the growth inhibitory potential of BMP4. Thus, we asked whether RB1-deficiency would affect responses to BMP4, which induces apoptosis and cell cycle arrest in other cell lines. It has been shown that BMP4 treatment of tumor cells increased apoptosis in OH-2, IH-1 myeloma cells and undifferentiated human cancer cells [20,30]. Results from our cell culture experiments likewise demonstrate an increase in apoptosis of WERI-Rb-1 retinoblastoma cells after BMP4 treatment. While analysing retinoblastoma cell lines, counting DAPI-positive pyknotic nuclei thereby provides an objective, reliable, but much more sensitive method evaluating subtle yet statistically highly significant apoptotic effects.
Bcl-2 family members play a critical role in the activation of cell death executors [51,52]. Members of the Bcl-2 family comprise pro-apoptotic (e.g. Bax, Bak) and anti-apoptotic (e.g. Bcl-2, Bcl-XL) proteins. It has been reported that BMP4 induces the expression of Bax in myeloma and hybridoma cells, whereas BMP2 has no effects on the expression of Bax [53,54]. Lagna et al. reported, however, that BMP4 mediates apoptosis of pulmonary artery smooth muscle cells by the activation of caspases, whereby Bax levels remained unchanged [55]. Results of our experiments showed that BMP4-mediated apoptosis in WERI-Rb-1 cells is mainly caspase independent and accordingly, Bax expression levels remained unchanged.
BMP4 inhibits proliferation in many cancer cell lines, e.g. in multiple myeloma cells and tumor-initiating precursors of glioblastoma cells [20]. In our experimental set up, cell proliferation was, however, not affected by BMP4 treatment irrespective of the culture time and concentration and one might think of different reasons for this result.
On the one hand, retinoblastoma cell lines lack the RB1 gene, but we demonstrated that the pRb-related proteins p107 and p130, contributing to the regulation of the same genes [56] are still expressed, at least in WERI-Rb1 cells. Besides, Rivadineira et al. reported that factors other than pRb can mediate E2F-dependent down-stream effects to regulate cell cycle control [57]. Sheahan et al. observed elevated levels of p21CIP1 and p53 in pRb-deficient hepatocytes and identified a pRb-independent-p53-dependent effector pathway of E2F inhibition in these cells [58]. In the present study, WERI-Rb1 cells likewise exhibited elevated endogenous levels of p21CIP1 and p53. In human cancer and non-cancer cell lines, BMPs - including BMP4 - have been reported to cause an up-regulation of p21CIP1 and p27KIP1, leading to cell cycle arrest [33,34,59-63]. This induction is mediated via the smad signalling pathway, which, according to our results, is still intact in WERI-Rb1 cells. We, however, could not detect changes in p21CIP1, p27KIP1or p53 expression levels in RB1 deficient WERI-Rb1 cells upon exogenous BMP4 administration, presumably due to the significantly elevated endogenous levels in these cells and/or concomitant Id induction (see below). We likewise did not detect any BMP4-dependent regulation of p107 or p130 levels. Thus, in WERI-Rb1 cells pRb loss is obviously not compensated for and prevents an anti-proliferative response to BMP4, which normally induces cell cycle arrest.
One might also have speculated that inhibitory Smad7 protein levels are constantly up-regulated in retinoblastoma cells as it has been shown for various cancers. Smad7 is overexpressed in 50% of human pancreatic cancers and it has been shown that these cancer cells are resistant to TGF-ß with respect to growth inhibition induction [35]. We, however, could not detect any changes in smad7 expression levels when comparing retinoblastoma cells to healthy human retina controls.
On the other hand, it has been reported that Id proteins, known to play a critical role in cancer [64,67], define the potency of cell proliferation responses to BMP [68]. Elevated levels of Id proteins have been found in tumor cell lines including neuroblastoma cells [64,69,70]. In the present study, we did not detect elevated endogenous Id levels in any of the retinoblastoma cell lines investigated. Id proteins become concomitantly up-regulated upon BMP4 signaling, e.g. in endothelial, epithelial and breast cancer cells [34,36,71], inhibiting the expression of the CDK inhibitor p21 [72]. BMP7 has been shown to highly induce p21 expression, but despite this, BMP7 only weakly suppresses epithelial cell proliferation as Id2 becomes concomitantly induced by BMP7 [37]. In the present study, we likewise found transcript levels of all three Id isoforms to be significantly up-regulated upon BMP4 administration in HER and WERI-Rb1 cells. Thus, the fact that in our experiments BMP4 administration had no effect on cell proliferation in the retinoblastoma cell line WERI-Rb1 can be interpreted in terms of a compensatory balance between high endogenous levels of the CDK inhibitor proteins p53 and p21, normally causing BMP4- induced cell cycle arrest and a strong induction of pro-proliferative Id proteins.
Acknowledgements
The authors would like to thank U. Laub, U. Gerster and D. Lanskowski for excellent technical assistance and D. Gioè for proofreading of the manuscript.
Conflict of Interests
There are no conflicts of interest and financial disclosures in the subject matter of this paper.
References
1. Massague J, Weis-Garcia F. Serine/threonine kinase receptors: mediators of transforming growth factor beta signals. Cancer Surv. 1996;27:41-64
2. Centrella M, Rosen V, Horowitz MC, Wozney JM, Mc-Carthy TL. Transforming growth factor-ß gene family members, their receptors, and bone cell function. Endocr Rev. 1995;4:211-226
3. Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. Growth factors. 2004;22:233-241
4. Roberts AB, Sporn MB. The transforming growth factors-ßs. In: (ed.) Sporn MB, Roberts AB. Handbook of Experimental Pharmacology; vol. 95. Heidelberg, Germany: Springer. 1990:419-472
5. Massague J. The transforming growth factor-beta family. Annu Rev Cell Biol. 1990;6:597-641
6. Hogan BLM. Bone morphogenetic proteins: multifunctional regulators of vertebrate development. Genes Dev. 1996;10:1580-1594
7. Mehler MF, Mabie PC, Zhang D, Kessler JA. Bone morphogenetic proteins in the nervous system. Trends Neurosci. 1997;20:309-317
8. Smith A, Graham A. Restricting BMP4-mediated apoptosis in hindbrain neural crest. Dev Dyn. 2001;220:276-283
9. Lein P, Drahushuk K, Higgins D. Effects of bone morphogenetic proteins on neural tissues. In: (ed.) Vukicevic S, Sampath KT. Bone Morphogenetic Proteins. Basel: Birkhauser-Verlag. 2002:289-319
10. Duenker N. The role of transforming growth factor beta (TGF-ß) in mediating programmed cell death in the vertebrate retina. Int Rev Cytol Survey Cell Biol. 2005;245:7-40
11. Trousse F, Esteve P, Bovolenta P. BMP4 mediates apoptotic cell death in the developing chick eye. J Neurosci. 2001;21:1292-1301
12. Belecky-Adams T, Adler R. Developmental expression patterns of bone morphogenetic proteins, receptors, and binding proteins in the chick retina. J Comp Neurol. 2001;430:562-572
13. Sakuta H, Takahashi H, Shintani T, Etani K, Aoshima A, Noda M. Role of bone morphogenic protein 2 in retinal patterning and retinotectal projection. J Neuoscience. 2006;26(42):10868-10878
14. Murali D, Yoshikawa S, Corrigan RR, Plas DJ, Crair MC, Oliver G, Lyons KM, Mishina Y, Furuta Y. Distinct developmental programs require different levels of Bmp signaling during mouse retinal development. Development. 2005;132:913-923
15. Furuta Y, Piston DW, Hogan BL. Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development. 1997;124:2203-2212
16. Mabie P, Mehler MF, Kessler JA. Multiple roles of bone morphogenetic protein signaling in the regulation of cortical cell number and phenotype. J Neurosci. 1999;19(16):7077-7088
17. Graham A, Francis-West P, Brickell P, Lumsden A. The signaling molecule BMP4 mediates apoptosis in the rhomboencephalic neural crest. Nature. 1994;372:684-686
18. Franke AG, Gubbe C, Beier M, Duenker N. Transforming growth factors beta and Bone morphogenetic proteins: Cooperative players in chick and murine programmed retinal cell death. J Comp Neurol. 2005;26:263-278
19. Ro TB, Holt RU, Brenne AT, Hjorth-Hansen H, Waage A, Hjertner O, Sundan A, Borset M. Bone morphogenetic protein-5, -6 and -7 inhibit growth and induce apoptosis in human myeloma cells. Oncogene. 2004;23:3024-3032
20. Hjertner Ö, Hjrth-Hansen H, Börset M, Seidel C, Waage A, Sundan A. Bone morphogenetic protein-4 inhibits proliferation and induces apoptosis of multiple myeloma cells. Blood. 2001;97:516-522
21. Kawamura C, Kizaki M, Yamato K, Uchida H, Fukuchi Y, Koseki T, Nishihara T, Ikeda Y. Bone morphogenetic protein-2 induces apoptosis in human myeloma cells with modulation of STAT3. Blood. 2000;96:2005-2011
22. Rothhammer T, Poser I, Soncin F, Bataille F, Moser M, Bosserhoff AK. Bone morphogenetic proteins are overexpressed in malignant melanoma and promote cell invasion and migration. Cancer Res. 2005;65(2):448-456
23. Deng H, Ravikumar TS, Yang WL. Overexpression of bone morphogenetic protein 4 enhances the invasiveness of Smad4-deficient human colorectal cancer cells. Cancer Lett. 2009;281(2):220-231
24. Deng H, Makizumi R, Ravikumar TS, Dong H, Yang WL. Bone morphogenetic protein-4 is overexpressed in colonic adenocarcinomas and promotes migration and invasion of HCT116 cells. Exp Cell Res. 2007;3313(5):1033-1044
25. Maegdefrau U, Amann T, Winkelmeier A, Braig S, Schubert T, Weiss TS, Schardt K, Warnecke C, Hellerbrand C, Bosserhoff AK. Bone morphogenetic protein 4 is induced in hepatocellular carcinoma by hypoxia and promotes tumour progression. J Pathol. 2009;218(4):520-529
26. Chen RH, Ebner R, Derynck R. Inactivation of type II receptor reveals two receptor pathways for the diverse TGF-ß activities. Science. 1993;260:1335-1338
27. Kimchi A, Wang XF, Weinberg RA, Cheifertz S, Massague J. Absence of TGF-ß receptors and growth inhibitory response in retinoblastoma cells. Science. 1988;240:196-199
28. Horie K, Yamashita H, Mogi A, Takenoshita S, Miyazono K. Lack of Transforming growth factor-ß type II receptor expression in human retinoblastoma cells. J Cell Physiol. 1998;175:305-313
29. Howe JR, Sayed MG, Ahmed AF, Larsen-Haidle J, Merg A, Mitros FA, Vaccaro CA, Petersen GM; Giardiello FM; Timley ST, Aaltonen LA, Lynch HT. The prevalence of MADH4 and BMPRIA mutations in juvenile polyposis and absence of BMPR2 BMPR1B and ACVR1 mutations. J Med Genet. 2004;41:484-491
30. Nishanian TG, Kim JS, Foxworth A, Waldman T. Suppression of tumorigenesis and activation of wnt signaling by bone morphogenetic protein 4 in human cancer cells. Cancer Biol Therapy. 2004;3:667-675
31. Yamada N, Kato M, ten Dijke P, Yamashita TK, Heldin CH, Miyazono K, Funa K. Bone morphogenetic protein type IB receptor is progressively expressed in malignant glioma tumours. Brit J Cancer. 1996;73:624-629
32. Miyazono K, Maeda S, Imamura T. BMP receptor signaling: transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine Growth Factor Rev. 2005;16:251-263
33. Johnson MD, O'Connell MJ, Vito F, Pilcher W. Bone morphogenetic protein 4 and its receptors are expressed in the leptomeninges and meningiomas and signal via the Smad pathway. J Neuropathol Exp Neurol. 2009;68(11):1177-83
34. Hanyu A, Ishidou Y, Ebisawa T, Shimanuki T, Imamura T, Miyazono K. The N-domain of Smad 7 is essential for specific inhibition of transforming growth factor-ß signalling. J Cell Biol. 2001;155:1017-1028
35. Arnold NB, Korc M. Smad7 abrogates transforming growth factor-ß1-mediated growth inhibition in COLO-357 cells through functional inactivation of the retinoblastoma protein. J Biol Chem. 2005;280:21858-21866
36. Miyazaki H, Watabe T, Kitamura T, Miyazono K. BMP signals inhibit proliferation and in vivo tumor growth of androgen-insensitive prostate carcinoma cells. Oncogene. 2004;23:9326-9335
37. Pardali K, Kowanetz M, Heldin CH, Moustakas A. Smad pathway-specific transcriptional regulation of cell cycle inhibitor p21waf1/cip1. J Cell Physiol. 2005;204:260-272
38. Miyazawa K, Shinozaki M, Hara T, Furuya T, Miyazono K. Two major pathways in TGF-beta superfamily signaling. Genes Cells. 2002;7(12):1191-1204
39. Miyazono K, Miyazawa K. Id: a target of BMP signaling. Sci. STKE. 2002;151:pe40
40. Yokota Y, Mori S, Narumi O, Kitajima K. In vivo function of a differentiation inhibitor, Id2. IUMBM Life. 2001;51(4):207-214
41. McFall RC, Sery TW, Makadon M. Characterization of a new continuous cell line derived from a human retinoblastoma. Cancer Res. 1977;37:1003-1010
42. Gallie BL, Albert DM, Wong JJY, Buyukmihci N, Puliafito CA. Heterotransplantation of retinoblastoma into the athymic “nude” mouse. Invest Opthlamol Vis Sci. 1977;16:256-259
43. Griegel S, Hong C, Frötschl R, Hülser DF, Greger V, Horsthemke B, Rajewski MF. Newly established human retinoblastoma cell lines exhibit an “immortalized” but not an invasive phenotype in vitro. Int J Cancer. 1990;46:125-132
44. Deitch AD, Law H, deVere White R. A stable propidium iodide staining procedure for flow cytometry. J Histochem Cytochem. 1982;30(9):967-72
45. Liu J, Wilson S, Reh T. BMP receptor 1b is required for axon guidance and cell survival in the developing retina. Dev Biol. 2003;256:34-47
46. Wordinger R, Clark A. Bone morphogenetic potein and their receptors in the eye. Exp Biol Med. 2007;232:979-992
47. Shen W, Finnegan S, Lein P, Sullivan S, Slaughter M, Higgins D. Bone morphogenetic proteins regulate ionotropic glutamate receptors in human retina. Eur J Neurosci. 2004;20:2031-2037
48. Wordinger RJ, Agarwal R, Talati M, Fuller J, Lambert W, Clark AF. Expression of bone morphogenetic proteins (BMP), BMP receptors, and BMP associated proteins in human trabecular meshwork and optic nerve head cells and tissues. Mol Vis. 2002;15:241-250
49. Mohan RR, Kim WJ, Mohan RR, Chen L, Wilson SE. Bone morphogenetic proteins 2 and 4 and their receptors in the adult human cornea. Invest Ophthalmol Vis Sci. 1998;39:2626-2636
50. Andreev K, Zenkel M, Kruse F, Jünemann A, Schlötzer-Schrehardt U. Expression of bone morphogenetic proteins (BMPs), their receptors, and activins in normal and scarred conjunctiva: Role of BMP-6 and activin-A in conjunctival scarring? Exp Eye Res. 2006;83:1162-1170
51. Strasser A, O'Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem. 2000;69:217-245
52. Gross A, McDonell JM, Korsmeyer SJ. Bcl-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899-1911
53. Fukuda N, Saitoh M, Kobayashi N, Miyazono K. Execution of BMP-4-induced apoptosis by p53-dependent ER dysfunction in myeloma and B-cell hybridoma cells. Oncogene. 2006;25(25):3509-3517
54. Kawamura C, Kizaki M, Yamato K, Uchida H, Fukuchi Y, Hattori Y, Koseki T, Nishihara T, Ikeda Y. Bone morphogenetic protein-2 induces apoptosis in human myeloma cells with modulation of STAT3. Blood. 2000;96:2005-2011
55. Lagna G, Nguyen PH, Ni W, Hata A. BMP-dependent activation of caspase-9 and caspase-8 mediates apoptosis in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1059-L1067
56. Dannenberg JH, te Riele HP. The retinoblastoma gene family in cell cycle regulation and suppression of tumorigenesis. Results Probl Cell Differ. 2006;42:183-225
57. Rivadeneira DB, Mayhew CN, Thangavel C, Sotillo E, Reed CA, Grna X, Knudsen ES. Proliferative suppression by CDK4/6 inhibition: complex function of the retinoblastoma pathway in liver tissue and hepatoma cells. Gastroenterolgy. 2010;138:1920-1930
58. Sheahan S, Bellamy CO, Dunbar DR, Harrison DJ, Prost S. Deficiency of GI regulators P53, P21CipI and /or pRb decreases hepatocyte sensitivity to TGFß cell cycle arrest. BMC Cancer. 2007;7:215-24
59. Brubaker KD, Corey E, Brown LG, Vessella RL. Bone morphogenetic protein signaling in prostate cancer cell lines. J Cell Biol. 2004;91:151-160
60. Gosh-Choudhury N, Ghosh-Choudhury G, Celeste A, Ghosh PM, Moyer M, Abboud SL, Kreisberg J. Bone morphogenetic protein-2 induces cyclin kinase inhibitor p21 and hypophosphorylation of retinoblastoma protein in estradiol-treated MCF-7 human breast cancer cells. Biochica et Biophysica Acta. 2000;1497:186-196
61. Zhu DH, Wu J, Spee C, Ryan SJ, Hinton DR. BMP4 mediates oxidative stress-induced retinal pigment epithelial cell senescence and is overexpressed in age-related macular degeneration. J Biol Chem. 2009;284:9529-9539
62. SuD Zhu S, Han X Feng Y, Huang H Ren G, Pan L Zhang Y, Lu J Huang B. BMP4-smad signalling pathway mediates adriamycin.induced premature senescence in lung cancer cells. J Biol Chem. 2009;284:12153-12164
63. Chang SF, Chang TK, Peng HH, Yehr YT, Lee DY, Yeh CR, Zhou J, Cheng CK, Chang CA, Chiu JJ. BMP-4 induction of arrest and differentiation of osteoblast-like cells via p21CIP1 and p27KIP1 regulation. Mol Endocrinol. 2009;23:1827-1838
64. Sato Y, Kobayashi H, Sasaki T, Toyama T, Kondo S, Kiriyama M, Fujii Y. Expression of Id2 mRNA in neuroblastoma and normal ganglion. Eur J Surgical Oncol. 2003;29:284-287
65. Ruzinova MB, Benezra R. Id proteins in development, cell cycle and cancer. Trends Cell Biol. 2003;13:410-418
66. Perk J, Iavarone A, Benezra R. ID family of helix-loop-helix proteins in cancer. Nat Rev Cancer. 2005;5:603-614
67. Norton JD. ID helix-loop-helix proteins in cell growth, differentiation and tumorigenesis. J Cell Sci. 2000;113:3897-3905
68. Kowanetz M, Valcourt U, Bergström R, Heldin CH, Moustakas A. Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor ß and bone morphogenetic protein. Mol Cell Biol. 2004;24:4241-4254
69. Maruyama H, Kleeff J, Wildi S, Friess H, Büchler MW, Israel MA, Korc M. Id-1 and Id-2 are overexprressed in pancreatic cancer and in dysplastic lesions in chronic pancreatitis. Am J Pathol. 1999;155:815-822
70. Sikder H, Devlin MK, Dunlap S, Ryu B, Alani RM. Id proteins in cell growth and tumorigenesis. Cancer Cell. 2003;3:525-530
71. Clement JF, Marr N, Meissner A, Schwalbe M, Sebald W, Kliche KO, Höffken K, Wölfl S. Bone morphogenetic protein 2 (BMP-2) induces sequential changes of Id gene expression in the breast cancer cell line MCF-7. J Cancer Clin Oncol. 2000;126:271-279
72. Prabhu S, Ignatova A, Park ST, Sun XH. Regulation of the expression of cyclin-dependent kinase inhibitor p21 by E2A and Id proteins. Mol Cell Biol. 1997;17:5888-5896
Author contact
![]() Corresponding author: Prof. Dr. Nicole Dünker, Institute for Anatomy, Department of Neuroanatomy, University of Duisburg-Essen, Medical Faculty, 45122 Essen, Germany. Phone: ++49-(0)201-723-4922; fax: ++49-(0)201-723-5635; e-mail: nicole.duenkerde
Corresponding author: Prof. Dr. Nicole Dünker, Institute for Anatomy, Department of Neuroanatomy, University of Duisburg-Essen, Medical Faculty, 45122 Essen, Germany. Phone: ++49-(0)201-723-4922; fax: ++49-(0)201-723-5635; e-mail: nicole.duenkerde