Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2010; 6(7):716-718. doi:10.7150/ijbs.6.716 This issue Cite
Commentary
Role of clusters in insulin-regulated GLUT4 trafficking in adipose cells: A new paradigm?
Jian Yang ![]()
1. EDMNS, Diabetes Branch, NIDDK, National Institutes of Health, Bethesda, MD, USA.
2. Department of Physiology, University of South Alabama College of Medicine, Mobile, AL, USA.
Received 2010-11-4; Accepted 2010-11-24; Published 2010-11-25
Abstract
Insulin stimulates glucose transport in muscle and adipose cells by stimulating translocation of glucose transporter 4 (GLUT4) to the plasma membrane. In a recent Cell Metabolism paper, Stenkula et al. found that insulin controls the spatial distribution of GLUT4 on the surface of isolated adipose cells through regulation of their post-fusion dispersal. The presence of GLUT4 in plasma membrane-associated clusters is suggestive of a new paradigm in membrane protein recycling.
Keywords: Adipocyte, GLUT4, exocytosis, endocytosis, insulin resistance
One of the most important biological actions of insulin, a peptide hormone secreted from pancreas, is to facilitate glucose utilization and storage in muscle and adipose cells by increasing glucose transport. Ever since the discovery of insulin in 1921 by Banting, Best, and MacLeod, the cellular mechanism of insulin-regulated glucose transport has remained the center of investigation. A major breakthrough was made in 1980 by two independent groups [1,2] showing that insulin stimulates glucose transport in isolated rat adipose cells not by increasing the specific activity, but rather by increasing the total number of glucose transporters in the plasma membrane (PM). It was found that in non-stimulated adipose cells the majority (95%) of the glucose transporters are sequestered as intracellular storage vesicles; following insulin stimulation these intracellular glucose transporters are then translocated to PM, leading to a 10-20-fold increase of the number of glucose transporters on the cell surface and the same magnitude of increase in glucose uptake activity. This observation led to the hypothesis that insulin stimulates translocation of glucose transporters from intracellular storage compartments to PM. A hot pursuit of the identity of the insulin-regulatable glucose transporter led to the discovery of a previously undescribed transporter, which was subsequently termed glucose transporter 4 or GLUT4, by five different groups in 1989. The identification of GLUT4 permitted many additional studies confirming the translocation hypothesis.
Over the last two decades many aspects of the molecular machinery that controls GLUT4 trafficking have been elucidated and numerous molecules in the insulin signaling pathway that regulate GLUT4 translocation have been identified [3,4]. With the advent of high-resolution and powerful total internal reflection fluorescence (TIRF) microscopy technologies, researchers have further shown that insulin facilitates docking/tethering of intracellular GLUT4 storage vesicles (GSV) to PM and their subsequent fusion into PM in living isolated primary rat adipose cells [5] and 3T3-L1 cells [6-8]. However, exactly how insulin regulates GLUT4 fusion into PM, e.g., the action sites of insulin and spatial distribution of GLUT4 in PM after fusion, remained unresolved.
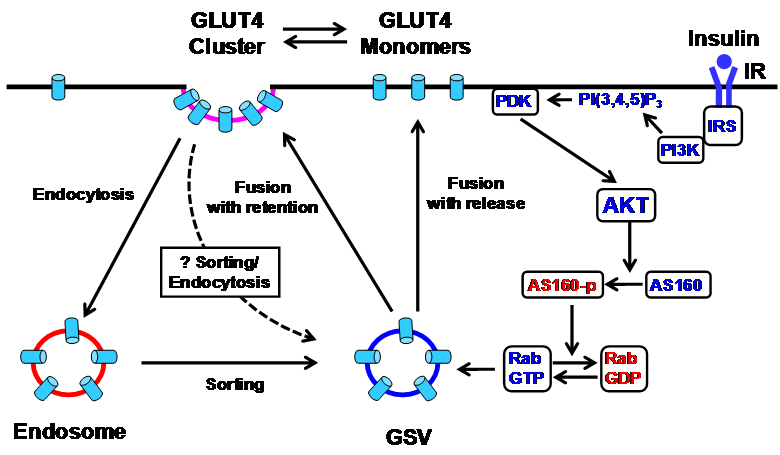
In the Sept 8 issue of Cell Metabolism, Stenkula et al. [9] presented solid evidence that insulin controls the spatial distribution of GLUT4 on the surface of isolated adipose cells through regulation of their post-fusion dispersal (Fig. 1). The authors describe several important findings. First, they identify two distinctive populations of GLUT4 in PM in adipose cells, clusters that are relatively stationary and monomers that are freely diffusible. In the basal state, the amounts of GLUT4 clusters and monomers in PM are equal; with insulin stimulation, the monomers increase 4-fold and the clusters 2.5-fold. Importantly the overall increase of the exposed GLUT4 on the surface of PM corresponds to the insulin-stimulated increase of glucose transport, indicating that both populations of GLUT4 in PM are functionally equivalent in transporting glucose. Second, they reported two types of GLUT4 exocytosis: fusion-with-release in which GLUT4 molecules are dispersed into PM and fusion-with-retention in which GLUT4 molecules are retained at the site of fusion. In the basal state, the majority (95%) of the fusion events are fusion-with-retention. Remarkably, within 2-3 minutes following insulin stimulation, the fusion-with-release of GLUT4 is increased more than 60-fold, whereas the fusion-with-retention is only moderately increased (~2-fold). Third, GLUT4 are internalized predominantly through the classical clathrin-mediated endocytosis pathway, not by sequestering cell-surface GLUT4 in coated pits, but rather by forming coated pits at the pre-existing clusters.
Schematic diagram of the insulin signaling cascade that regulates GLUT4 exocytosis in the plasma membrane in adipose cells. Insulin stimulation results in tyrosine phosphorylation of IRS and activation of PI3K, which catalyzes the formation of PI(3,4,5)P3 from PI(4,5)P2, leading to the action of PDK1 and 2. The PDK's phosphorylate and activate AKT, which in turn phosphorylates and inactivates AS160, a Rab GTPase-activating protein (GAP). AS160 negatively regulates GLUT4 translocation by converting the active GTP-bound form of Rab protein to the inactive GDP-bound form. This removes the inhibition of AS160 and permits GSV translocation to the plasma membrane for fusion. In the basal state, GLUT4 molecules are inserted into the plasma membrane primarily by fusion-with-retention in which GLUT4 molecules are retained at the site of fusion. Insulin modestly stimulates this fusion with retention, but markedly stimulates a second type of exocytosis, fusion-with-release, in which GLUT4 molecules are dispersed into the plasma membrane. In addition to the main PI3K pathway, the Rho-family GTPase TC10 pathway (not shown) has also been proposed to mediate insulin signaling in regulating GLUT4 translocation [4]. AKT, protein kinase B; AS160, AKT substrate 160; GLUT4, glucose transporter 4; GSV, GLUT4 storage vesicles; IR, insulin receptor; IRS, insulin receptor substrate; PDK, phosphoinositide-dependent kinase; PI3K, phosphatidylinositol 3 kinase; PI (4,5)P2, phosphatidylinositol (4,5) bisphosphate; PI (3,4,5)P3, phosphatidylinositol (3,4,5) trisphosphate; Rab, Ras-related small GTP-binding protein.
While Stenkula et al. [9] describes many important observations regarding the action sites of insulin in PM and the novel role of clusters in insulin-regulated GLUT4 trafficking in adipose cells, molecular insights remain to be determined. Moreover, the experiments are conducted solely in transfected primary rat adipocytes; it has not yet been established whether these same events occur in vivo. It will be important to substantiate these conclusions in a transgenic animal model with adipose cell-specific expression of GLUT4 fused with a reporter gene.
Despite these limitations, the finding that GLUT4 exist in distinctive clusters in PM is exciting. One could hypothesize that the GLUT4 cluster functions as a molecular organization that directs both GLUT4 exposure and internalization in PM in adipose cells. It can be speculated that the GLUT4 clusters are functionally equivalent to intracellular organelles, or in a more general term, “molecular post offices”. Indeed, one can even imagine that sorting and processing of GLUT4 could take place in the clusters so that endocytosis contributes directly to the regeneration of GSV. GLUT4 was previously found to be inhomogeneous and associated with caveolae-rich regions in PM [10], but Stenkula et al. [9] did not find evidence that GLUT4 clusters are associated with caveolin, the main structural protein of caveolae. It is warranted that identification and functional characterization of new molecular components, especially those that are targeted by insulin signaling, in the GLUT4 clusters in normal adipose cells will not only add fundamental knowledge to cell biology, but also help understand the molecular mechanisms of impaired glucose uptake in adipose tissue in insulin resistance and type 2 diabetes.
Conflict of Interests
No conflict of interests exists.
References
1. Cushman S.W, Wardzala L.J. Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. Apparent translocation of intracellular transport systems to the plasma membrane. J Biol Chem. 1980;255:4758-4762
2. Suzuki K, Kono T. Evidence that insulin causes translocation of glucose transport activity to the plasma membrane from an intracellular storage site. Proc Natl Acad Sci U S A. 1980;77:2542-2545
3. Bryant N.J, Govers R, James D.E. Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol. 2002;3:267-277
4. Watson R.T, Pessin J.E. Bridging the GAP between insulin signaling and GLUT4 translocation. Trends Biochem Sci. 2006;31:215-222
5. Lizunov V.A, Matsumoto H, Zimmerberg J, Cushman S.W, Frolov V.A. Insulin stimulates the halting, tethering, and fusion of mobile GLUT4 vesicles in rat adipose cells. J Cell Biol. 2005;169:481-489
6. Bai L, Wang Y, Fan J. et al. Dissecting multiple steps of GLUT4 trafficking and identifying the sites of insulin action. Cell Metab. 2007;5:47-57
7. Koumanov F, Jin B, Yang J, Holman G.D. Insulin signaling meets vesicle traffic of GLUT4 at a plasma-membrane-activated fusion step. Cell Metab. 2005;2:179-189
8. Gonzalez E, McGraw T.E. Insulin signaling diverges into Akt-dependent and -independent signals to regulate the recruitment/docking and the fusion of GLUT4 vesicles to the plasma membrane. Mol Biol Cell. 2006;17:4484-4493
9. Stenkula K.G, Lizunov V.A, Cushman S.W, Zimmerberg J. Insulin controls the spatial distribution of GLUT4 on the cell surface through regulation of its postfusion dispersal. Cell Metab. 2010;12:250-259
10. Parton R.G, Molero J.C, Floetenmeyer M, Green K.M, James D.E. Characterization of a distinct plasma membrane macrodomain in differentiated adipocytes. J Biol Chem. 2002;277:46769-46778
Author contact
![]() Corresponding author: Jian Yang, Ph.D., EDMNS, Diabetes Branch, NIDDK, National Institutes of Health, Bethesda, MD 20892, USA. Phone: (301) 496-7354; Fax: (301) 451-6989; Email: jian.yanggov or jyangedu
Corresponding author: Jian Yang, Ph.D., EDMNS, Diabetes Branch, NIDDK, National Institutes of Health, Bethesda, MD 20892, USA. Phone: (301) 496-7354; Fax: (301) 451-6989; Email: jian.yanggov or jyangedu