Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(1):28-40. doi:10.7150/ijbs.7.28 This issue Cite
Research Paper
Functional Genomics of Rice Pollen and Seed Development by Genome-wide Transcript Profiling and Ds Insertion Mutagenesis
Shu-Ye Jiang, Srinivasan Ramachandran ![]()
Rice Functional Genomics Group, Temasek Life Sciences Laboratory, 1 Research Link, Singapore 117604
Received 2010-11-12; Accepted 2010-12-27; Published 2010-12-30
Abstract
Rice pollen and seed development are directly related to grain yield. To further improve rice yield, it is important for us to functionally annotate the genes controlling pollen/seed development and to use them for rice breeding. Here we first carried out a genome-wide expression analysis with an emphasis on genes being involved in rice pollen and seed development. Based on the transcript profiling, we have identified and functionally classified 82 highly expressed pollen-specific, 12 developing seed-specific and 19 germinating seed-specific genes. We then presented the utilization of the maize transposon Dissociation (Ds) insertion lines for functional genomics of rice pollen and seed development and as alternative germplasm resources for rice breeding. We have established a two-element Activator/Dissociation (Ac/Ds) gene trap tagging system and generated around 20,000 Ds insertion lines. We have subjected these lines for screens to obtain high and low yield Ds insertion lines. Some interesting lines have been obtained with higher yield or male sterility. Flanking Sequence Tags (FSTs) analyses showed that these Ds-tagged genes encoded various proteins including transcription factors, transport proteins, unknown functional proteins and so on. They exhibited diversified expression patterns. Our results suggested that rice could be improved not only by introducing foreign genes but also by knocking out its endogenous genes. This finding might provide a new way for rice breeder to further improve rice varieties.
Keywords: Activator/Dissociation (Ac/Ds), flanking sequence tags, gene trap, germplasm, grain yield, Pollen and seed development
Introduction
Rice is one of the main staples in the world and is cultivated mainly in Asia, Africa, and Latin America, accounting for 50-80% of the daily diet of approximately half the world's population. With the growing population and decreasing cultivable land, it is estimated that 40% more rice has to be produced in 2030 [1]. Therefore, it is important for us to genome-widely identify and functionally characterize rice genes related to rice yield traits. Pollen and/or seed development is directly related to grain production. Among the yield-related genes, many of them have been identified to be related to pollen or grain development and knockout of pollen/seed-specific genes might lead to male sterility or abnormal seed development, thus, reducing seed yield [2-7]. Therefore, genome-wide identification of pollen/seed-specifically expressed genes could significantly contribute to the better understanding of biological mechanisms controlling grain yield, which is also a prerequisite for the functional genomics of rice pollen and seed development.
On the other hand, it is compulsory for rice breeders to develop new rice varieties with higher yield. Two ways may contribute to producing more rice seeds by breeding strategy. One of them is to develop new conventional rice varieties and another one is to develop new hybrid rice. However, elite germplasms are a prerequisite for both breeding strategies. In rice, three kinds of elite germplasm resources have been significantly contributed to higher rice yield. One of them is the utilization of dwarf germplasm Dee-geo-woo-gen from China and the release of rice variety IR8, which was developed from the dwarf line [8]. The second type of important germplasm is the cytoplasmic male sterile (CMS) rice lines and their restorers of fertility, which are widely utilized to produce hybrid rice seeds by three-line hybrid rice technology because they eliminate the need for laborious hand emasculations [9]. More than 20% yield advantage over improved conventional varieties has been achieved by releasing of various three-line hybrid rice combinations [10]. The third type of important germplasm is photoperiod (temperature)-sensitive male sterile lines. These lines can be used to produce hybrid rice seeds by two-line hybrid rice technology since these lines can be used not only as male-sterile lines but also as maintainer lines. Thus, the heterosis between subspecies can be used and higher yield can be achieved [9]. The yield advantage of two-line hybrid rice is 5-10% higher than that of the existing three-line hybrid rice [10]. Therefore, it is very important for us to develop these elite germplasms.
A useful germplasm can be developed from a natural mutation population, which is important for current breeding. Besides natural variation, physical or chemical mutation is also an efficient method to produce diverse variations for crop breeding [11]. More than 2250 varieties have been released that were developed from direct mutants or their progenies [12]. Variations from various tissue cultures also contributed to the collection of germplasm resources and a few cultivars have been developed including rice [13, 14]. On the other hand, transgenic techniques have been employed to produce various germplasm improved in specific traits [15]. However, in order to face severe challenges of rapid population growth and reduced cropland area, it is necessary to develop new ways to produce more elite germplasms for development of higher-yield varieties in rice breeding.
With the complete of both japonica and indica rice genome sequences [16, 17], assigning a function to unknown or predicted genes has become the major work of functional genomics. Knockout of a gene is a direct way to achieve this purpose. Insertion mutagenesis with either T-DNA or transposon has been successfully used in functional genomics of plants [18]. Various insertion mutation populations have been produced [19-26]. Therefore, it is very important how to use these resources for the functional annotations of rice genes. Among them, we are interested in these genes related to rice pollen and seed development since they may contribute to the higher rice yield by molecular breeding. In this study, we first identify the highly expressed pollen/seed-specific genes based on genome-wide transcript profiling in rice. On the other hand, since we have generated around 20,000 rice insertion lines using maize Ac/Ds transposon system [27], we also investigated the variations in their pollen and seed development among these insertion lines. We then analyzed their flanking sequence tags (FSTs) to anchor the genes with Ds insertions. We also carried out the expression analyses to better understand their functions in pollen and seed development. Finally, we evaluated these lines in their potential in rice breeding.
Materials and Methods
Plant materials
Japonica rice (Oryza sativa, cv. Nipponbare) was used for all of our experiments. Both wild-type (WT) and mutant plants were grown under both greenhouse and natural field conditions.
Activator/Dissociation (Ac/Ds) tagging system and franking sequence tags (FSTs)
Establishment of Ac/Ds tagging system was carried out according to Kolesnik et al. (2004) [27]. Homozygous lines at the sixth generation were used to screen for grain yield, tolerance / resistance or more sensitivity to biotic stresses. Screens for lines with higher or lower yield were carried out in both Singapore and China. In Singapore, only small scale of screenings was conducted with 12 plants for each line under greenhouse with natural light and temperature conditions. In china, field trials were carried out with around 300 (30 cm × 10 cm) individuals for each line according to the description by Jiang et al. (2007) [28]. The investigated agronomic traits included seed weight per plant and seed setting rate per plant according to the standard evaluation system for rice available from International Rice Research Institute (IRRI) resource (http://www.knowledgebank.irri.org/extension/index.php/ses). To evaluate viability, pollen grains were stained with 1% Iodine Potassium Iodide (I2/KI) solution. The I2/KI solution is widely used for staining starch and the starch content in pollen grains can serve as an indicator of viability ([29]).
Sequences flanking Ds element of the putative candidate lines obtained from various screens were amplified by TAIL-PCR (Thermal Asymmetric Interlaced-PCR) as described by Liu et al. (1995) [30]. Tagged genes were obtained via BLAST searches by submitting Ds flanking sequences to TIGR (The Institute of Genome Research, http://tigrblast.tigr.org/euk-blast/index.cgi?project=osa1) databases. The locus numbers were retrieved from TIGR database and were used for searching expression patterns of tagged genes through public plant MPSS (Massively Parallel Signature Sequencing) database ([31]; http://mpss.udel.edu/). Based on the database, MPSS identifies short sequence signatures produced from a defined position within an mRNA, and the relative abundance of these signatures in a given library represents a quantitative estimate of expression of that gene; the MPSS signatures are 17 or 20 bp in length, and can uniquely identify >95% of all genes in rice [31]. Signatures are normalized to transcripts per million (TPM) to facilitate comparisons among different tissues or under different treatments. Based on default setting in the database, a summary of signatures from class 1 (TPM was detected using probes inside annotated open reading frame, ORF), class 2 (within 500 bp 3' of annotated ORF), class 5 (within annotated intron, sense strand) and class 7 (spans intron splice site) was used for expression analyses.
Genome-wide expression analyses of rice genes
Genome-wide expression of rice genes were also carried out by analyzing the expression data from the rice MPSS database ([32]; http://mpss.udel.edu/rice/). In the database, expression data from total of 70 different libraries are available. We are interested in 7 libraries. One of them is from mature pollens and the second is from germinating seeds. The remaining 5 libraries are from developing seeds. Expression data from all libraries were downloaded from the MPSS database. The expression abundance from the 7 selected libraries was compared with those from the remaining libraries. Pollen/seed-specific genes were identified if the transcript abundance in the pollen/seed libraries were higher than the sum of the remaining libraries. The highly expressed pollen/seed-specific genes were selected if their expression abundance was higher than 1000 TPM, as determined by the MPSS database.
GO annotations and categories
GO annotations for rice pollen/seed-specific genes were downloaded from the the TIGR rice genome annotation database (now moved to Michigan State University (MSU); http://rice.plantbiology.msu.edu/; [33, 34]). We used plant-related GO Slim terms [35] to explore the functions of these genes. Each gene can be associated with several GO Slim terms in the molecular function (MF), cellular component (CC) and biological process (BP) GO functional categories [36]. We studied each GO Slim term category independently.
Results
Pollen/seed-specific genes in rice genome by genome-wide transcript profiling
To identify and characterize the genes related to rice pollen/seed development, we first investigated the genome-wide expression profiling. The investigation was carried out by using the publicly available expression database, the MPSS database. The database contains the expression data from 70 libraries constructed from various developmental stages of tissues. Based on our analyses, we have identified more than 1000 putative pollen-specific genes. These genes were preferentially expressed in pollens as their expression abundance was higher than the sum of the remaining tissues. Among them, we have identified 82 highly expressed pollen-specific genes with more than 1000 TPM in their expression abundance. The MSU locus names of these genes and their transcript abundance have been listed in Table 1. Similarly, we have identified 12 developing seed-specific and 19 germinating seed-specific genes with highly expressed signals at developing and germinating seeds, respectively (Table 2 and 3). We have identified relatively low numbers of seed-specific genes due to that genes with low expression abundance were not included in our analyses.
82 Pollen-specific Genes in the Rice Genome and Their Expression Profiling

12 Developing Seed-specific Genes in the Rice Genome and Their Expression Profiling
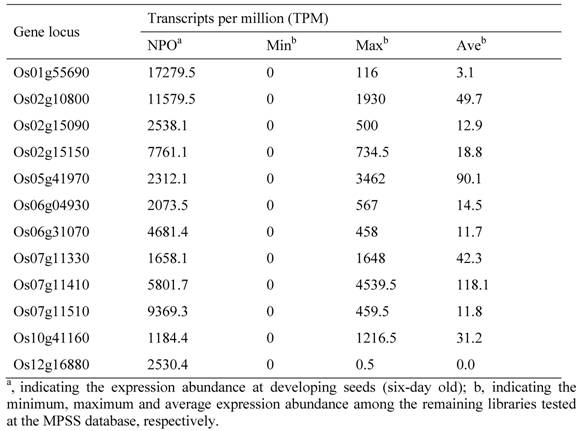
19 Germinating Seed-specific Genes in the Rice Genome and Their Expression Profiling
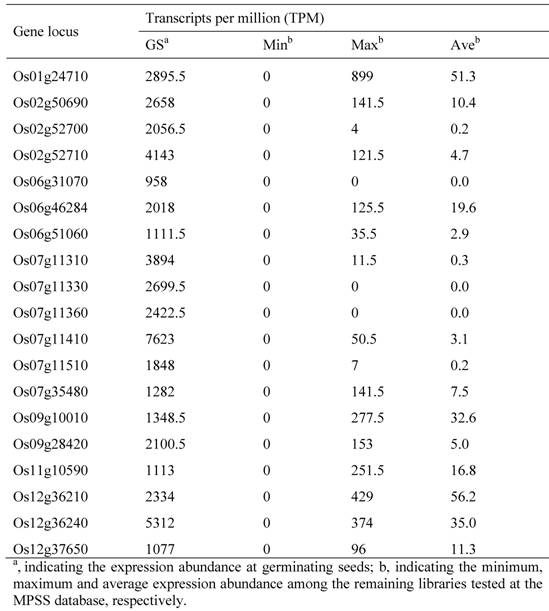
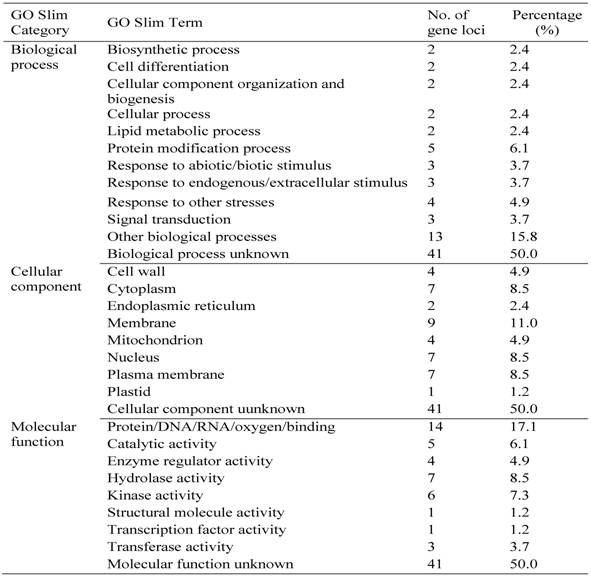
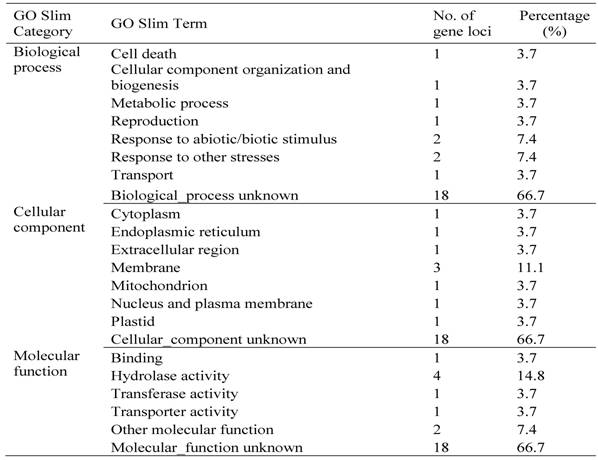
To examine the functional specificities of these pollen/seed-specific genes, we identified Gene Ontology (GO) terms (Materials and methods). For each term, we identified GO-slim terms in three categories: MF, BP and CC. For pollen-specific genes, half of them contain no GO annotation in all three categories (Table 4). We have listed GO slim terms for all remaining genes as shown in the table. Various biological functions have been assigned by these pollen-specific genes, suggesting that pollen development is a complex biological process being involved in genes and their proteins located in various cellular components with different molecular functions. Similarly, we have identified GO terms of seed-specific genes. The analysis shows that no GO term can be assigned for two-third of these genes (Table 5). The remaining seed-specific genes have been involved in various biological processes and their proteins were located different cellular components with multiple molecular functions (Table 5).
Classification of Some of Rice Pollen-specific Genes in GO Slim Categories
Classification of Some of Rice Seed-specific Genes in GO Slim Categories
Pollen and seed development is a very comprehensive process. It is involved in not only pollen/seed-specific genes but also other genes with different expression patterns. On the other hand, besides these genes related to pollen/seed development, other genes may also contribute to the improvement of the rice yield traits. In order to dissect the genes contributing to the higher yield, other strategies have to be employed. In the following section, we reported the establishment of Ac/Ds gene tagging system and its application on gene function annotation and rice breeding.
Two-element Ac/Ds gene trap system
We have developed an efficient two-element maize Ac/Ds gene trap system [27]. In this system, we use two different parental lines for crossing. One parental line is transgenic Ac plant, in which the transposase is immobilized and provides only Ac transposase under the control of 35S promoter. Another parental line is transgenic Ds plant, in which only two wings of Ds element (5' Ds and 3' Ds) is present. This element carries a bar gene conferring resistance to herbicide Basta, which serves as a positive selection (transposition) marker. The promoter-less gusA gene encodes a β-glucuronidase as a reporter gene (expression marker) for detecting gene expression patterns of tagged rice genes (gene trap). The green fluorescence protein (gfp) under maize ubiquitin promoter serves as a negative selection marker in both the Ac (to obtain stable transposants) and Ds (to enrich unlinked transposants) plants. In these starter lines both Ac and Ds elements are incapable of transposition. The Ds element can be mobilized and transposed into different genome positions in F1 generation after crossing with Ac plants. The F2 seeds are generated from these F1 plants by self-crosses. The putative unlinked, stable transposants can be selected by screening the GFP negative and Basta resistant F2 seedlings. Since Ac locus also contains the GFP as a negative selection marker, the GFP negative F2 seedlings will be stable. These stable Ds lines were self-crossed to obtain F3, F4 and F5 generation to obtain homozygous lines and to screen for phenotype.
Our general goal is to generate a large numbers of Ds insertional lines using this system. The large numbers of Ds lines are used to screen for various agriculturally important phenotypes for commercial release. Now we have generated more than 20,000 Ds insertional lines of which around 18,000 are homozygous. In addition to this more than 3,000 Ds flanking sequences were obtained from their corresponding lines.
High yield screening of Ds insertion lines and characterization of some putative yield-related Ds-tagged genes
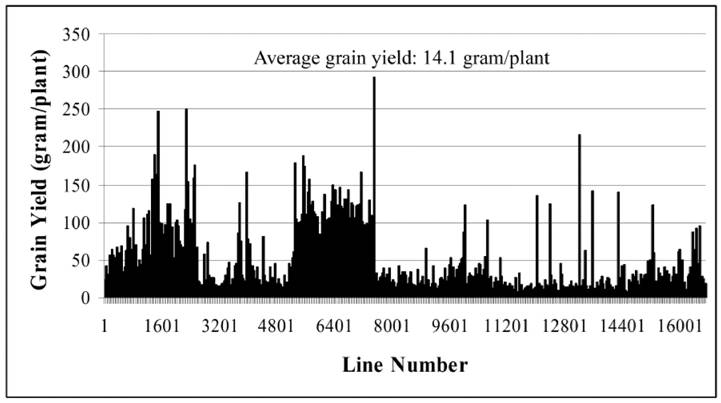
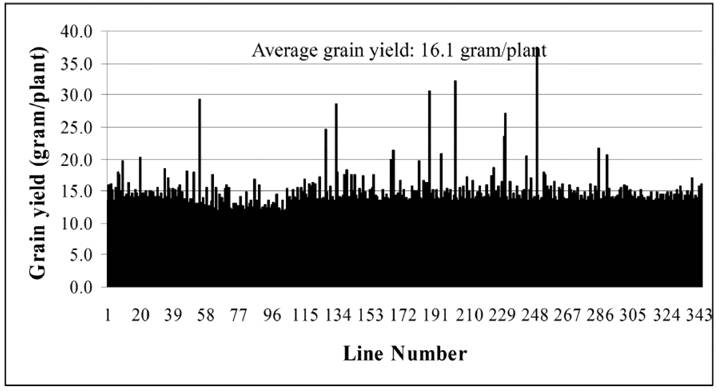
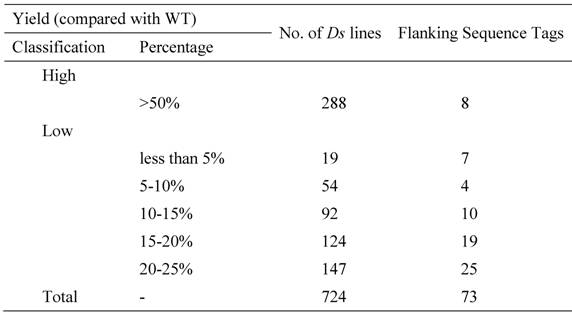
Until now, we have carried out two batches of high yield screens. In the preliminary screen, only around 10 plants for each line were used for the screen since around 20,000 Ds lines were subjected to the screen. Among all screened Ds lines, yield data were collected from 16,700 Ds lines. These lines showed significantly difference in their yield performance (Fig. 1), suggesting that Ds lines might be used for yield screening and subsequent breeding practice. Most of lines have similar yield while compared with WT plants. The average yield of these Ds lines is 14.1 gram per plant, similar to WT plant. Some of them showed lower yield and the remaining lines exhibited higher yield (Fig. 1). Based on this screen, 343 lines were selected with higher yield. These lines were subjected to further screening. In this screen, around 300 plants per line were planted. The data were presented in Fig. 2. In this screen, most of Ds lines showed higher yield compared with WT. The average yield also increases to 16.1 gram per plants. Based on these screen, we have selected 288 Ds lines with more than 50% higher yield than WT (Table 6).
Grain yield screen of 16705 Ds insertion lines. The screen was carried out in south of China. This is a small-scale screening with around 10 plants for each line.
Grain yield screen of 343 Ds insertion lines. The screen was carried out in south of China. This is a middle-scale screening with around 300 plants for each line.
A Summary of Yield Screening from Around 17000 Ds Lines
Phenotype investigation showed that, besides the higher grain yield, these Ds lines also exhibited additional agronomic traits such as slightly delayed growth period, higher tiller numbers and more strongly growth. We have also observed some higher yield Ds lines with slightly shorter growth period with erected leaf structure. Among these higher yield Ds lines, one of them was displayed in Fig. 3. This Ds line showed very strong and nearly double size of its original WT plant (Fig. 3A). As a result, the mutant produced 25% more seeds compared to WT. Cross-section of tillers showed less starch and more fibers in the mutant (Fig. 3B). Therefore, the mutant can be used for multiple purposes, not only for harvesting seeds but also collecting rice straw for other purposes for example for paper making or the artificial culture of eatable fungi. This mutant contained no Ds element and the footprint retained by Ds remobilization is the cause for the phenotype without the presence of Ds element. This approach of remobilizing the Ds element from the exons of genes may result in plants with mutant phenotype with no Ds element.
An example of high yield line. (A) A high yield plant (right) compared with WT (left). (B) Cry-SEM images of the WT (up) and the Ds line showing the difference in their starch content.

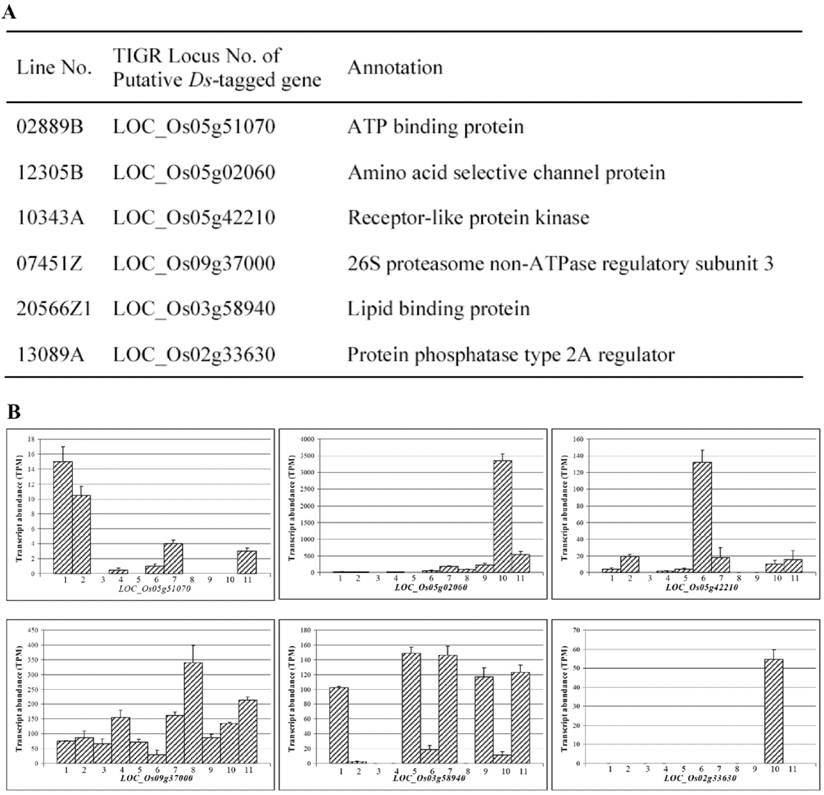
Among 288 Ds lines, DNA samples from some Ds lines were subjected to TAIL-PCR [30] to obtain their flanking sequences tagged by Ds element. Flanking sequence analysis revealed that knockout of many rice genes by Ds insertion may contribute to higher yield. These genes included those that encoded various transcription factors, sucrose transport proteins, hormone regulated proteins, and so on. Some representative genes were listed in Fig. 4A. These genes included those encoding ATP binding protein, amino acid selective channel protein, receptor-like protein kinase, 26S proteasome non-ATPase regulatory subunit 3, lipid binding protein, protein phosphatase type 2A regulator. Furthermore, the expression data from rice MPSS database were used to analyze the expression patterns of these genes using TIGR locus numbers. Totally, data from 11 different tissues were retrieved and analyzed for evaluating their expression patterns including young leaf, mature leaf, young and mature root, stem, merismatic tissue, immature panicle, ovary and mature stigma, mature pollen, developing seed, and germinating seed. These analyses showed that the candidate genes for high yield phenotype were expressed in different tissues (Fig. 4B). They were sometimes detected in multiple tissues with varying expression levels. Such expression patterns were observed in those genes including LOC_Os09g37000 and LOC_Os03g58940 (Fig. 4B). However, sometimes, they were expressed in some specific tissues. For example, LOC_Os05g51070 was mainly expressed in young and mature leaves; both LOC_Os05g02060 and LOC_Os02g33630 were mainly expressed in developing seeds; and LOC_Os05g42210 was mainly expressed in merismatic tissues (Fig. 4B). These diversified expression patterns suggested that high yield might be controlled by multiple genes with various pathways.
Yield-related lines and their putative Ds-tagged genes as well as their expression. (A) A list of some putative Ds-tagged genes and their annotations. (B) Expression patterns of some tagged genes shown by MPSS expression database. Numbers in X axis indicate different tissues. 1, young leaf; 2, mature leaf; 3, young root; 4, mature root; 5, stem; 6, merismatic tissue; 7, immature panicle; 8, ovary and mature stigma; 9, mature pollen; 10, developing seed and 11, germinating seed. Y axis indicates transcript abundance shown by TPM signals.
Low yield screening of Ds insertion lines and types of Ds-mediated rice sterility
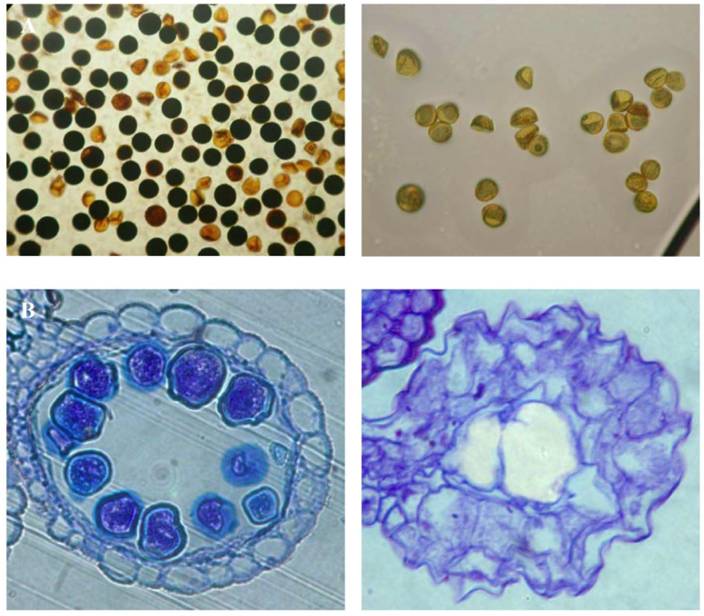
Besides the screen of rice high yield Ds insertion lines, the low yield screening was also carried out with the same population of Ds insertion lines. Among the 16,700 Ds insertion lines, we found that at least 436 lines showed low yield phenotype with at least 25% less in their grain yield (Table 6). Among them, some Ds lines exhibited completely sterility. Further characterization was carried out for these lines. The investigation showed that some of them were completely male sterile with 100% inviability of mature pollens (Fig. 5A). Another type of sterility is the lack of mature pollens at the flowering stage (Fig. 5B). In addition to these, more than 500 lines did not segregate homozygous plants even in F5/F6 generation, indicating that these could be putative homozygous lethal lines propagated only as heterozygous plants. Interestingly, we have observed another type of sterility, ie. photoperiod sensitive male sterility. This line exhibited male sterility under short day length conditions and the sterility was recovered under long day length conditions [37]. Therefore this line may be useful for developing two-line hybrid rice varieties.
Examples of low yield lines. (A) Pollen viability shown by starch content in WT (left) compared with a Ds line (right). (B) Section of anthers from WT (left) and a Ds line (right).
Discussion
Pollen/seed development related genes
In this manuscript, we have genome-widely identified 82 highly expressed pollen-specific, 12 developing seed-specific and 19 germinating seed-specific genes. Recently, Fujita et al (2010) and Wei et al (2010) also reported the expression atlas of rice genes in reproductive developmental stage and they identified more genes specifically expressed in the stage [38, 39]. This may be due to the difference in the employed methods and the criteria used for identifying the tissue-specific genes. On the other hand, our expression analysis may also provide the basis to screen pollen/seed specific promoters, which should be useful for engineering genetically modified rice varieties. In fact, some of seed-specific promoters such as the promoters from some glutelin genes have been used for the production of transgenic rice. For example, Akama et al (2009) have employed the seed-specific promoter GluB-1 to produce gamma-aminobutyric acid (GABA) enriched rice grains that influence a decrease in blood pressure [40]. One of seed-specific promoters has also been used for exploring the potential in producing rice seed-based edible vaccines [41].
Pollen/seed development and grain yield
Grain yield in rice is a complex trait multiplicatively determined by its three component traits: number of panicles, number of grains per panicle, and grain weight; all of which are typical quantitative traits [42]. Grain yield will be decreased if pollens/seeds can not be properly developed since viable pollen, receptive stigma and well developed ovule are required for successful seed set in rice. Transcript profiling of pollen/seed development will significantly contribute to the identification of genes for grain yield [43]. Not all pollen/seed-specific genes may directly contribute to grain production. However, some of them have been proven to be related to grain yield as shown in this study. Thus, our study may provide some information for further improving grain yield by genetically modifying pollen/seed related genes.
Functional genomics of pollen/seed development and crop improvement
Currently, several yield-related genes have been isolated [44-49]. However, only a few of them have been functionally characterized. Since considerable pollen/seed development related genes may contribute to grain production, studies on functional genomics of rice pollen/seed development may speed up the identification of yield-related genes. Since we have identified a batch of pollen/seed-specific genes, these genes can be used for reverse genetics screening to obtain corresponding Ds insertion lines. Thus, their biological functions can be annotated by characterizing these Ds lines. On the other hand, since we have identified several hundreds of Ds insertion lines with changed grain production, yield related genes could be identified and annotated from these Ds tagged lines. Upon the identification and functional characterization, these yield related genes will be employed to further improve rice yield by over-expressing or suppressing these genes through marker-free transgenic strategies [28]. In the mean time, tagged Ds lines may be directly used for developing non-transgenic rice varieties with higher yield according to our strategies [28].
Acknowledgements
We thank Drs. Ildiko Szeverenyi, Tatiana Kolesnik and Doris Bachmann for their help in generation of transposant lines. We take this opportunity to thank Zhigang Ma, Rengasamy Ramamoorthy and Hongfen Luan for their technical assistance.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Khush GS. What it will take to feed 5.0 billion rice consumers in 2030. Plant Mol Biol. 2005;59:1-6
2. Tzafrir I, Dickerman A, Brazhnik O, Nguyen Q, McElver J, Frye C, Patton D, Meinke D. The Arabidopsis Seed Genes Project. Nucleic Acids Res. 2003;31:90-93
3. Tzafrir I, Pena-Muralla R, Dickerman A, Berg M, Rogers R, Hutchens S, Sweeney TC, McElver J, Aux G, Patton D, Meinke D. Identification of genes required for embryo development in Arabidopsis. Plant Physiol. 2004;135:1206-1220
4. Itoh J, Nonomura K, Ikeda K, Yamaki S, Inukai Y, Yamagishi H, Kitano H, Nagato Y. Rice plant development: from zygote to spikelet. Plant Cell Physiol. 2005;46:23-47
5. Kurata N, Miyoshi K, Nonomura K, Yamazaki Y, Ito Y. Rice mutants and genes related to organ development, morphogenesis and physiological traits. Plant Cell Physiol. 2005;46:48-62
6. Borg M, Brownfield L, Twell D. Male gametophyte development: a molecular perspective. J Exp Bot. 2009;60:1465-1478
7. Wilson ZA, Zhang DB. From Arabidopsis to rice: pathways in pollen development. J Exp Bot. 2009;60:1479-1492
8. International Rice Research Institute. Annual Report for 1966. Los Banos, Philippines: International Rice Research Institute. 1967:59-82
9. Virmani SS, Sun ZX, Mou TM, Jauhar Ali A, Mao CX. Two-line hybrid rice breeding manual. Los Banos, Philippines: International Rice Research Institute. 2003
10. Yuan L. Hybrid rice technology for food security in the world. Rome, Italy: FAO Rice Conference. 2004
11. Brock RD. Quantitatively inherited variation in Arabidopsis thaliana induced by chemical mutagens. Environ. Exp. Bot. 1976;16:241-253
12. Ahloowalia BS, Maluszynski M, Nichterlein K. Global impact of mutation-derived varieties. Euphytica. 2004;135:187-204
13. Jain SM. Tissue culture-derived variation in crop improvement. Euphytica. 2001;118:153-166
14. Larkin PJ, Scowcroft WR. Somaclonal variation — a novel source of variability from cell cultures for plant improvement. Theor Appl Genet. 1981;60:197-214
15. Bajaj S, Mohanty A. Recent advances in rice biotechnology—towards genetically superior transgenic rice. Plant Biotechnol J. 2005;3:275-307
16. International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature. 2005;436:793-800
17. Yu J, Hu S, Wang J, Wong GK, Li S, Liu B, Deng Y, Dai L, Zhou Y, Zhang X, Cao M, Liu J, Sun J, Tang J, Chen Y, Huang X, Lin W, Ye C, Tong W, Cong L, Geng J, Han Y, Li L, Li W, Hu G, Huang X, Li W, Li J, Liu Z, Li L, Liu J, Qi Q, Liu J, Li L, Li T, Wang X, Lu H, Wu T, Zhu M, Ni P, Han H, Dong W, Ren X, Feng X, Cui P, Li X, Wang H, Xu X, Zhai W, Xu Z, Zhang J, He S, Zhang J, Xu J, Zhang K, Zheng X, Dong J, Zeng W, Tao L, Ye J, Tan J, Ren X, Chen X, He J, Liu D, Tian W, Tian C, Xia H, Bao Q, Li G, Gao H, Cao T, Wang J, Zhao W, Li P, Chen W, Wang X, Zhang Y, Hu J, Wang J, Liu S, Yang J, Zhang G, Xiong Y, Li Z, Mao L, Zhou C, Zhu Z, Chen R, Hao B, Zheng W, Chen S, Guo W, Li G, Liu S, Tao M, Wang J, Zhu L, Yuan L, Yang H. A draft sequence of the rice genome (Oryza sativa L. ssp. Indica). Science. 2002;296:79-92
18. Upadhyaya NM, Wing R, Yu SM. Rice Functional Genomics---Challenges, Progress and Prospects. New York: Springer. 2007:181-271
19. Izawa T, Ohnishi T, Nakano T, Ishida N, Enoki H, Hashimoto H, Itoh K, Terada R, Wu C, Miyazaki C, Endo T, Iida S, Shimamoto K. Transposon tagging in rice. Plant Mol Biol. 1997;35:219-229
20. Hirochika H, Guiderdoni E, An G, Hsing YI, Eun MY, Han CD, Upadhyaya N, Ramachandran S, Zhang Q, Pereira A, Sundaresan V, Leung H. Rice mutant resources for gene discovery. Plant Mol Biol. 2004;54:325-334
21. Kim CM, Piao HL, Park SJ, Chon NS, Je BI, Sun B, Park SH, Park JY, Lee EJ, Kim MJ, Chung WS, Lee KH, Lee YS, Lee JJ, Won YJ, Yi G, Nam MH, Cha YS, Yun DW, Eun MY, Han CD. Rapid, large-scale generation of Ds transposant lines and analysis of the Ds insertion sites in rice. Plant J. 2004;39:252-263
22. Kumar CS, Wing RA, Sundaresan V. Efficient insertional mutagenesis in rice using the maize En/Spm elements. Plant J. 2005;44:879-897
23. Jeong DH, An S, Park S, Kang HG, Park GG, Kim SR, Sim J, Kim YO, Kim MK, Kim SR, Kim J, Shin M, Jung M, An G. Generation of a flanking sequence-tag database for activation-tagging lines in japonica rice. Plant J. 2006;45:123-132
24. Hsing YI, Chern CG, Fan MJ, Lu PC, Chen KT, Lo SF, Sun PK, Ho SL, Lee KW, Wang YC, Huang WL, Ko SS, Chen S, Chen JL, Chung CI, Lin YC, Hour AL, Wang YW, Chang YC, Tsai MW, Lin YS, Chen YC, Yen HM, Li CP, Wey CK, Tseng CS, Lai MH, Huang SC, Chen LJ, Yu SM. A rice gene activation/knockout mutant resource for high throughput functional genomics. Plant Mol Biol. 2007;63:351-364
25. Miyao A, Iwasaki Y, Kitano H, Itoh J, Maekawa M, Murata K, Yatou O, Nagato Y, Hirochika H. Large-scale collection of phenotypic data describing an insertional mutant population to facilitate functional analysis of rice genes. Plant Mol Biol. 2007;63:625-635
26. Park SH, Jun NS, Kim CM, Oh TY, Huang J, Xuan YH, Park SJ, Je BI, Piao HL, Park SH, Cha YS, Ahn BO, Ji HS, Lee MC, Suh SC, Nam MH, Eun MY, Yi G, Yun DW, Han CD. Analysis of gene-trap Ds rice populations in Korea. Plant Mol Biol. 2007;65:373-384
27. Kolesnik T, Szeverenyi I, Bachmann D, Kumar CS, Jiang SY, Ramamoorthy R, Cai M, Ma ZG, Sundaresan V, Ramachandran S. Establishing an efficient Ac/Ds tagging system in rice: large-scale analysis of Ds flanking sequences. Plant J. 2004;37:301-314
28. Jiang SY, Bachmann D, La H, Ma Z, Venkatesh PN, Ramamoorthy R, Ramachandran S. Ds insertion mutagenesis as an efficient tool to produce diverse variations for rice breeding. Plant Mol Biol. 2007;65:385-402
29. Song ZP, Lu BR, Chen JK. A study of pollen viability and longevity in Oryza rufipogon, O sativa, and their hybrids. International Rice Research Newsletter. 2001;26:31-32
30. Liu YG, Whittier RF. Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics. 1995;25:674-681
31. Brenner S, Johnson M, Bridgham J, Golda G, Lloyd DH, Johnson D, Luo S, McCurdy S, Foy M, Ewan M, Roth R, George D, Eletr S, Albrecht G, Vermaas E, Williams SR, Moon K, Burcham T, Pallas M, DuBridge RB, Kirchner J, Fearon K, Mao J, Corcoran K. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat Biotechnol. 2000;18:630-634
32. Nobuta K, Venu RC, Lu C, Beló A, Vemaraju K, Kulkarni K, Wang W, Pillay M, Green PJ, Wang GL, Meyers BC. An expression atlas of rice mRNAs and small RNAs. Nat Biotechnol. 2007;25:473-477
33. Yuan Q, Ouyang S, Wang A, Zhu W, Maiti R, Lin H, Hamilton J, Haas B, Sultana R, Cheung F, Wortman J, Buell CR. The institute for genomic research Osa1 rice genome annotation database. Plant Physiol. 2005;138:18-26
34. Ouyang S, Zhu W, Hamilton J, Lin H, Campbell M, Childs K, Thibaud-Nissen F, Malek RL, Lee Y, Zheng L, Orvis J, Haas B, Wortman J, Buell CR. The TIGR Rice Genome Annotation Resource: improvements and new features. Nucleic Acids Res. 2007;35(Database Issue):D846-D851
35. Berardini TZ, Mundodi S, Reiser L, Huala E, Garcia-Hernandez M, Zhang P, Mueller LA, Yoon J, Doyle A, Lander G, Moseyko N, Yoo D, Xu I, Zoeckler B, Montoya M, Miller N, Weems D, Rhee SY. Functional annotation of the Arabidopsis genome using controlled vocabularies. Plant Physiol. 2004;135:745-755
36. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genetics. 2000;25:25-29
37. Jiang SY, Cai M, Ramachandran S. ORYZA SATIVA MYOSIN XI B controls pollen development by photoperiod-sensitive protein localizations. Dev Biol. 2007;304:579-592
38. Fujita M, Horiuchi Y, Ueda Y, Mizuta Y, Kubo T, Yano K, Yamaki S, Tsuda K, Nagata T, Niihama M, Kato H, Kikuchi S, Hamada K, Mochizuki T, Ishimizu T, Iwai H, Tsutsumi N, Kurata N. Rice expression atlas in reproductive development. Plant Cell Physiol. 2010;51:2060-2081
39. Wei LQ, Xu WY, Deng ZY, Su Z, Xue Y, Wang T. Genome-scale analysis and comparison of gene expression profiles in developing and germinated pollen in Oryza sativa. BMC Genomics. 2010;11:338
40. Akama K, Kanetou J, Shimosaki S, Kawakami K, Tsuchikura S, Takaiwa F. Seed-specific expression of truncated OsGAD2 produces GABA-enriched rice grains that influence a decrease in blood pressure in spontaneously hypertensive rats. Transgenic Res. 2009;18:865-876
41. Yang L, Suzuki K, Hirose S, Wakasa Y, Takaiwa F. Development of transgenic rice seed accumulating a major Japanese cedar pollen allergen (Cry j 1) structurally disrupted for oral immunotherapy. Plant Biotechnol J. 2007;5:815-826
42. Xing Y, Zhang Q. Genetic and molecular bases of rice yield. Annu Rev Plant Biol. 2010;61:421-442
43. Deshmukh R, Singh A, Jain N, Anand S, Gacche R, Singh A, Gaikwad K, Sharma T, Mohapatra T, Singh N. Identification of candidate genes for grain number in rice (Oryza sativa L.). Funct Integr Genomics. 2010;10:339-347
44. Li X, Qian Q, Fu Z, Wang Y, Xiong G, Zeng D, Wang X, Liu X, Teng S, Hiroshi F, Yuan M, Luo D, Han B, Li J. Control of tillering in rice. Nature. 2003;422:618-621
45. Ashikari M, Sakakibara H, Lin S, Yamamoto T, Takashi T, Nishimura A, Angeles ER, Qian Q, Kitano H, Matsuoka M. Cytokinin oxidase regulates rice grain production. Science. 2005;309:741-745
46. Song XJ, Huang W, Shi M, Zhu MZ, Lin HX. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet. 2007;39:623-630
47. Xue W, Xing Y, Weng X, Zhao Y, Tang W, Wang L, Zhou H, Yu S, Xu C, Li X, Zhang Q. Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat Genet. 2008;40:761-767
48. Shomura A, Izawa T, Ebana K, Ebitani T, Kanegae H, Konishi S, Yano M. 2008. Deletion in a gene associated with grain size increased yields during rice domestication. Nat Genet. 2008;40:1023-1028
49. Huang X, Qian Q, Liu Z, Sun H, He S, Luo D, Xia G, Chu C, Li J, Fu X. Natural variation at the DEP1 locus enhances grain yield in rice. Nat Genet. 2009;41:494-497
Author contact
![]() Corresponding author: E-mail: sriorg.sg; Telephone: 65-68727480; Fax: 65-68727007
Corresponding author: E-mail: sriorg.sg; Telephone: 65-68727480; Fax: 65-68727007