Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(3):347-363. doi:10.7150/ijbs.7.347 This issue Cite
Research Paper
MiR-21 plays an Important Role in Radiation Induced Carcinogenesis in BALB/c Mice by Directly Targeting the Tumor Suppressor Gene Big-h3
Cong Liu1#, Bailong Li1#, Ying Cheng1#, Jing Lin1, Jun Hao2, Shuyu Zhang3, R.E.J. Mitchel4, Ding Sun1, Jin Ni1, Luqian Zhao1, Fu Gao1 ![]() , Jianming Cai1
, Jianming Cai1 ![]()
1. Department of Radiation Medicine, Second Military Medical University, Xiangyin Road, Shanghai 200433, PR China;
2. Department of Pancreatic Surgery, Changhai Hospital, Second Military Medical University, Xiangyin Road, Shanghai 200433, PR China;
3. School of Radiation Medicine and Public Health, Medical College of Soochow University, Suzhou 215123, China;
4. Radiation Protection Research and Instrumentation Branch, Atomic Energy of Canada Limited, Chalk River Laboratories, Chalk River, ON, K0J 1J0, Canada.
# These authors contributed equally to this work.
Received 2011-2-15; Accepted 2011-3-29; Published 2011-4-1
Abstract
Dysregulation of certain microRNAs (miRNAs) in cancer can promote tumorigenesis, metastasis and invasion. However, the functions and targets of only a few mammalian miRNAs are known. In particular, the miRNAs that participates in radiation induced carcinogenesis and the miRNAs that target the tumor suppressor gene Big-h3 remain undefined. Here in this study, using a radiation induced thymic lymphoma model in BALB/c mice, we found that the tumor suppressor gene Big-h3 is down-regulated and miR-21 is up-regulated in radiation induced thymic lymphoma tissue samples. We also found inverse correlations between Big-h3 protein and miR-21 expression level among different tissue samples. Furthermore, our data indicated that miR-21 could directly target Big-h3 in a 3′UTR dependent manner. Finally, we found that miR-21 could be induced by TGFβ, and miR-21 has both positive and negative effects in regulating TGFβ signaling. We conclude that miR-21 participates in radiation induced carcinogenesis and it regulates TGFβ signaling.
Keywords: Radiation-induced carcinogenesis, Radiation induced thymic lymphoma, MicroRNA, miR-21, Big-h3, TGFβ.
Introduction
MicroRNAs (miRNAs, miRs) are a novel class of small noncoding RNAs, about 22 nucleotides in length, that have been shown to play important roles in many physiological and developmental pathways in a multitude of different organisms [1-4]. These RNAs are produced from endogenous transcripts that serve as substrates for Drosha/Phasa and Dicer, members of the RNase III enzyme family [4, 5]. Drosha and Dicer then work sequentially to generate mature 20-25 nucleotide miRNAs from the hairpin precursors [5, 6]. In animal cells, single-stranded miRNAs bind to specific target mRNAs through partly complementary sequences that are predominantly in the 3′ untranslated region (3′UTR) [3, 7, 8]. The bound mRNAs either remain untranslated, resulting in a decrease in the proteins they encode, or are degraded by the RNA-induced silencing complex (RISC) which results in a decreased number of transcripts[9-11]. Studies have shown that miRNAs are involved in numerous cellular processes including development, differentiation, proliferation, apoptosis, and the immune response [12-16]. Studies from the last ten years have demonstrated that there is altered expression of miRNA genes in several human malignancies, including Burkitt's lymphoma, large cell lymphoma, chronic lymphocytic leukemia, gastric cancer, lung cancer, and liver cancer [12, 17, 18]. In cancer biology, miRNAs function as both oncomiR (oncogene like miRNA) and tumor suppressor genes (anti-oncomiR) depending on the miRNAs and the tumor type [19, 20]. However, the miRNAs that participate in radiation-induced carcinogenesis and that target the tumor suppressor gene, Big-h3, are still unknown.
Radiotherapy is now an important treating method for many types of cancers like breast cancer [21] and lung cancer [22-24]. However, Ionizing radiation (IR) is a well-known carcinogen for various human tissues and a complete carcinogen that is able to initiate and promote neoplastic progression [25, 26]. The mechanism for this promotion is still poorly understood, but studies of mouse thymic lymphomas may provide some hints [27, 28]. Indeed, studies of radiation-induced mouse thymic lymphomas, one of the classic models in radiation-induced carcinogenesis, demonstrated that multi-steps and many factors, like oncogene Ras, tumor suppressor gene Pten and p53, and many cell types like dendritic cells and bone marrow cells were involved in radiation induced carcinogenesis [28-30]. Our previous studies have found that ERK1/2, SHP-2 and STAT3 are also involved in γ-ray Induced thymic lymphoma in BALB/c mice [31]. Then we focused on studying another tumor suppressor gene Big-h3 and its' potential role in radiation-induced carcinogenesis this time.
Tumor suppressor gene Big-h3, also known as TGFBI (TGFβ induced, hereafter Big-h3), was found ubiquitously expressed in human normal tissues, however, down-regulation or lost expression of this gene has been found in a variety of human tumors including lung, breast, prostate, liver and leukemia [32-34]. In addition, Big-h3 expression has been shown to correlate inversely with the tumorigenic phenotype in many types of human cancer cells [32, 35, 36]. Big-h3 expression is also associated with a better response to chemotherapy and other therapeutic effects [37, 38]. However, role of Big-h3 in radiation-induced carcinogenesis is still not clear.
Here in this study, using bioinformatics methods and radiation induced thymic lymphoma model in BALB/c mice, we identified that miR-21 could directly targeting the tumor suppressor gene Big-h3, which may also play an important role in radiation-induced carcinogenesis.
Materials and Methods
Mice and treatments
Male wild-type BALB/c mice, 5-6 weeks of age, were purchased from Chinese academy of science (Shanghai, China). All mice were housed in a pathogen-free facility for all experiments. All animal experiments were undertaken in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals, with the approval of the Laboratory Animal Center of Second Military Medical University, Shanghai. The approval ID for this study was 20071010. All efforts were made to minimize the number of animals used as well as their suffering. A fraction of the mice (about 100) were left as controls. Other mice (about 400) were subjected to whole body irradiation split into four weekly sub-lethal doses of 1.75 Gy (Dose rate: 0.58 Gy/min). Thymic lymphomas were observed to develop at regular intervals of upto 15 months and we find that in our experimental conditions, a global incidence of 30.4% was observed after 6 month or more as described previously [31].
Total Body Irradiation (TBI)
A 60Co irradiator was introduced for total-body ionizing irradiation as described in our previous work [31, 39-42]. In detail: un-anaesthetized mice were placed in well-ventilated plastic boxes and exposed to the 60Co irradiator at a distance of 5 m from the source. Four weekly sub-lethal doses of 1.75 Gy gamma-ray irradiation was delivered at the dose rate of 0.58Gy/min. The mice were then released from the plastic box and allowed free access to food and drinking water [31, 40].
Plasmids
The luciferase-3' UTR reporter constructs were generated by introducing the full length murine Big-h3 3'-UTR into pGL3 promoter vector (Promega) in a method similar to our published paper [43, 44]. We first amplified the wild type Big-h3 3'-UTR sequence by PCR using primers: Big-h3.F (5' ttatctagaCAGGAAGACCGAGGAGGAGA 3') and Big-h3.R (5' ggctctagaTTATTACAAATGCACCATGGGTC 3') and the murine genome cDNA were as a template. The PCR product was ligased into pGL3 promoter vector by the Xba1 site. The Position 429-431 mutated mouse Big-h3 3' UTR plasmid was directly synthesized by Jierui company (Jierui, shanghai, China).
The murine TGFBI/Big-h3 over-expression plasmid was generated in collaboration with Dr. Pu You from the department of cell biology of our university. Briefly, the murine TGFBI/Big-h3 over-expression vector was generated by introducing the full length murine Big-h3 cDNA into the pcDNA3.1 vector (Invitrogen) in a method similar to many published papers [43, 45]. The primers used in this experiment to get the length murine Big-h3 cDNA were Big-h3 Upper Primer Sequence: (5' GATCggatccGCCACCATGGCGCTCCTCATGCGACTGCT 3') (BamHI) and Big-h3 Lower Primer Sequence: (5' GATCtctagaCTAATGCTTCATCCTCTCCAGTAACC 3') (XbaI) and the murine genome cDNA as a template. All PCR products were verified by DNA sequencing.
Luciferase assay
Luciferase assays were carried out in NIH3T3 cells as described previously [43]. First, cells were transfected with appropriate plasmids in 24 -well plates. Then, the cells were harvested and lysised for luciferase assay 48h after transfection as described previously [43, 44]. Luciferase assays were performed using a luciferase assay kit (E1910, Promega) according to the manufacturer's protocol. TK renilla luciferase was used for normalization in Dual Luciferase assays.
FACS assays
FACS assays were used for detection of Treg cells, MDSC cells, Apoptosis rate and intracellular Big-h3 protein level similar to our published methods[42]. Generally, prior to fluorescent antibody staining, all cells were incubated with CD16/CD32 antibody at a concentration of 1µg/106cells/100µl for 15 minutes at 4oC to block Fc receptors. Fluorescent antibodies and respective isotype controls were then added at a concentration of 1µg/106cells/100µl, and cells incubated for a further 30 minutes at 4°C. The cells were washed once with ice-cold PBS (pH 7.2) containing 0.1% NaN3 and 0.5% BSA, and re-suspended in 300µl PBS. Flow Cytometry was performed using a FACS Calibur and data analyzed using Cell-Quest software (both from Becton Dickinson, Mountain View, CA) as described previously [42, 46-48].
For detection of MDSC cells
The CD11b+Gr-1+ cells were regarded as myeloid-derived suppressor cells [49]. Mice were sacrificed and each organ was removed for isolation of CD11b+Gr-1+ cells using a protocol described below. The percentage of CD11b+Gr-1+ myeloid cells or others analyzed in the total number of leukocytes isolated from each organ were determined by FACS.
For detection of Treg cells
The CD4+CD25+ T cells were regarded as Treg-like cells [50]. To detect CD4+CD25+ T cells, spleens of different mice were prepared, then stained with FITC-conjugated anti-CD4 and PE-labeled anti-CD25 and analyzed by FACS as described previously [48].
For detection of Big-h3 protein
Thymus cells and EL4 cells were fixed and Permeabilizated by eBioscience Fixation and Permeabilizationfixed kit (eBioscience) according to the manufacturer's instructions. Then cocultured with antibodies specific for murine Big-h3 (sc-14742, Santa Cruz) and then labeled with FITC labeled Donkey anti-goat IgG Ab as secondary antibodies (Protein Tech Group, 00003-3) in PBS for 30 min, and then washed, analyzed by FACS according to the manufacturer's instructions as described previously [48].
Apoptosis assay
After different treatments, cells were labeled with annexin V-FITC and propidium iodide (PI) provided by BIPEC, following the manufacturer's instructions as described previously [42, 44]. Samples were examined by Fluorescence Activated Cell Sorting (FACS) analysis, and the results were analyzed using CellQuest software (Becton Dickinson, San Jose, CA) as described previously in our published paper [39, 43].
RNA extraction and real-time PCR
RNA from different radiation induced thymic lymphoma tissues and normal thymus tissues was homogenized in Trizol (Invitrogen) and isolated according to the manufacturer's instructions. Reverse Transcription and Real-time PCR was subsequently performed in triplicate using the miScript RT Kit and miScript PCR system (Qiagen) as described detail in previous studies [43, 44]. All microRNA quantification data were normalized to GAPDH. The primer used in this study for detection of mmu-miR-21 was Universal Reverse primer and mmu-miR-21 specific Forward primer provided by the Qiagen Company. While, the primers used in this study for detection of mouse GAPDH was GAPDH F (CCCCACACACATGCACTTACC) and GAPDH R (CCTACTCCCAGGGCTTTGATT).
Western blot analysis
Western blot analysis was performed according to a published method with minor revisions [44, 48]. A BCA protein Assay Reagent Kit (Pierce) was used to measure protein concentration. Samples containing equal amounts of protein were separated by 12% SDS-PAGE and transferred to Protran nitrocellulose membranes (Schleicher and Schuell). Blots were probed with antibodies specific for murine Big-h3 (sc-14742, Santa Cruz) with appropriate donkey anti-goat IgG-horse radish peroxidase-conjugated antibodies as secondary antibodies (Cell Signaling). Supersignal West Femto Maximum Sensitivity substrate (Pierce) was used for the chemiluminescent visualization of proteins.
ELISA Assays
To detect TGFβ productions, the clarified serum or cell supernant from different treatment were subjected to ELISA Assays according to the manufacturer's instructions (R&D System, R&D, Minneapolis, MN) as reported previously [46-48].
Transfection
Cells were cultured and transfected as described previously [43]. NIH3T3 cells and MCF7 cells were seeded at 6×104 cells per well in 24-well plates and transfected with synthetic miRNAs (mimics/inhibitors, genepharm, shanghai, china) at a final concentration of 50nM or 100nM using Lipofectamine 2000 (Invitrogen, ON, Canada) transfection reagent according to the manufacturer's instructions as described previously [44]. Transfection in EL4 cell line was generated with Amaxa electroporation apparatus (Amaxa Biosystems, Koln, Germany) using manufacturer's protocols for each individual cell line (Amaxa Cell Line Nucleofector Kit L) as described previously with minor modifications [51-53]. Cells were infected 48 h later and subjected to further experiments. Three anti-Big-h3 siRNA (siRNA-1, siRNA-2 and siRNA-3) and a control non-targeting siRNA (si-NC) were purchased from genepharm company (genepharm, shanghai, china) and they were transfected into cells similar to miRNA. Cells were infected 48 h later and subjected to further experiments.
Cell proliferation assay
Cells were seeded in 96-well plate at 4 000 cells per well the day before transfection. The cells were transfected with miR-21 mimics or negative control mimics or other vectors as described. CCK8 assay was used to measure the viable, proliferating cells at 24, 48, and 72 h after transfection. The absorbance at 450 nm was measured using a thermo Spectrophotometer as described in our previous work [39, 40].
MicroRNA target prediction
MiRNA targets were predicted using the algorithms TargetScan (http://www.targetscan.org) [54], miRanda (http://www.cbio.mskcc.org/mirnaviewer) [55], PicTar (http://pictar.mdc-berlin.de) [8], miRGen [56, 57] and miRBase (http://www.mirbase.org) [58]. To identify the genes commonly predicted by these different algorithms, results of predicted targets were Interpreted using our own bioinformatics methods and in collaboration with Capitalbio Company (http://www.capitalbio.com).
Statistical analysis
Comparisons between experimental groups and relevant controls were performed by Student's t-test. P< 0.05 was considered a statistically significant difference. The inverse correlation of Big-h3 protein and miR-21 expression levels was examined by Spearman correlation analysis.
Results
Big-h3 is down-regulated and MiR-21 is up-regulated in radiation induced thymic lymphoma tissues
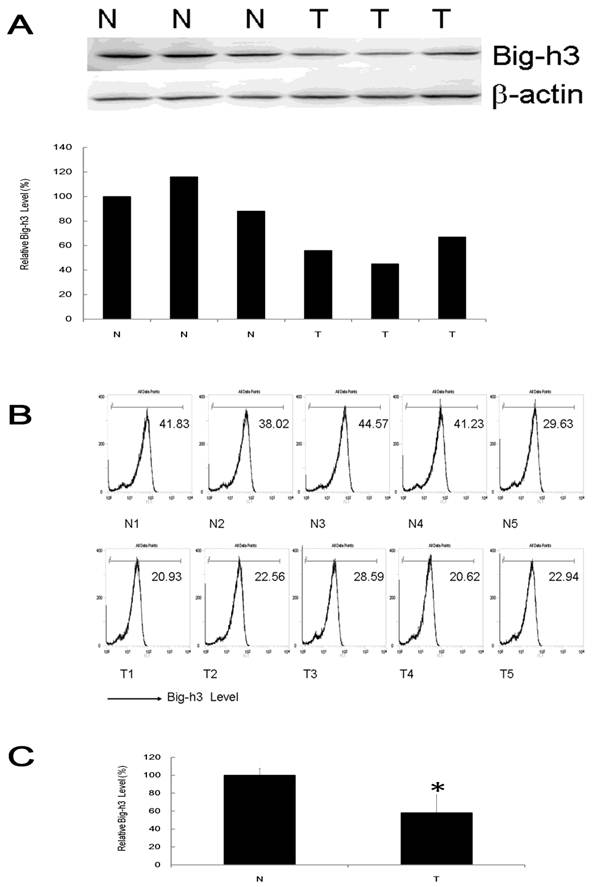
To test the expression of tumor suppressor gene Big-h3 in radiation induced thymic lymphoma tissue samples (shorted as T, tumor) and normal control non-irradiated thymus tissue samples (shorted as N, Non-Irradiated normal control), western blot assays and FACS analysis were performed in 9 pairs and 15 pairs of radiation induced thymic lymphoma tissue samples with normal control non-irradiated thymus tissues of the same strain (BALB/c), sex (male) and age (7-month-old, 6-month post last IR). It was shown that Big-h3 expression level was generally and significantly lower in radiation induced thymic lymphoma tissue samples than in matched normal control non-irradiated thymus tissue samples (Figure 1A, 1B and 1C).
Big-h3 was down-regulated in split radiation induced lymphoma tissue samples. (A) The expression of Big-h3 protein in radiation induced thymic lymphoma tissue sample and normal control non irradiated thymus tissue samples detected by western blot assay. (B) The relative expression of Big-h3 protein in radiation induced lymphoma tissue sample and normal control thymus tissue samples by FACS assay. Data are shown the Mean fluorescence intensity from a representative data of at least three independent experiments. The Mean fluorescence intensity of a proper negative control IgG is 5. (C) Statistical analysis of 1A and 1B. T: Tumor, radiation induced lymphoma tissue samples; N: Normal control thymus tissue samples, regarded as 1(100%). *: P < 0.05.
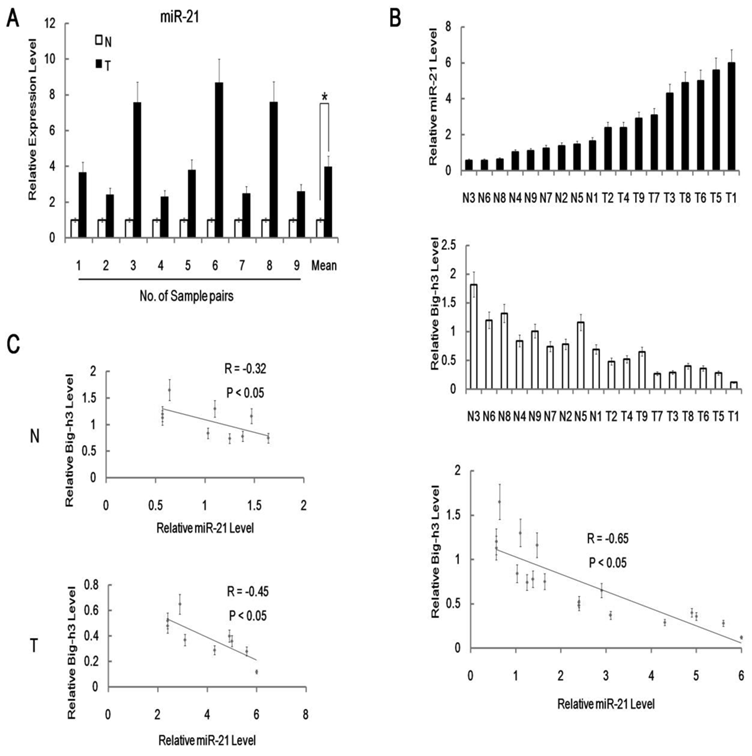
In addition, qRT-PCR anslysis of miR-21 expression level was performed in 9 pairs of radiation induced carcinogenesis tissue samples with normal control non-irradiated thymus tissues. It was shown that miR-21 expression level was significantly higher in radiation induced thymic lymphoma tissues than in matched normal control non-irradiated thymus tissues (Figure 2A). Furthermore, we also found inverse correlations between Big-h3 protein and miR-21 expression level among 9 pairs of different tissues (Figure 2B). We found that inverse correlations between Big-h3 protein and miR-21 expression were stronger in tumor samples(R=-0.45), while a little weaker for non-irradiated normal control tissues (R=-0.32).
MiR-21 was up-regulated in radiation induced lymphoma tissues and inversely correlated with Big-h3. (A) The relative expression of miR-21 in 9 pairs of radiation induced lymphoma tissue samples and normal control thymus tissue samples were detected using real-time PCR. Not all normal samples showed the same expression level of miR-21 in this experiment. The relative expression level of miR-21 in each normal control thymus tissue samples were normalized as 1(100%) in each pair. Mean Data are shown as the means ± standard deviation from triplicate experiments. (B/C) Big-h3 protein and miR-21 expression levels are inversely correlated in radiation induced thymic lymphoma tissue sample and normal control non irradiated thymus tissue samples. (B) Real-time PCR analyses of the relative expression levels of miR-21 and FACS analysis of the relative expression levels of Big-h3 protein in 9 pairs of radiation induced thymic lymphoma tissues and normal control non irradiated thymus tissue samples (The same tissue sample was subjected to both Real-time PCR analyses and FACS analysis). The inverse correlation of Big-h3 protein and miR-21 expression levels was examined by Spearman correlation analysis (R=-0.65, P<0.05). (C) Big-h3 protein and miR-21 expression levels are inversely correlated within the radiation induced thymic lymphoma tissue samples groups (T) and the normal control non irradiated thymus tissue samples (N). *: P < 0.05.
Suppression of radiation induced apoptosis by over-expression of miR-21 in lymphoma cells
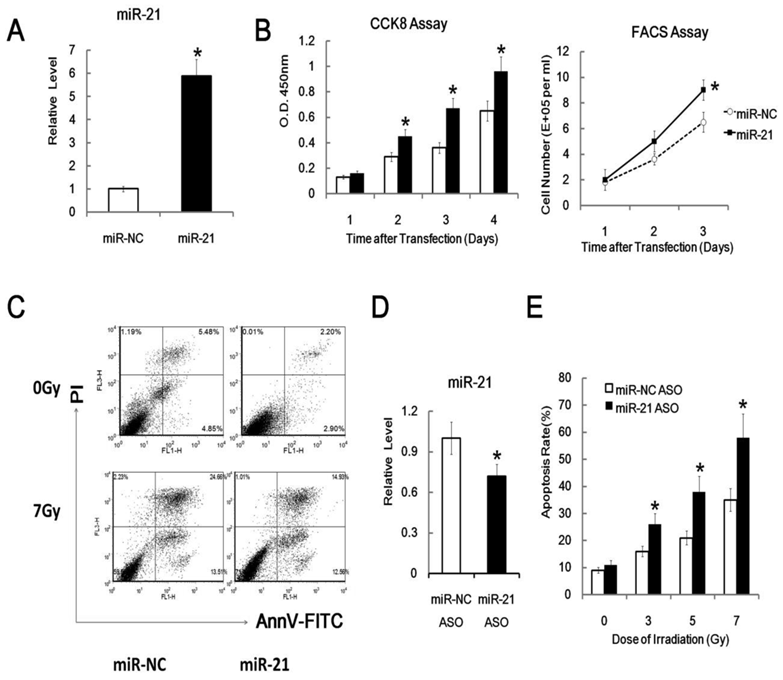
To determine the role of miR-21 in cell proliferation, a miR-21 mimics was used for transfection into the murine thymic lymphoma cell line EL4 cells. The validity of miR-21 ectopic expression was confirmed by quantitative RT-PCR, which revealed a 5.9-fold increase of miR-21 expression in miR-21 transfected cells than in the negative control (NC) transfected cells EL4 group (Figure 3A). To test the effects of miR-21 over-expression on EL4 cell proliferation, we investigated cell growth by CCK8 assay and found that miR-21 could enhance EL4 cell growth (Figure 3B). To further confirm the effect of miR-21 over-expression on the cell growth of EL4 cells, FACS assay was performed for cell counting. The total cell number of EL4 cells transfected with miR-21 mimics was significantly higher than the control group (Figure 3B). Furthermore, the effects of ectopic expression of miR-21 on radiation induced EL4 cell apoptosis were also investigated by FACS analysis. Compared with the blank or scrambled oligonucleotides control group, cell apoptosis was significantly decreased after irradiation in cells transfected with miR-21 mimics (Figure 3C). The natural apoptosis rate of EL4 was 5.0±1.3%. After transfected with miR-21 mimics for 24h and irradiated with 7Gy gamma-ray (Dose rate: 1Gy/min) irradiation and subjected to FACS assay for additional 24 hours later, the apoptosis rate of EL4 were 14.5±5.5 %, which were significantly lower than those of transfected with negative control miR-NC mimics groups (24.7±8.3%). These results indicated that the enhanced expression of miR-21 by gene transfer showed a pro-proliferative and oncogenic effect. Furthermore, miR-21 specific antisense oligo (miR-21 ASO) could down-regulate the expression of miR-21 in EL4 cells and upregulate the apoptosis rate of EL4 cells after irradiation (Figure 3D and 3E). These results indicated a potential therapeutic target of miR-21 in radiation induced thymic lymphoma.
The effects of ectopically expressed miR-21 on cell function. (A) Validation of miR-21 over-expression mimics (miR-21). miR-21 mimics and nonspecific control miRNA mimics (miR-NC) were transfected into cells EL4 cells by Amaxa electroporation apparatus (Amaxa Biosystems, Koln, Germany) using Amaxa Cell Line Nucleofector Kit L as described in the material and method parts, then 24 hours later relative levels of miR-21 were analyzed by qRT-PCR. (B) miR-21 promoted cell proliferation as assessed by the CCK8 assay and FACS analysis different time after transfection of the miR-21 mimics or miRNA negative control mimics (NC) in EL4 cells. (C) MiR-21 protected cells from radiation induced apoptosis as analyzed using flow cytometry method. EL4 cells were transfected with miR-21 mimics or miRNA negative control (NC) and treated with or without 7Gy of gamma-ray irradiation as described in the Materials and Methods. Figure showed a representative data of at least three independent experiments. (D) Validation of miR-21 specific inhibitor antisense oligo (miR-21 ASO). (E) miR-21 specific inhibitor antisense oligo (miR-21 ASO) enhanced radiation induced apoptosis as analyzed using flow cytometry. *: P < 0.05.
Big-h3/TGFBI is a direct target of miR-21
MiRNAs can function as tumor suppressors or oncogenes, depending on whether they specifically target oncogenes or tumor suppressor genes [19, 20]. OncomiRs, such as miR-21, has high expression levels in glioblastoma, breast cancer and tumors of the gastrointestinal tract compared with normal tissues [59-61]. In addition, it could counteract the expression of putative tumor-suppressive targets, such as phosphatase and tensin homolog deleted on chromosome 10 (PTEN), programmed cell death 4 (Pdcd4), tropomyosin 1, maspin and reversion-inducing cysteine-rich protein with kazal motifs [59, 61]. On the basis of these findings, miR-21 has been proposed to play a pivotal role in the onset of several tumor types. However, the potential novel target gene of miR-21 in radiation induced carcinogenesis is still unclear. Therefore, identification of novel miR-21 regulated targets is a necessary step to understand miR-21 functions in radiation-induced carcinogenesis.
We used a three-step consequential approach to identify miR-21 target genes. First, target genes were predicted by bioinformatics analysis; second, target genes were validated by fluorescent reporter assay; and finally, the correlation of miRNA expression and target protein expression were detected to confirm the specific miRNA:mRNA interactions. The target genes of miR-21 were predicted using four programs, known as Targets [55], PicTar [8] , miRGen [56, 57] and miRBase [58]. Of the predicted target genes, the murine tumor suppressor gene Big-h3, whose mRNA 3'UTR contained a putative binding site of miR-21, was identified. This analysis is consistent with the model in which oncomiR play an important role in tumor development by targeting and negatively regulating tumor suppressor genes.
The wild type of mouse Big-h3 mRNA 3'UTR carries a putative miR-21 binding site while the 429-431 mutated mouse Big-h3 3' UTR failed to bind miR-21 (Figure 4.A). To validate the miRNA-target interactions, the wild type and the 429-431 mutated Big-h3 complementary sites were cloned into the 3′UTR of the firefly luciferase gene and co-transfected with miR-21 mimics or negative control RNA (NC) in NIH3T3 cells. As shown in Figure 4B, miR-21 significantly reduced the luciferase activity of the wild type Big-h3 3'UTR with respect to the negative control. The miR-21 mimics at a final concentration of 50 nM and 100 nM both reduced the luciferase activity, but there were no significant difference between the two groups (Data not shown). Therefore, miRNA at a final concentration of 50 nM was transfected into cells in the following experiment. However, miR-21 has no effect on the luciferase activity of the 429-431 mutated Big-h3 3′UTR (Figure 4B). These data indicated that miR-21 may target Big-h3 in a 3′UTR dependent manner.
miR-21 targets Big-h3/TGFBI in a 3'UTR dependent manner. (A) The predicted miR-21 binding site on murine Big-h3/TGFBI mRNA 3'UTR and the mutation strategy is shown. (B) A dual luciferase assay of NIH3T3 cells co-transfected with the firefly luciferase constructs containing the wild type or 429-431 mutated Big-h3/TGFBI 3'-UTR and miR-21 mimics or scrambled negative control (NC). (C) miR-21 mimics inhibit endogenous Big-h3 expression in EL4 cells as detected by western blot assay. *: P < 0.05; NS: No Significant difference detected.
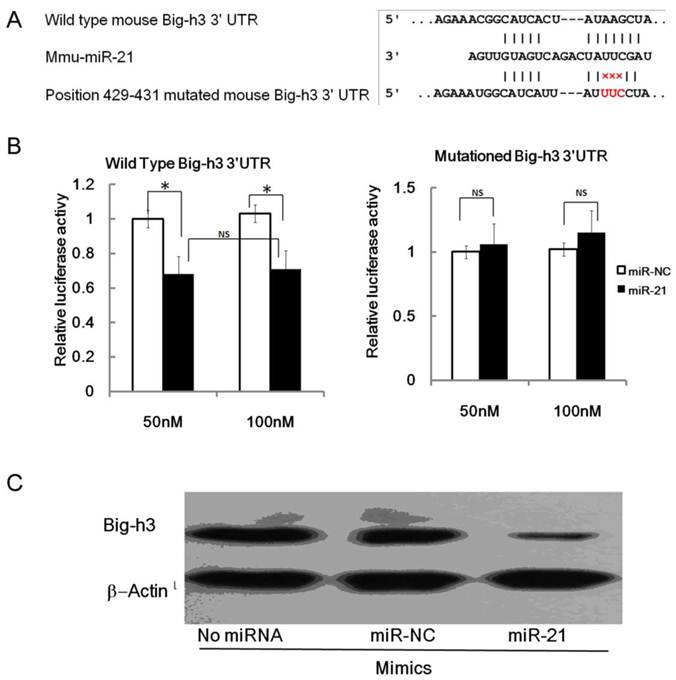
Translational repression is regarded as a major mechanism for miRNA regulation of target gene expression [1-4]. To determine whether miR-21 suppresses endogenous Big-h3 through translational repression, EL4 cells were transfected with miR-21 and the expression of Big-h3 protein was examined by Western blot. It was shown that the amount of Big-h3 protein was decreased after over-expression of miR-21 (Figure 4C), suggesting that miR-21 negatively regulates endogenous Big-h3 protein expression through translational repression mechanism.
Big-h3 is a mediator of miR-21 functions and plays a role in cell proliferation and radiation-induced apoptosis
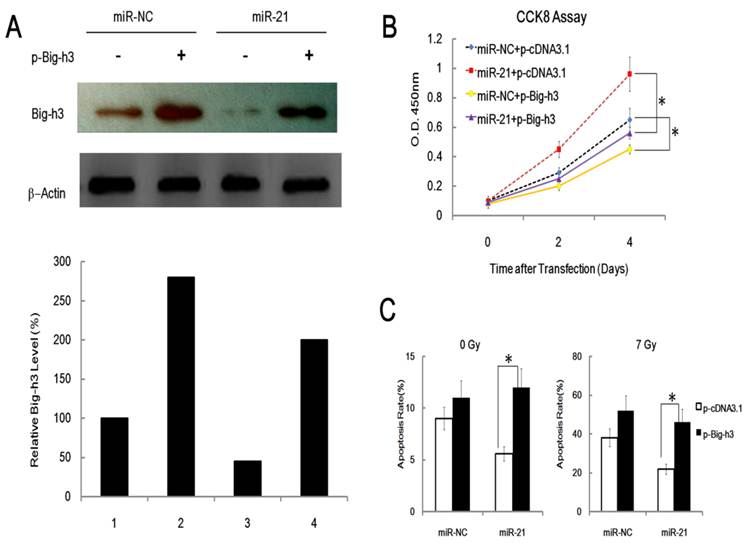
To explore the relevance of Big-h3 in miR-21 related functions, we first performed rescue experiments of Big-h3 over-expression in cells ectopically expressing miR-21. As can be seen from Figure 5A and 5B, over-expression of Big-h3 attenuated the pro-proliferation effect of miR-21 significantly. Furthermore, over-expression of Big-h3 also enhanced the apoptosis rate in cells with or without miR-21 over-expression with or without IR (Figure 5C). These data indicate that Big-h3 does play a role in cell proliferation and radiation-induced apoptosis, while over-expression of Big-h3 rescued the pro-proliferation effect of miR-21. These experiments demonstrate that Big-h3 mediates miR-21 function.
Rescue effects of Big-h3 over-expression in cells ectopically expressing miR-21. (A). EL4 cells were transfection with Big-h3 over-expression vector (p-Big-h3) or control vector (p-cDNA3.1) with (miR-21) or without (miR-NC) miR-21 by Amaxa electroporation apparatus (Amaxa Biosystems, Koln, Germany) using Amaxa Cell Line Nucleofector Kit L as described in the material and method parts and 24 hours after transfection the cell lysis was made and subjected to Western Blot assay for the detection of Big-h3 protein. (B). Over-expression of Big-h3 attenuated the pro-proliferation effect of miR-21; CCK8 Assay. (C).Over-expression of Big-h3 enhanced the apoptosis rate in cells with or without miR-21 over-expression with or without IR. Ann-V/PI double staining assay and detected by FACS Assay; *:P < 0.05.
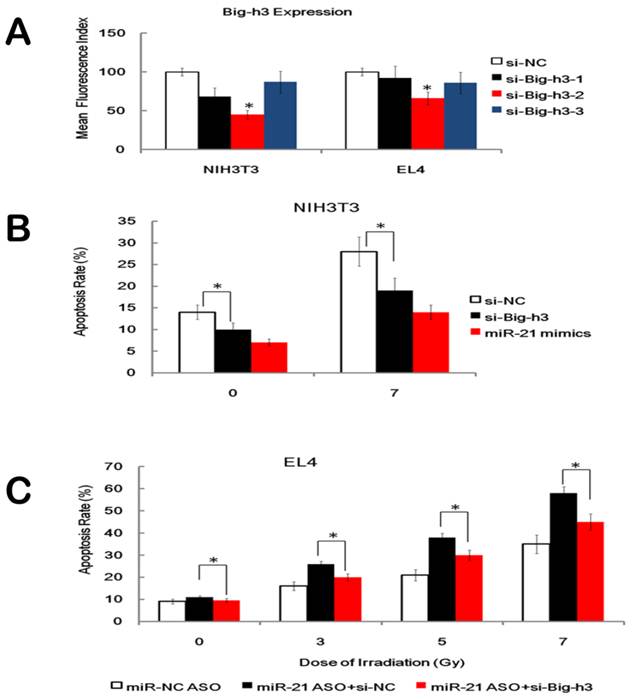
Furthermore, three anti-Big-h3 siRNA (siRNA-1, siRNA-2 and siRNA-3) and a control non-targeting siRNA (siRNA-NC) were transfected into NIH3T3 cells and EL4 cells and we found that anti-Big-h3 siRNA-2 showed the highest Big-h3 knockdown capacity as detected by FACS assay (Figure 6A). We also found that KD of Big-h3 (si-Big-h3), but not KD of nonspecific control (si-NC), down-regulated the apoptosis rate of NIH3T3 cells after IR, which mimics the effects of miR-21 over-expression (Figure 6B). Furthermore, KD of Big-h3 also attenuated, but not fully wiped off the pro-apoptosis role of miR-21 inhibitor in EL4 cells (Figure 6C). These data, taken together, indicate that although other possible mediators existed, one possible downstream target of miR-21 that played a role in cell proliferation and radiation-induced apoptosis was Big-h3.
Effects of Big-h3 Knockdown on cell proliferation and radiation induced apoptosis. (A). Validation of three anti-Big-h3 siRNA (siRNA-1, siRNA-2 and siRNA-3) and a control non-targeting siRNA (si-NC) in transfected NIH3T3 cells and EL4 cells 48 hours after transfection. Data showed the Mean fluorescence intensity level. (B). Knockdown of Big-h3 (si-Big-h3), but not KD of nonspecific control (si-NC), attenuated the apoptosis rate of the NIH3T3 cells after IR, which mimics the effects of miR-21 over-expression. (C). Knockdown of Big-h3 (si-Big-h3) attenuated, but not fully wiped off, the pro-apoptosis role of miR-21 inhibitor (miR-21 ASO) in EL4 cells. *: P < 0.05.
Up-regulation of Tregs, MDSCs and TGFβ production in radiation induced lymphoma mice
We next focused our attention on TGFβ since Big-h3 is a TGFβ induced protein [35]. More recently studies have shown that TGFβ play critical roles in induction of regulatory subset (Tregs) and Myeloid-derived suppressor cells (MDSCs) [49, 50]. Studies also indicated that Tregs and MDSCs are expanded in large numbers during tumor growth, and accumulated in secondary lymphoid organs, blood, and tumor tissue. These expanded Tregs and MDSCs contribute to tumor progression, providing supporting stroma and immune evasion[50]. Important as they are, however, the changes of Tregs, MDSCs and TGFβ in radiation induced thymic lymphoma mice remain poorly defined.
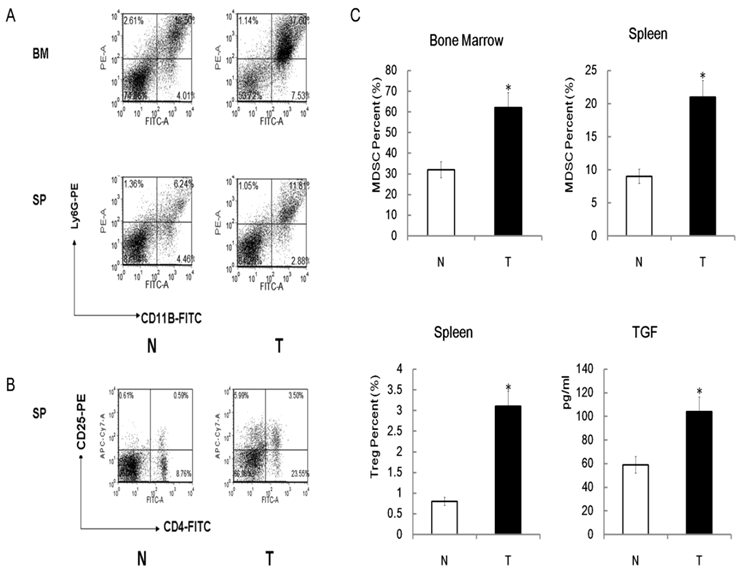
To investigate role of Tregs, MDSCs and TGFβ pathway in radiation-induced carcinogenesis further, we determine the level of Tregs, MDSCs and TGFβ among different mice by FACS and ELISA assays. Similar to other tumor bearing mice, radiation induced lymphoma mice also been found to have higher number of MDSCs in the spleens and bone marrows when compared with normal control mice (Figure 7A). Up-regulation of Tregs in the spleens, but not in bone marrows, of radiation-induced lymphoma mice also been found compared with normal control mice (Figure 7B). Interestingly, we found that although Big-h3 protein level is down-regulated in radiation induced lymphoma tissues, serum from tumor bearing mice showed a higher TGFβ production than normal mice (Figure 7C).
Up-regulation of Tregs, MDSC and TGFβ production in radiation induced lymphoma mice. (A) Bone marrow cells and spleen cell from the radiation-induced lymphoma mice and normal control mice were subjected to FACS assay for detection of MDSCs as described in the Materials and Methods. Figure showed a representative data of at least three independent experiments. (B) Spleen cell from the radiation-induced lymphoma mice and normal control mice were subjected to FACS assay for detection of Tregs as described in the Materials and Methods. Figure showed a representative data of at least three independent experiments. (C) Quantitative analysis of Tregs, MDSC and TGFβ in radiation-induced lymphoma mice and control mice. Serum from the radiation-induced lymphoma mice and normal control mice were subjected to ELISA assays for detection of TGFβ as described in the Materials and Methods. The TGFβ level was calculated and is shown. While, quantitative analysis of Tregs and MDSC were the statistical analysis of 7A and 7B.Data are representative of three independent experiments (n=3~6).T:Tumor, Tissues or serum from radiation induced thymic lymphoma mice; N: Normal, Tissues or serum from normal control mice. *:P < 0.05).
MiR-21 plays different feedback roles on regulation of TGFβ and Big-h3
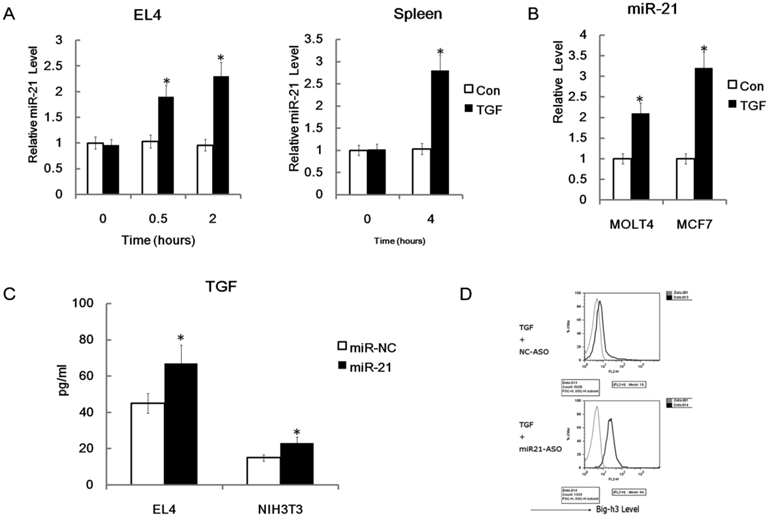
Down-regulation of Big-h3 protein level and up-regulation of TGFβ production have been found in our radiation induced thymic lymphoma tissues and many other cancer cells [62]. However, the crosstalk between Big-h3, TGFβ and miR-21 is still not clear. We hypothesized that miR-21 may have regulating roles in TGFβ signaling. To test our hypothesis, we treated EL4 cells with recombinant murine TGFβ (rmTGFβ). We found that miR-21 could be enhanced rapidly by rmTGFβ (Figure 8A). We also found that miR-21 could be enhanced by rmTGFβ in the primary mouse spleen cells (Figure 8A). Furthermore, we treated human thymic lymphoma cell line MOLT4 cells and human breast cancer cell line MCF7 cells with recombinant human TGFβ1. We found that that miR-21 could also be enhanced by recombinant human TGFβ1 (rhTGFβ1) in these two cell lines (Figure 8B). These data indicated that miR-21 could be induced by TGFβ in many cell types especially the lymphoma cells. While, we also find that miR-21 mimics could enhance the secretion of TGFβ in EL4 cells and NIH3T3 cells (Figure 8C). These data indicate that miR-21 may play a positive feedback role in regulation of TGFβ signaling by up-regulation of TGFβ. However, since Big-h3 was a direct target of miR-21, miR-21 may also play a negative feedback role in regulation of TGFβ signaling by down-regulation of TGFβ inducible protein Big-h3. Moreover, miR-21 specific inhibitors could significantly enhance the induction of Big-h3 by rmTGFβ in EL4 cells (Figure 8D). This data also indicates that miR-21 may play a negative feedback role in regulation of TGFβ signaling by down-regulation of TGFβ inducible protein Big-h3.
Possible crosstalks between miR-21, TGFβ and Big-h3. (A) miR-21 could be induced quickly by recombinant mice TGFβ in EL4 cells and primary murine spleen cells. Left: A time-course expression of miR-21 in murine thymic lymphoma cell line stimulated with 400pm of recombinant mice TGFβ for 0.5-4 hr as indicated; Right: The whole murine spleen cells were stimulated with or without 400pm of rmTGFβ and 4 hours later RNA was made and subjected to Q-RT-PCR assay for the detection of miR-21. Average of three experiments each performed in triplicate with standard errors is presented. (B) miR-21 could be induced in human cell lines by rhTGFβ (recombinant human TGFβ). Human MOLT4 cells and MCF7 cells were stimulated with or without 400pm of rhTGFβ and 4 hours later RNA was made and subjected to Q-RT-PCR assay for the detection of miR-21. (C) miR-21 enhanced TGFβ production in EL4 cells and NIH3T3 cells significantly; ELISA Assay. (D) miR-21 inhibitors could enhance the induction of Big-h3 by recombinant mice TGFβ in EL4 cells. FACS assay. Data are representative of three independent experiments. *: P < 0.05
Discussion
In the last few years, several studies have shown the dysregulation of miRNAs in various types of cancers [4, 63, 64]. Identification of cancer-specific miRNAs and their targets is critical for understanding their role in tumorigenesis and may be important for defining novel therapeutic targets [1, 5, 14, 65]. Studies have shown that ionizing radiation is a well-known carcinogen for various human tissues and a complete carcinogen that is able to initiate and promote neoplastic progression [25]. Here, by studying the radiation-induced thymic lymphomas in BALB/c mice, one of the classic models in radiation-induced carcinogenesis, we focused on the role of miR-21 in the pathogenesis of radiation-induced carcinogenesis. We identified that the miR-21 may also play an important role in radiation-induced carcinogenesis by directly targeting the tumor suppressor gene Big-h3. To the best of our knowledge, this may be the first report of miR-21 and the tumor suppressor gene Big-h3 in radiation induced thymic lymphoma in BALB/c mice.
Big-h3 was firstly identified in a human lung adenocarcinoma cell line (A549) treated with TGF-β [32, 35, 36]. Many studies have previously shown that Big-h3 is ubiquitously expressed in human/mice normal tissues. However, down-regulation or lost expression of this gene has been found in various human tumor tissues including lung, breast, prostate, and leukemia [32]. In addition, down-regulation of Big-h3 expression correlates with tumorigenic phenotype in many cancer cells [35, 36]. Our results indicated that Big-h3 protein down-regulated in radiation induced lymphoma tissues were thus consistent with these findings and Big-h3 may also play important role in radiation-induced carcinogenesis. Our data also indicate that the tumor suppressor gene Big-h3 maybe a potential therapeutic target of radiation induced carcinogenesis.
Furthermore, we also found inverse correlations between Big-h3 protein and miR-21 expression among different tissues and our data also strongly indicated that miR-21 participate in radiation induced carcinogenesis by directly targeting the tumor suppressor gene Big-h3.
However, although the TGF-β inducible protein Big-h3 was down-regulated in radiation induced lymphoma tissues, up-regulation of Tregs, MDSCs and TGFβ production in radiation-induced lymphoma mice was also observed. Studies have shown that TGFβ played critical roles in induction of regulatory subset (Tregs) and Myeloid-derived suppressor cells (MDSCs). In mice, removal of the primary tumor results in reduction of the number of systemic MDSCs, revealing a causal role for MDSCs in tumor growth [49]. Purified MDSCs have been shown to inhibit both CD4+ and CD8+ T cell responses in vitro [49]. Furthermore, studies imply that these cells can down-regulate T cell functions in vivo and promote tumor metastasis by releasing a number of chemokines [49]. Our results that Tregs, MDSCs and TGFβ production was up-regulation in radiation-induced lymphoma mice were thus consistent with these findings [49, 50].
Ionizing radiation has been shown to release TGFβ in vitro and in vivo [66]. In normal tissues however, the binding of TGFβ to cell-surface receptors leads to growth inhibition, apoptosis, inhibition of angiogenesis, induction of senescence to maintain normal tissue architecture [67]. The situation in tumors seems to be different. In malignancy, TGFβ expression is often significantly increased, and yet these tumor cells do not respond to TGFβ due to multiple mechanisms [67]. This switches the balance in favor of the pro-oncogenic functions of TGFβ, such as increased motility/invasiveness, growth stimulation, enhanced angiogenesis and, importantly, immunosuppression. Furthermore, we also found that miR-21 could be induced by TGFβ and miR-21 has both positive and negative effects in regulating TGFβ signaling.
Since recent work suggests that TGFβ could increase miR-21 expression in human primary pulmonary artery smooth muscle cells (PASMCs) [68], it would be important because it may be a novel model to explain the bi-functional role of TGFβ in carcinogenesis[69]. On the one hand, TGFβ could induce Big-h3 to exert its tumor suppressing function. On the other hand, TGFβ could also induce miR-21 to down-regulate Big-h3 to exert its pro-tumor function (Figure 9).
A possible model for the bi-functional effects of TGFβ in carcinogenesis.
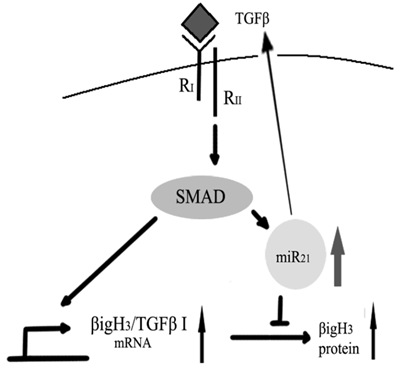
In conclusion, we identified that the miR-21 could directly target tumor suppressor gene Big-h3, which may also play an important role in radiation-induced carcinogenesis. More importantly, our data also strongly indicated that miR-21 has both positive and negative feedback effects in regulating TGFβ signaling.
Acknowledgements
We thank Dr. Zihai Li, Dr. Liren Qian, Dr. Cindy Savage and Trip Barthel M.A for help editing of the manuscript. Special thanks to Dr. Xuetao Cao, Dr. Sheng Xu, Dr. Yushi Yao and Dr. Weimin Sun from National Key Laboratory of Medical Immunology of our university for providing additional helps. We wish to thank Dr. Yiping Hu, Dr. Pu You, Dr. Zhiying He and Dr. Bing Yu from department of cell biology of our university for help designing the Big-h3 over-expression experiments. We wish to thank Dr. Min Li, Dr. Yiling Cai and Dr. Junqin Wang from the department of hygienics of our university for help doing the q-RT-PCR assays. We wish to thank Dr. Shengsheng Yang from the department of biochemistry and Dr. Yue Wang from the department of genetics of our university for their helpful advice on the Dual Luciferase assays. We wish to thank Dr. Xiaoyan Zhu and Dr. Lu Gao from the department of physiology of our university for their helps.
Funding: This work was supported in part by the grants from National Natural Science Foundation of China (No. 31070761) and by the grants from Natural Science Foundation of Shanghai, China (No.09ZR1439400). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests
The authors have declared that no competing interests exist.
References
1. Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215-33
2. Lanford R.E. et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327(5962):198-201
3. Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350-5
4. Melton C, Judson R.L, Blelloch R. Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. Nature. 2010;463(7281):621-6
5. Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281-97
6. Ruby J.G, Jan C.H, Bartel D.P. Intronic microRNA precursors that bypass Drosha processing. Nature. 2007;448(7149):83-6
7. Lewis B.P, Burge C.B, Bartel D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15-20
8. Lewis B.P. et al. Prediction of mammalian microRNA targets. Cell. 2003;115(7):787-98
9. Yekta S, Shih I.H, Bartel D.P. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004;304(5670):594-6
10. Farh K.K. et al. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 2005;310(5755):1817-21
11. Baek D. et al. The impact of microRNAs on protein output. Nature. 2008;455(7209):64-71
12. Mayr C, Hemann M.T, Bartel D.P. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315(5818):1576-9
13. Giraldez A.J. et al. MicroRNAs regulate brain morphogenesis in zebrafish. Science. 2005;308(5723):833-8
14. Chen C.Z. et al. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303(5654):83-6
15. Hornstein E. et al. The microRNA miR-196 acts upstream of Hoxb8 and Shh in limb development. Nature. 2005;438(7068):671-4
16. Marson A. et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134(3):521-33
17. Calin G.A, Croce C.M. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6(11):857-66
18. Calin G.A. et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl Med J. 2005;353(17):1793-801
19. Esquela-Kerscher A, Slack F.J. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6(4):259-69
20. Hammond S.M. MicroRNAs as tumor suppressors. Nat Genet. 2007;39(5):582-3
21. Pierce L.J, Haffty B.G. Radiotherapy in the treatment of hereditary breast cancer. Semin Radiat Oncol. 2011;21(1):43-50
22. Barcellos-Hoff M.H, Park C, Wright E.G. Radiation and the microenvironment - tumorigenesis and therapy. Nat Rev Cancer. 2005;5(11):867-75
23. Thoms J. et al. Neoadjuvant radiotherapy for locally advanced and high-risk prostate cancer. Nat Rev Clin Oncol. 2011Feb;8(2):107-13
24. Bewley D.K. Pions and heavy ions in radiotherapy. Nature. 1972;237(5349):17-9
25. Little J.B. Radiation carcinogenesis. Carcinogenesis. 2000;21(3):397-404
26. Trott K.R, Rosemann M. Molecular mechanisms of radiation carcinogenesis and the linear, non-threshold dose response model of radiation risk estimation. Radiat Environ Biophys. 2000;39(2):79-87
27. Dange P. et al. Radiation-induced incidence of thymic lymphoma in mice and its prevention by antioxidants. J Environ Pathol Toxicol Oncol. 2007;26(4):273-9
28. Villa-Morales M, Santos J, Fernandez-Piqueras J. Functional Fas (Cd95/Apo-1) promoter polymorphisms in inbred mouse strains exhibiting different susceptibility to gamma-radiation-induced thymic lymphoma. Oncogene. 2006;25(14):2022-9
29. Utsuyama M, Hirokawa K. Radiation-induced-thymic lymphoma occurs in young, but not in old mice. Exp Mol Pathol. 2003;74(3):319-25
30. Santos J. et al. A new locus for resistance to gamma-radiation-induced thymic lymphoma identified using inter-specific consomic and inter-specific recombinant congenic strains of mice. Oncogene. 2002;21(43):6680-3
31. Fu Z. et al. Expression changes of ERK1/2, STAT3 and SHP-2 in bone marrow cells from gamma-ray induced leukemia mice. J Radiat Res (Tokyo). 2006;47(2):121-30
32. Zhao Y.L, Piao C.Q, Hei T.K. Downregulation of Betaig-h3 gene is causally linked to tumorigenic phenotype in asbestos treated immortalized human bronchial epithelial cells. Oncogene. 2002;21(49):7471-7
33. Kang S, Dong S.M, Park N.H. Frequent promoter hypermethylation of TGFBI in epithelial ovarian cancer. Gynecol Oncol. 2010;118(1):58-63
34. Shah J.N. et al. Methylation screening of the TGFBI promoter in human lung and prostate cancer by methylation-specific PCR. Cancer BMC. 2008;8:284
35. Zhang Y. et al. TGFBI deficiency predisposes mice to spontaneous tumor development. Cancer Res. 2009;69(1):37-44
36. Zhao Y, El-Gabry M, Hei T.K. Loss of Betaig-h3 protein is frequent in primary lung carcinoma and related to tumorigenic phenotype in lung cancer cells. Mol Carcinog. 2006;45(2):84-92
37. Ahmed A.A. et al. The extracellular matrix protein TGFBI induces microtubule stabilization and sensitizes ovarian cancers to paclitaxel. Cancer Cell. 2007;12(6):514-27
38. Irigoyen M. et al. TGFBI expression is associated with a better response to chemotherapy in NSCLC. Mol Cancer. 2010;9:130
39. Qian L. et al. Radioprotective effect of hydrogen in cultured cells and mice. Free Radic Res. 2010;44(3):275-82
40. Qian L. et al. The potential cardioprotective effects of hydrogen in irradiated mice. J Radiat Res (Tokyo). 2010;51(6):741-7
41. Liu C. et al. Hydrogen therapy may be an effective and specific novel treatment for acute radiation syndrome. Med Hypotheses. 2010;74(1):145-6
42. Liu C. et al. Gamma-ray Irradiation Impairs Dendritic Cell Migration to CCL19 by Down-regulation of CCR7 and Induction of Cell Apoptosis. Int J Biol Sci. 2011;7(2):168-79
43. Hao J. et al. MicroRNA 483-3p suppresses the expression of DPC4/Smad4 in pancreatic cancer. Lett FEBS. 2011;585(1):207-13
44. Hao J. et al. MicroRNA 421 suppresses DPC4/Smad4 in pancreatic cancer. Biochem Biophys Res Commun. 2011;406(4):552-7
45. An H. et al. Phosphatase SHP-1 promotes TLR- and RIG-I-activated production of type I interferon by inhibiting the kinase IRAK1. Nat Immunol. 2008;9(5):542-50
46. Ohl L. et al. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity. 2004;21(2):279-88
47. Zhang M. et al. Splenic stroma drives mature dendritic cells to differentiate into regulatory dendritic cells. Nat Immunol. 2004;5(11):1124-33
48. Chen T. et al. Cyclosporin A impairs dendritic cell migration by regulating chemokine receptor expression and inhibiting cyclooxygenase-2 expression. Blood. 2004;103(2):413-21
49. Hiratsuka S. et al. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006;8(12):1369-75
50. Bluestone J.A, Abbas A.K. Natural versus adaptive regulatory T cells. Nat Rev Immunol. 2003;3(3):253-7
51. Zhang Y. et al. Interaction of Mint2 with TrkA is involved in regulation of nerve growth factor-induced neurite outgrowth. J Biol Chem. 2009;284(18):12469-79
52. Su S. et al. Proteinase-activated receptor 2 expression in breast cancer and its role in breast cancer cell migration. Oncogene. 2009;28(34):3047-57
53. Rui Y. et al. PECAM-1 ligation negatively regulates TLR4 signaling in macrophages. Immunol J. 2007;179(11):7344-51
54. Krek A. et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37(5):495-500
55. John B. et al. Human MicroRNA targets. PLoS Biol. 2004;2(11):e363
56. Megraw M. et al. miRGen: a database for the study of animal microRNA genomic organization and function. Nucleic Acids Res. 2007;35(Database issue):D149-55
57. Alexiou P. et al. miRGen 2.0: a database of microRNA genomic information and regulation. Nucleic Acids Res. 2010;38:D137-41
58. Griffiths-Jones S. et al. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36(Database issue):D154-8
59. Selaru F.M. et al. MicroRNA-21 is overexpressed in human cholangiocarcinoma and regulates programmed cell death 4 and tissue inhibitor of metalloproteinase 3. Hepatology. 2009;49(5):1595-601
60. Vinciguerra M. et al. Unsaturated fatty acids inhibit the expression of tumor suppressor phosphatase and tensin homolog (PTEN) via microRNA-21 up-regulation in hepatocytes. Hepatology. 2009;49(4):1176-84
61. Meng F. et al. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133(2):647-58
62. Qian B. et al. High miR-21 expression in breast cancer associated with poor disease-free survival in early stage disease and high TGF-beta1. Breast Cancer Res Treat. 2009;117(1):131-40
63. He L. et al. microRNAs join the p53 network--another piece in the tumour-suppression puzzle. Nat Rev Cancer. 2007;7(11):819-22
64. Nicoloso M.S. et al. MicroRNAs--the micro steering wheel of tumour metastases. Nat Rev Cancer. 2009;9(4):293-302
65. Hammond S.M, Sharpless N.E. HMGA2, microRNAs, and stem cell aging. Cell. 2008;135(6):1013-6
66. Barcellos-Hoff M.H. et al. Transforming growth factor-beta activation in irradiated murine mammary gland. J Clin Invest. 1994;93(2):892-9
67. Derynck R, Akhurst R.J, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29(2):117-29
68. Davis B.N. et al. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454(7200):56-61
69. Bachman K.E, Park B.H. Duel nature of TGF-beta signaling: tumor suppressor vs. tumor promoter. Curr Opin Oncol. 2005;17(1):49-54
Author contact
![]() Corresponding author: Jianming Cai and Fu Gao. Address: Department of Radiation Medicine, Faculty of Naval Medicine, Second Military Medical University; 800, Xiangyin Road 200433, Shanghai; P.R. China. Fax: +86-21-81871150. E-mail: caijianming882003com and gaofu_2002com.cn.
Corresponding author: Jianming Cai and Fu Gao. Address: Department of Radiation Medicine, Faculty of Naval Medicine, Second Military Medical University; 800, Xiangyin Road 200433, Shanghai; P.R. China. Fax: +86-21-81871150. E-mail: caijianming882003com and gaofu_2002com.cn.