Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
STAT3 signaling in liver...
STAT3 signaling in liver...
STAT3 in fatty liver and glucose...
STAT3 in liver injury and...
STAT3 and liver regeneration
STAT3 in liver fibrosis
STAT3 and liver cancer
STAT3 as a therapeutic target to...
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(5):536-550. doi:10.7150/ijbs.7.536 This issue Cite
Review
Signal Transducer and Activator of Transcription 3 in Liver Diseases: A Novel Therapeutic Target
Hua Wang1, Fouad Lafdil2,3, Xiaoni Kong1, Bin Gao1 ![]()
1. Laboratory of Liver Diseases, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, MD 20892, USA;
2. Laboratory of Liver Pathophysiology, INSERM, U955, Créteil, F-94000 France;
3. Université Paris-Est, Faculté de Médecine, UMR-S955, Créteil, F-94000 France.
Received 2011-2-28; Accepted 2011-4-18; Published 2011-4-27
Abstract
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor that is activated by many cytokines and growth factors and plays a key role in cell survival, proliferation, and differentiation. STAT3 activation is detected virtually in all rodent models of liver injury and in human liver diseases. In this review, we highlight recent advances of STAT3 signaling in liver injury, steatosis, inflammation, regeneration, fibrosis, and hepatocarcinogenesis. The cytokines and small molecules that activate STAT3 in hepatocytes may have therapeutic benefits to treat acute liver injury, fatty liver disease, and alcoholic hepatitis, while blockage of STAT3 may have a therapeutic potential to prevent and treat liver cancer.
Keywords: STAT3, STAT3 signaling, liver injury, liver diseases
Introduction
Alcoholic hepatitis, nonalcoholic steatohepatitis, and viral hepatitis are the 3 major causes of chronic liver diseases, leading to liver fibrosis, cirrhosis, and the end-stage of hepatocellular carcinoma. It is generally accepted that inflammation, characterized by an infiltration of inflammatory cells, plays a key role in promoting the progression of liver diseases. These inflammatory cells can modulate the progression of liver diseases by producing a wide variety of cytokines that activate multiple signaling pathways in liver cells and subsequently affect these cells. Among these pathways, the roles of nuclear factor-kappa B (NF-κB) and signal transducer and activator of transcription 3 (STAT3) in the pathogenesis of liver diseases have been extensively investigated. It has been shown that NF-κB plays complex and multicellular roles in hepatic inflammation, injury, fibrosis, regeneration, and hepatocarcinogenesis, which have been summarized in several excellent reviews [1-5]. In 2004, we summarized the role of STAT3 in liver injury, inflammation, and regeneration [6]. Since then, cell-type specific STAT3 knockout mice and gp130 knockout mice have been made available and used extensively to investigate the functions of STAT3 in many murine liver injury models. In this review, we highlight recent advances in STAT3 signaling in liver pathophysiology and discuss the potential application of STAT3 as a therapeutic target to treat liver diseases.
STAT3 signaling in liver parenchymal cells (hepatocytes)
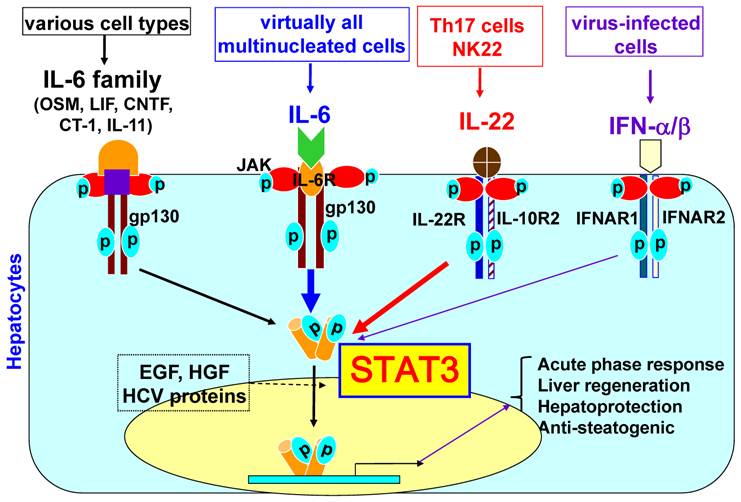
The liver, the largest solid organ in the body, plays a key role in metabolism, detoxification, and innate immunity [7]. Eighty percent of the liver volume, or seventy percent of liver cells, is composed of hepatocytes that are responsible for the metabolic and detoxifying functions of the liver. The remaining cells are composed of Kupffer cells (liver resident macrophages), sinusoidal endothelial cells, stellate cells, and lymphocytes. It has been shown that a wide variety of cytokines can activate STAT3 in hepatocytes. These cytokines include interleukin-6 (IL-6), the IL-6 family of cytokines (such as leukemia inhibitory factor, ciliary neurotrophic factor, oncostatin M, cardiotrophin-1, and IL-11), and IL-22 [6]. IFN-α/β and IFN-γ mainly induce STAT1 activation in hepatocytes, but also significantly stimulate STAT3 activation in hepatocytes [8]. As shown in Fig. 1, the ligation of these cytokines to their receptors induces receptor dimerization, which is followed by activation of the receptor-associated Janus tyrosine kinases (JAKs). This receptor-kinase complex interacts with and phosphorylates the SH2-containing cytoplasmic protein STAT3. The phosphorylated STAT3 then forms a dimer and translocates to the nucleus to activate the transcription of many target genes that play important roles in inducing the acute phase response, promoting hepatocyte survival and liver regeneration, and ameliorating steatosis. In addition, hepatocyte growth factor, epidermal growth factor, and hepatitis viral proteins can also activate STAT3 in hepatocytes to a lesser extent.
STAT3 signaling in hepatocytes. Hepatocytes express high levels of gp130, which is a common signal chain for IL-6 and IL-6 family cytokines, high levels of IL-6 receptors and various corresponding receptors for IL-6 family cytokines. IL-6 family cytokines include leukemia inhibitory factor (LIF), ciliary neurotrophic factor (CNTF), oncostatin M (OSM), cardiotrophin-1 (CT-1), and IL-11. The ligation of these cytokines (IL-6 and IL-6 family cytokines) with their corresponding receptors leads to the dimerization of gp130, followed by dimerization of gp130-associated Janus kinases (JAKs) and phosphorylation of JAKs and gp130. This receptor-kinase complex then recruits and phosphorylates cytoplasmic protein STAT3. Phosphorylated STAT3 forms a dimer, translocates into the nuclei and subsequently induces transcription of many genes. Hepatocytes also express high levels of IL-22R1 and IL-10R2 for IL-22 signaling. IL-6, IL-6 family cytokines, and IL-22 predominantly activate STAT3, but also induce a weak activation of other STATs and MAP kinases. Human hepatocytes express high levels of IFNAR1 and functional IFNAR2c (while mouse and rat hepatocytes predominantly express inhibitory IFNAR2a and poorly respond to IFN-α stimulation). IFN-α/β predominately induce STAT1 activation in primary human hepatocytes but also induce strong STAT3 activation. Activated STAT3 induces transcription of many genes that play important roles in inducing acute phase responses, promoting hepatocyte survival and liver regeneration, and ameliorating fatty liver.
STAT3 signaling in liver non-parenchymal cells
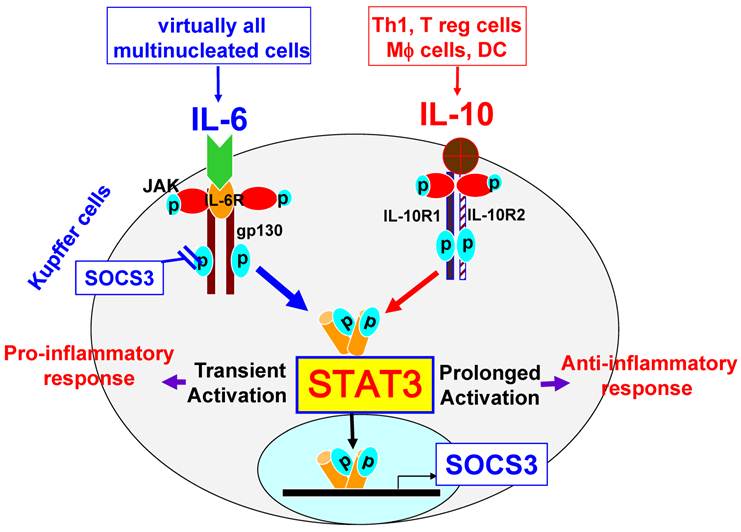
Kupffer cells are liver resident macrophages that account for 80% to 90% of the total population of fixed tissue macrophages in the body. Kupffer cells are the major source not only of proinflammatory cytokines such as IL-6, but also of anti-inflammatory cytokines such as IL-10. In addition, Kupffer cells can also respond to IL-6 and IL-10. Notably, both IL-6 and IL-10 induce STAT3 activation in macrophages, but exert opposing functions. While IL-10 induces STAT3 activation and subsequently inhibits LPS-induced inflammatory responses in macrophages and Kupffer cells [9-11], IL-6 also activates STAT3 but potentiates proinflammatory responses in peritoneal macrophages [12]. Although the effect of IL-6 on Kupffer cells has not been investigated, it is plausible to speculate that IL-6 may also promote proinflammatory responses in Kupffer cells. The reason why STAT3 activation by IL-10 or IL-6 inhibits or promotes, respectively, the macrophage inflammatory response remains unclear. It has been shown that both IL-6 and IL-10 activate STAT3 and subsequently upregulate expression of suppressor of cytokine signaling 3 (SOCS3) in macrophages [12]. SOCS3 then, in turn, inhibits IL-6 signaling via binding to gp130, resulting in IL-6 induction of transient STAT3 activation, while SOCS3 does not block IL-10 activation of STAT3; therefore, IL-10 induces prolonged STAT3 activation (Fig. 2) [12]. It is believed that IL-6 induction of transient STAT3 activation is responsible for its pro-inflammatory response, while IL-10 induces prolonged STAT3 activation and subsequently inhibits inflammatory responses in macrophages [12]. In addition, the effects of other IL-6 family cytokines on STAT3 activation and pro-inflammatory responses in Kupffer cells remain largely unknown.
STAT3 signaling in Kupffer cells. Kupffer cells express high levels of IL-10R1 and IL-10R2. The ligation of IL-10 with IL-10R1 and IL-10R2 leads to prolonged activation of STAT3, thereby inhibiting inflammatory responses. In contrast, the ligation of IL-6 with IL-6R and gp130, which are expressed at high levels on Kupffer cells, leads to transient activation of STAT3, followed by the induction of inflammatory responses. STAT3 activation induces expression of SOCS3, which in turns inhibits IL-6 activation of STAT3, but does not inhibit IL-10 signaling.
STAT3 signaling in hepatic stellate cells. Hepatic stellate cells express high levels of the long form of the leptin receptor (OBRL). The ligation of leptin with OBRL induces activation of STAT3, leading to stellate cell activation, proliferation, and survival. IL-6 can also stimulate stellate cell survival, proliferation, and activation via binding of IL-6R and gp130 on stellate cells.
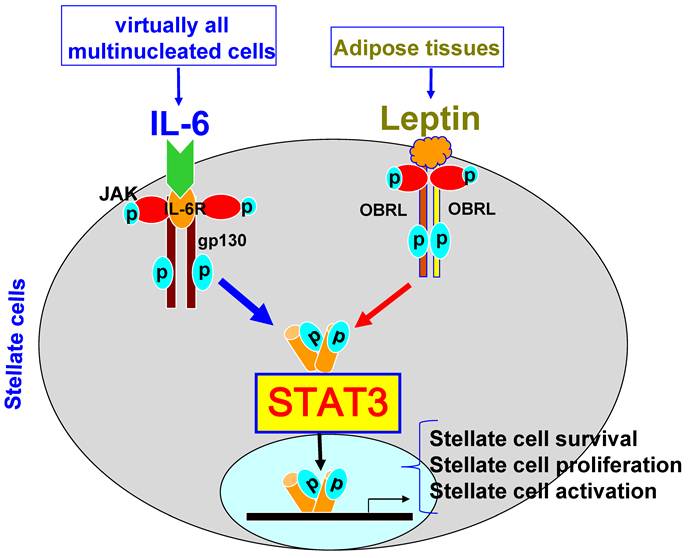
Hepatic stellate cells are located in the space of Disse (a small area between the endothelial cells and hepatocytes) and store 75% of the body's supply of vitamin A. Activation of hepatic stellate cells plays a key role in the pathogenesis of liver fibrosis via producing extracellular matrix proteins [13,14]. IL-6 and leptin have been shown to activate STAT3 in hepatic stellate cells and promote their survival and proliferation, thereby contributing to liver fibrogenesis [15,16]. However, the exact role of STAT3 in hepatic stellate cells remains to be determined.
Liver sinusoidal endothelial cells lack a basement membrane and form a fenestrated monolayer, separating hepatocytes from the passing blood. It has been shown that IL-6 treatment activates STAT3 and protects against cell death in liver sinusoidal endothelial cells [17,18].
STAT3 in fatty liver and glucose metabolism
Fatty liver (accumulation of fat in hepatocytes) is mainly caused by alcohol consumption and obesity, but can also be caused by metabolic diseases, nutritional factors, drugs, toxins, genotype 3 hepatitis C infection, and alpha 1-antitrypsin deficiency. The presence of a fatty liver was considered to be a benign feature in the past, but is now known to be prone to steatohepatitis, fibrosis, and hepatocellular carcinoma [19-21]. Excess fat accumulation in hepatocytes may lead to hepatocellular injury via direct cellular cytotoxicity mediated by FFAs, oxidative stress, lipid peroxidation, mitochondrial impairment, and cytokine-induced hepatoxicity [19-23]. For example, both IL-1β and TNF-α have been shown to promote hepatocellular damage and the development of fatty liver [24-26]. In contrast, several cytokines that activate STAT3 in hepatocytes are known to ameliorate fatty liver. IL-6 deficient-mice are more susceptible to alcohol- and high-fat diet-induced fatty liver [27,28], while treatment with IL-6 ameliorates fatty liver [29]. Recent studies also showed that treatment with IL-22 induces STAT3 activation in the liver and alleviates steatosis induced by alcohol or high-fat diet feeding [30,31]. The protective effect of IL-22 on alcoholic fatty liver is diminished in hepatocyte-specific STAT3 knockout mice [30], suggesting that IL-22 ameliorates steatosis through activation of STAT3 in hepatocytes. Furthermore, disruption of gp130 (upstream of STAT3) or STAT3 in hepatocytes exacerbates fatty liver induced by a choline-deficient, ethionine-supplemented diet [32], alcohol-containing diet [33], or high-fat diet [34], whereas overexpression of constitutively activated STAT3 ameliorates high-fat diet-induced fatty liver [34]. The anti-steatogenic effect of hepatocyte STAT3 is mediated, at least in part, via inhibition of SREBP-1, a master transcription factor that controls lipid synthesis, and subsequent suppression of hepatic lipogenesis [33,34]. However, the mechanism by which STAT3 represses SREBP-1 remains to be determined.
In addition to controlling lipid metabolism, STAT3 signaling also plays an important role in maintaining normal glucose homeostasis via downregulation of hepatic gluconeogenic genes, including glucose-6-phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase (PEPCK), in hepatocytes [34,35]. STAT3 activation inhibits expression of G6Pase and PEPCK in both cultured hepatocytes and mouse liver; such inhibition is mediated via STAT3 binding to the promoters of the G6Pase and PEPCK genes [35].
STAT3 in liver injury and inflammation
STAT3 in hepatocytes: It has been reported that treatment with IL-6, IL-6 family cytokines, or IL-22 protects against liver injury in many rodent models [6,17,29,36-39], while blockage of IL-22 with a neutralizing antibody exacerbates liver injury induced by Con A injection [37,40] or by viral infection [41]. Transgenic mice with overexpression of IL-22 in the liver are completely resistant to T cell hepatitis-induced hepatocellular damage [42]. Moreover, disruption of the IL-6/gp130, OSM, or IL-22 genes, or hepatocyte STAT3, increases the susceptibility of mice to liver injury in most animal models tested so far [6,43-53]. All of these findings clearly demonstrate that STAT3 plays a key role in protecting against hepatocellular damage. In contrast to the well-documented hepatoprotection, hepatocyte STAT3 has a more complex role in liver inflammation. Compared to wild-type mice, hepatocyte-specific STAT3 knockout mice have reduced liver inflammation in models of liver injury induced by acute carbon tetrachloride injection [50] or alcohol feeding [33] but elevated liver inflammation in Con A-induced T cell hepatitis (Lafdil and Gao, unpublished data) or in LPS-induced liver injury [54], suggesting that hepatocyte STAT3 acts as either a pro-inflammatory or an anti-inflammatory signal according to the models. The pro-inflammatory effect of hepatocyte STAT3 is likely mediated via its induction of acute phase proteins and chemokines in the liver [33,50]. In contrast, activation of hepatocyte STAT3 protects against hepatocellular damage and subsequently reduces liver necrosis-associated inflammation. Hepatocyte STAT3 also inhibits IFN-γ-activated STAT1, a key proinflammatory signal in the liver [55], thereby playing an important role in attenuating inflammatory responses. In both Con A-induced T cell hepatitis and LPS-induced liver injury models, IFN-γ is markedly elevated, and STAT1 is highly activated in hepatocytes [54,56], whereas such activation is not observed in CCl4- or alcohol-induced liver injury models. Therefore, activation of STAT3 inhibits STAT1 activation in the liver and subsequently represses liver inflammation in both Con A- and LPS-induced liver injury models [54,56]. However, such anti-inflammatory effects of STAT3 may not occur in CCl4- or alcohol-induced liver injury models, as the IFN-γ/STAT1 signaling pathway is not upregulated in these models.
STAT3 in macrophages/Kupffer cells: The anti-inflammatory effect of STAT3 in myeloid cells, including macrophages/Kupffer cells, has been well-documented in various models of organ injury, including liver injury [33,50,57-59], and in cultured Kupffer cells [33]. However, the role of STAT3 in myeloid cells in hepatocellular damage remains obscure. Interestingly, a specific deletion of STAT3 in myeloid cells enhances the susceptibility of mice to Con A-induced T cell hepatitis [58] and alcohol-induced liver injury [33], but reduces CCl4-induced hepatocellular damage [50]. This opposing effect could be attributable to the deletion of STAT3 in myeloid cells, preferentially augmenting IFN-γ production that subsequently enhances liver injury in Con A-induced T cell hepatitis [58], while preferentially enhancing IL-6 production that consequently protects against hepatocellular damage in CCl4-induced liver injury [50]. Collectively, STAT3 in myeloid cells not only inhibits expression of pro-inflammatory cytokines, such as TNF-α and IFN-γ, that induce liver damage and inflammation, but also represses production of hepatoprotective cytokines, such as IL-6 and IL-22, that ameliorate liver injury [58]. Thus, the outcome of myeloid STAT3 on hepatocellular damage is likely determined by the balance between detrimental and hepatoprotective cytokines produced during liver injury.
STAT3 in endothelial cells: Previous studies suggest that STAT3 signaling in endothelial cells plays an important role in inhibiting inflammation and protecting against LPS-induced inflammation [60-62], but the role of endothelial cell STAT3 in liver injury and inflammation has not been extensively studied. We have previously reported that endothelial cell-specific STAT3 knockout mice were more susceptible to alcohol-induced liver injury and inflammation [63], suggesting that endothelial cell STAT3 plays important dual functions of attenuating hepatic inflammation and sinusoidal endothelial cell apoptosis during alcoholic liver injury. However, the role of STAT3 in liver injury and inflammation in other models remains to be determined.
STAT3 and liver regeneration
The liver is the only solid organ in mammals with amazing regenerative capabilities after loss of tissue or injury [64-66]. Emerging evidence suggests that liver regeneration is controlled by a wide variety of cytokines, growth factors, hormones, and their downstream signaling pathways [64-66]. Among them, the roles of IL-6 and its downstream signaling molecule STAT3 in liver regeneration have been extensively investigated [64-66]. In addition, several other IL-6 family cytokines and IL-22, which activate STAT3 in hepatocytes, are also reported to promote liver regeneration [47,67,68], while the anti-inflammatory cytokine IL-10, which activates STAT3 in immune cells, has been shown to inhibit liver regeneration via suppression of the proinflammatory response during liver regeneration [69].
Two-third partial hepatectomy (PHx) is a widely used model to study liver regeneration. It is well-established that after PHx, the ability of the remnant liver to detoxify endotoxin (LPS) decreases, leading to elevation of hepatic LPS levels [70,71]. LPS stimulates Kupffer cells to produce inflammatory cytokines, including TNF-α and IL-6, that subsequently initiate liver regeneration. This priming phase usually takes place shortly after PHx with a transient increase in low inflammation. This notion was supported by studies using several strains of knockout mice. For example, genetic deletion of the type I TNF receptor (TNFR-1) resulted in an increase of the mortality rate in mice after PHx, and was accompanied by reduced hepatocyte proliferation [72]. Similarly, IL-6 deficient mice, reported initially by Cressman et al. [73], had increased mortality, blunted hepatocyte DNA synthesis, and repressed AP-1, Myc, and cyclin D with an absence of STAT3 activation after PHx. Treatment with a single preoperative dose of IL-6 restored STAT3 binding and hepatocyte proliferation and prevented liver damage in IL-6-deficient mice [73]. This study strongly posited an important role for IL-6 in hepatocyte proliferation via activation STAT3. However, several follow-up studies using IL-6 knockout mice generated conflicting data on the role of IL-6 on liver regeneration. Sakamoto et al. [74] reported that the peak of hepatocyte proliferation was only reduced by 20-30% in IL-6-deficient mice compared with wild-type mice after PHx, and similar mortality was observed in both groups, while Blindenbacher et al. [75] reported that IL-6-deficient mice had higher mortality after PHx but that the surviving IL-6-deficient mice had similar hepatocyte proliferation compared to wild-type mice. In addition, two later studies demonstrated that IL-6 knockout mice and wild-type mice had similar hepatocyte proliferation after PHx [76,77]. Systemic delivery of high levels of IL-6 using CHO cell tumors in nude mice resulted in a dramatic induction of hepatocyte proliferation and hepatomegaly via activation of the MAPK/ERK and STAT3 signaling pathways [78]. However, hyperstimulation with IL-6 in transgenic mice with overexpression of the human soluble IL-6 receptor/gp80 in hepatocytes before PHx resulted in delayed and inhibited cell cycle progression after PHx in mice [79]. The reasons for these controversial reports on the role of IL-6 in liver regeneration are not clear and may be due to using different surgery techniques, strains of mice, or research environments. Despite of these controversial reports, most investigators still accepted the notion that IL-6 plays an important role in liver regeneration [80-82]. Furthermore, several IL-6 family cytokines, such as oncostatin M [47] and cardiotrophin-1 [36], have also been shown to promote liver regeneration in various models. IL-22 was also shown to play a role in promoting liver regeneration after PHx [83].
In contrast to the cytokines that activate STAT3 in hepatocytes and promote liver regeneration discussed above, the anti-inflammatory cytokine IL-10, which activates STAT3 in immune cells (e.g., macrophages), but not in hepatocytes, may negatively regulate liver regeneration. Expression of IL-10 in the liver is upregulated after PHx [69]. Disruption of IL-10 enhances liver inflammation and regenerative responses with increased STAT3 activation in the liver [69]. An additional deletion of hepatocyte STAT3 reduced liver regeneration in IL-10-deficient mice [69], suggesting that enhanced liver regeneration in IL-10 knockout mice is due to elevated IL-6/STAT3 activation in the liver.
Rapid activation of STAT3 has been well-documented during liver regeneration after PHx or liver injury [59,84-86]. After PHx, activation of STAT3 was first detected in Kupffer cells and in sinusoidal endothelial cells and later detected in hepatocytes localized in the periportal zones of hepatic lobules in rats [86]. As global disruption of the STAT3 gene leads to early embryonic death, tissue-specific STAT3 knockout mice were generated to investigate the biological impact of STAT3 in liver regeneration. Studies from our lab and other groups showed that deletion of STAT3 in hepatocytes only moderately reduced liver regeneration without induction of hepatocyte apoptosis after PHx [59,85,87]. Several STAT3 targeted genes have been identified; these include cyclin D and c-myc, which induce the cell-cycle transition from the G1 to the S phase, and bcl-2, bcl-xl, mcl-1, and c-Flip, which protect against hepatocyte apoptosis [85]. STAT3 in immune cells was also markedly activated by PHx, and conditional deletion of STAT3 in myeloid linage cells resulted in enhanced inflammatory response and increased liver regeneration [59]. Combined conditional ablation of STAT3 in both hepatocytes and myeloid cells resulted in a dramatic reduction in survival with elevated activation of STAT1 and hepatocyte apoptosis after PHx, as compared to wild-type or single knockout animals [59]. These findings suggest that the interplay of STAT3 in myeloid cells and hepatocytes plays an important role in ensuring normal liver regeneration via tempering systemic and hepatic innate inflammatory responses [59]. Activation of STAT3 also induces expression of SOCS3, which in turn terminates STAT3 signaling and negatively regulates liver regeneration [88]. Interestingly, recent studies from Dr. Ozaki's group suggest that PDK1/Akt signaling also contributes to liver regeneration by regulating cell size [89,90].
In summary, the role of hepatocyte STAT3 in liver regeneration has been extensively investigated, and it is generally accepted that activation of STAT3 in hepatocytes promotes hepatocyte proliferation after PHx or loss of tissue, while myeloid cell STAT3 activation seems to suppress liver regeneration via inhibiting inflammatory responses. In contrast, the roles of STAT3 in other immune cells and sinusoidal endothelial cells in liver regeneration have not been explored and deserved further studies. In addition, the interplay of STAT3 from different cell types in liver regeneration likely plays a critical role in ensuring normal liver regeneration, which also needs further investigation.
STAT3 in liver fibrosis
Liver fibrosis, or cirrhosis, is a common end-stage condition of many chronic liver diseases after incomplete recovery from hepatocyte damage. During fibrosis progression, inflammation and liver injury trigger complex cellular events that result in collagen deposition and disruption of the normal liver architecture. Generally, activated hepatic stellate cells are considered the most important cell type for the production of collagens [14,91]. More recent research suggests that myofibroblasts, bone marrow-derived progenitor cells, and hepatocytes may also contribute to fibrogenesis via production of collagens [14,91,92], and immune cells may regulate fibrogenesis via production of a wide variety of cytokines; among them, TGF-β has been shown to promote liver fibrosis via stimulating hepatic stellate cell activation, while IFN-γ is known to inhibit liver fibrosis via inducing hepatic stellate cell apoptosis and cell cycle arrest [14,91]. Although the hepatoprotection ability of IL-6, one of the most important cytokines to activate STAT3 in the liver, has been well-documented, the data on the role of IL-6 in liver fibrogenesis from animal models remain controversial.
Initial experiments showed that IL-6 knockout mice were more susceptible to CCl4-induced liver injury and fibrosis [93], while other reports demonstrated reduced liver fibrosis in IL-6-deficient mice after CCl4 treatment [94,95]. The reasons for the discrepancy between these studies are not clear. In addition, deletion of gp130 (the common signal transducer of IL-6 family cytokines) in hepatocytes had no effect on liver fibrosis, while deletion of gp130 in nonparenchymal liver cells aggravated fibrosis progression [45]. In vitro culture experiments showed that Kupffer cell-derived IL-6 promotes hepatic stellate cell survival and proliferation [15]. Clinical studies showed that hepatic IL-6 expression was upregulated and correlated positively with the degree of liver fibrosis in opisthorchiasis periductal fibrosis and in nonalcoholic steatohepatitis [96,97]. Furthermore, genetic polymorphisms of IL-6 were reported to modulate fibrosis progression in patients with mild chronic hepatitis C (HCV) [98]. Because IL-6 receptors are expressed ubiquitously on all types of liver cells, it is plausible to speculate that IL-6 may positively and negatively regulate liver fibrosis via targeting different types of liver cells. For example, IL-6 protects against hepatocellular damage, thereby reducing liver fibrosis, while IL-6 may also directly promote hepatic stellate cell survival and proliferation, followed by enhancing liver fibrosis. The final effect of IL-6 on liver fibrosis is likely determined by the balance between these inhibitory and stimulatory effects and is dependent on the stage and etiology of liver fibrosis.
The initial clue for the role of the IL-6 downstream signaling molecule STAT3 in liver fibrogenesis is emerging from the clinical observations of Dr. Starkel's group [99]. This study documented that STAT3 expression and phosphorylation were not altered in HCV-fibrosis patients and alcoholic cirrhosis, while STAT3-DNA binding was markedly suppressed in all alcoholic and most HCV fibrotic patients when compared with to normal healthy livers. This impaired STAT3-DNA binding might contribute to disturbed liver regeneration and repair in these patients. Further studies from the same group later showed that fibrosis progression in HCV-infected patients correlated positively with a continuous decline in STAT3-DNA binding activity. This decreased STAT3 activity also correlated with reduced hepatocyte proliferation and a positive anti-apoptotic balance in infiltrating inflammatory cells that are known mediators of cell damage in HCV [100]. Taken together, these clinical studies indicate that STAT3 may ameliorate HCV and alcohol-related liver fibrosis via protecting against hepatocellular damage.
Recent studies from animal models suggest the STAT3 in hepatocytes plays a protective role in preventing liver fibrosis, mainly because of STAT3's hepatoprotective and proliferative functions [50,53,101]. The hepatoprotective function of STAT3 in the liver has been well-documented in many murine models of liver injury as discussed in above sections. Thus, it is probable that activation of STAT3 in hepatocytes may suppress liver fibrosis via preventing hepatocyte damage. This speculation is, indeed, supported by the studies from several animal models. In the models of Sclerosing Cholangitis, which use mice lacking the multidrug resistance gene 2 (mdr2-/-), conditionally inactivated STAT3 in hepatocytes and cholangiocytes leads to strongly aggravated hepatocellular damage and fibrosis [53]. The hepatoprotective role of STAT3 is likely due to activation of EGFR and IGF-1 signaling pathways in hepatocytes, thereby preventing bile acid-induced liver injury and fibrosis [53]. Similarly, another study showed that lack of gp130/STAT3-mediated signaling in hepatocytes resulted in enhanced chronic cholestatic liver injury and fibrosis progression induced by 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) [101]. Deletion of the gp130/STAT3 pathway in hepatocytes leads to more severe liver injury and an enhanced inflammatory response with increased TNF-α expression and subsequent cytotoxicity. This greater chronic liver injury leads to enhanced hepatic stellate cell activation and a profibrogenic response. In contrast, Ogata et al. [102] reported that deletion of SOCS3 (a protein with a strong inhibitory effect on STAT3 signaling) in the liver, via injection of adenovirus dominant STAT3 or in the SOCS3flox/flox mice via injection of adenovirus Cre, resulted in hyperactivation of STAT3 in the liver and enhanced hepatic fibrosis through the production of TGF-β1. Although adenovirus mainly infects hepatocytes, it may also infect other liver cells including hepatic stellate cells and Kupffer cells. Thus, it is not clear whether enhanced liver fibrosis in the models reported by Ogata et al. [102] is due to deletion of SOCS3 in hepatocytes or in other nonparenchymal cells.
Until now, the functions of STAT3 in nonparenchymal liver cells and inflammatory cells during liver fibrogenesis are largely unknown. Several studies provide indirect evidence of profibrogenic and survival roles of STAT3 in hepatic stellate cells. Leptin is an important pro-fibrogenic cytokine that activates STAT3 in hepatic stellate cells [103,104]. Inhibition of JAK/STAT3 activation by a specific JAK2 inhibitor AG490 prevents hepatic stellate cell early activation [105]. In addition, STAT3 is involved in leptin-mediated production of TIMP-1, an important survival factor for hepatic stellate cells [106]. Other mechanisms leading to fibrogenesis via STAT3 signaling pathway activation in hepatic stellate cells, such as phagocytosis of apoptotic bodies, have been shown to promote their survival [107]. Kupffer cells also play an important role in the pathogenesis of leptin-induced liver fibrosis [108]. Treatment with leptin potentiated activation of STAT3, Akt, and extracellular signal-related kinase 1/2 phosphorylation in Kupffer cells and increased AP-1 and nuclear factor-kappaB DNA binding. Blockage of STAT3 activation by a STAT3 inhibitor attenuated TGFβ1 production by Kupffer cells [108]. These observations indicate that leptin may promote liver fibrogenesis via activation of STAT3 and subsequent production of TGF-β in Kupffer cells. Additional studies are required to clarify the roles of STAT3 in hepatic stellate cells, Kupffer cells, sinusoidal cells, and inflammatory cells in the pathogenesis of liver fibrosis.
STAT3 and liver cancer
Aberrantly hyperactivated IL-6-STAT3 signaling in cancer cells and the tumor microenvironment has been detected in a wide variety of human cancers and is considered to be an important factor for cancer initiation, development, and progression [109-112]. The interplay of STAT3 in cancer cells and other cells around cancer cells likely plays an important role in modulating tumor growth via several steps. First, it is well known that IL-6-STAT3 signaling is crucial for tumor cell proliferation and survival via induction a variety of protooncogenes [113-115]. In contrast to normal cells, in which STAT3 activation is rapid and transient, cancer cells routinely harbor persistently activated STAT3 proteins that promote a permanent alteration of some genes that control cellular processes. Constitutively activated STAT3 promotes tumorigenesis through the upregulation of cell survival proteins (Bcl-xl and Bcl-2), cell cycle regulators (c-Myc and cyclin D) [112,114,116,117], anti-oxidant genes (Mn-SOD, ferritin, and catalse), and tissue repair genes (RegIIIβ, RegIIIγ, and Tff3) [114,118,119]. Second, persistent activation of STAT3 also increases angiogenesis and metastasis via upregulation of vascular endothelial growth factor (VEGF), hypoxia inducible factor 1 alpha (HIF1α) and basic fibroblast growth factor (bFGF), all of which are important mediators of angiogenesis during tumor formation [120-122]. Third, the persistent activation of STAT3 in immune/inflammatory cells is also very important in the control of tumor promotion and progression through tumor-promoting inflammation and suppressing anti-tumor immunity [123]. Recently, a key novel molecule, sphingosine-1-phosphate receptor-1 (S1PR1), that is induced by STAT3 has been uncovered to play an important role in inducing persistent STAT3 activation in tumor cells and the tumor microenvironment [124]. In summary, STAT3 can broadly and profoundly affect tumor growth via stimulation of tumor cell survival and proliferation, induction of tumor angiogenesis, and suppressing anti-tumor immunity. Thus, constitutively activated STAT3 both in tumor cells and tumor stromal immune cells is an attractive target for cancer treatment [125].
Hepatocellular carcinoma (HCC) is the most common primary malignancy of the liver in adults and the third leading cause of cancer deaths worldwide, with few effective therapeutic options for this advanced disease [126-128]. Most HCC appears in cirrhotic livers after years of chronic liver inflammation caused by viral hepatitis and alcoholic and non-alcoholic steatohepatitis [127,129]. Several cytokines (such as IL-6, IL-6 family cytokines, IL-22, etc) that activate STAT3 in hepatocytes have been shown to promote HCC growth in vitro and in vivo [37,42,130]. Clinical studies reported that serum IL-6 concentrations were elevated in patients with chronic liver inflammation, including alcoholic hepatitis, viral hepatitis, and nonalcoholic steatohepatitis, and in patients with HCC [131]. Notably, men are about three to five times more likely to develop HCC than women [132]. A similar gender disparity was also seen in a murine model of HCC induced by diethylnitrosamine (DEN). It is believed that higher serum levels of IL-6 in male mice contributed to the higher susceptibility to DEN-induced liver cancer compared to female mice [130]. In addition to IL-6, IL-22 and leptin, which also activate STAT3 in liver cancer cells, have also been implicated in liver tumorigenesis. IL-22, a recently identified IL-10 family cytokine, also activates STAT3 in hepatocytes and has been shown to promote HCC in vitro and in vivo [37]. In vitro treatment with IL-22 or overexpression of IL-22 promotes cell growth and survival of human hepatoma HepG2 cells. Stable overexpression of IL-22 in HepG2 cells constitutively activates STAT3 and upregulates expression of a variety of anti-apoptotic (e.g., Bcl-2, Bcl-xl, and Mcl-1) and mitogenic (e.g., c-myc, cyclin D1, Rb2, and CDK4) proteins. Overexpression of IL-22 in HepG2 and Hep3B cells also promotes tumor formation of these cells in nude mice [37]. Transgenic mice with overexpression of IL-22 in the liver are more susceptible to DEN-induced liver tumorigenesis [42]. Leptin is an adipose-derived hormone that plays an important role in food intake and energy expenditure. Recent studies showed that leptin also activates STAT3 in liver cancer cells and promotes liver cancer cell survival and proliferation [133-135].
STAT3 is the major downstream signaling molecule of IL-6, IL-22, and leptin in hepatocytes. Several lines of evidence suggest that STAT3 plays an important role in the development of liver cancer. First, constitutively activated STAT3 is detected in human hepatoma cells and human liver tumor tissues [136]. In HCC tissues, strong STAT3 immunostaining was observed in the cytoplasm, and pY705STAT3 immunostaining was observed in the nucleus [136]. In addition, blockage of STAT3 using chemical inhibitors or siRNA induced liver cancer cell apoptosis and cell cycle arrest in vitro and inhibited growth of transplanted liver cancer cells in vivo [136-138]. Second, altered p-STAT3 expression was positively correlated with the histological grading and intra-tumor microvessel density in HCC [139]. Third, deletion or methylation silencing of hepatic SOCS3, an inhibitor for STAT3, resulted in enhanced STAT3 activation in the liver and accelerated DEN-induced liver tumorigenesis [88,140], while overexpression of SOCS3 inhibited HCC cell growth [141]. Finally, the conclusive evidence for an important role of STAT3 in liver cancer development is from the fact that conditional deletion of STAT3 in hepatocytes prevented DEN-induced liver cancer development in mice [142]. Notably, recent studies suggest that STAT3 activation is also implicated in HCV- and obesity-mediated hepatocarcinogenesis [143,144]. Another important line of evidence for the role of STAT3 in liver cancer development is that constitutively activated STAT3 is detected in cancer stem cells from HCC and likely contributes to liver cancer stem cell proliferation and survival [145]. Collectively, activation of STAT3 plays an important role in liver tumorigenesis. Blockage of STAT3 may have a therapeutic potential in preventing and treating liver cancer.
STAT3 as a therapeutic target to treat liver disease
Because the hepatoprotection and anti-steatogenic effects of hepatocyte STAT3 are well-established, the cytokines and small molecules that activate STAT3 in hepatocytes may have a therapeutic potential to treat acute liver injury and fatty liver diseases. IL-6 has been shown to effectively ameliorate liver injury and fatty liver diseases in animal models [17,29,38]. However, the clinical application of IL-6 in patients has been halted owing to many adverse effects of IL-6 that are likely caused by ubiquitous expression of the IL-6 receptor, IL-6 receptor signal chain gp130, and the existence of circulating soluble IL-6 receptor. In addition, patients with liver diseases are always associated with highly elevated levels of IL-6 [96,97,146,147], resulting in resistance to IL-6 treatment. Interestingly, we have recently demonstrated that ex vivo treatment of fatty donor liver with IL-6 prevents fatty liver transplant failure via improving microcirculation and reducing liver cell apoptosis [17]. Application of IL-6 ex vivo could be a magic potion to prevent fatty liver transplant failure in humans [148].
Since IL-22 receptor expression is restricted to epithelial cells, including hepatocytes, in vivo IL-22 treatment will likely have few side effects [149]. Thus, IL-22 may have a better therapeutic potential than IL-6 in treating patients with fatty liver disease or acute liver failure induced by acute viral hepatitis, ischemia/reperfusion, liver transplantation, and alcoholic hepatitis. Alcoholic hepatitis is a severe disease with up to 40% mortality within 6 months after the onset of the clinical syndrome [150]. Corticosteroids are widely used to treat alcoholic hepatitis, but such treatment only improves the short-term survival rate [150]. This may be because treatment with corticosteroids inhibits inflammation, which is beneficial for alcoholic hepatitis, but can also suppress liver regeneration and increase the rate of bacterial infection. Corticosteroids plus IL-22 treatment appear to have multiple beneficial effects on alcoholic hepatitis such as IL-22 preventing hepatocellular damage, promoting hepatocyte proliferation, and inhibiting bacterial infection. Clinical trials examining such therapeutic combinations for patients with severe alcoholic hepatitis should be tested in the future.
Like many other types of cancer, hepatocellular carcinoma and cholangiocarcinoma also have elevated levels of STAT3 activation [4]. Blockage of STAT3 activation has been shown to effectively reduce hepatocellular carcinoma and cholangiocarcinoma growth in vitro and in in vivo animal models [135,136,151]. During last several years, extreme efforts have been made to develop STAT3 inhibitors. These inhibitors have been extensively used in preclinical studies in various types of cancer, and some of these inhibitors have also been tested in Phase 1 clinical trials [152]. We anticipate that these STAT3 inhibitors will have beneficial effects in treating liver cancer in the future.
Acknowledgements
The work described here from the authors' laboratory was supported by the intramural program of NIAAA, NIH.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Elsharkawy AM, Mann DA. Nuclear factor-kappaB and the hepatic inflammation-fibrosis-cancer axis. Hepatology. 2007;46:590-597
2. Robinson SM, Mann DA. Role of nuclear factor kappaB in liver health and disease. Clin Sci (Lond). 2010;118:691-705
3. Sun B, Karin M. NF-kappaB signaling, liver disease and hepatoprotective agents. Oncogene. 2008;27:6228-6244
4. He G, Karin M. NF-kappaB and STAT3 - key players in liver inflammation and cancer. Cell Res. 2011;21:159-168
5. Luedde T, Schwabe RF. NF-kappaB in the liver-linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8:108-118
6. Gao B. Cytokines, STATs and liver disease. Cell Mol Immunol. 2005;2:92-100
7. Gao B, Jeong WI, Tian Z. Liver: An organ with predominant innate immunity. Hepatology. 2008;47:729-736
8. Radaeva S, Jaruga B, Hong F, Kim WH, Fan S, Cai H, Strom S. et al. Interferon-alpha activates multiple STAT signals and down-regulates c- Met in primary human hepatocytes. Gastroenterology. 2002;122:1020-1034
9. Mandal P, Park PH, McMullen MR, Pratt BT, Nagy LE. The anti-inflammatory effects of adiponectin are mediated via a heme oxygenase-1-dependent pathway in rat Kupffer cells. Hepatology. 2010;51:1420-1429
10. Zhang M, Xu S, Han Y, Cao X. Apoptotic cells attenuate fulminant hepatitis by priming Kupffer cells to produce interleukin-10 through membrane-bound TGF-beta. Hepatology. 2011;53:306-316
11. Knolle PA, Loser E, Protzer U, Duchmann R, Schmitt E, zum Buschenfelde KH, Rose-John S. et al. Regulation of endotoxin-induced IL-6 production in liver sinusoidal endothelial cells and Kupffer cells by IL-10. Clin Exp Immunol. 1997;107:555-561
12. Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, Hanada T. et al. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol. 2003;4:551-556
13. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125-172
14. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209-218
15. Nieto N. Oxidative-stress and IL-6 mediate the fibrogenic effects of [corrected] Kupffer cells on stellate cells. Hepatology. 2006;44:1487-1501
16. Handy JA, Saxena NK, Fu P, Lin S, Mells JE, Gupta NA, Anania FA. Adiponectin activation of AMPK disrupts leptin-mediated hepatic fibrosis via suppressors of cytokine signaling (SOCS-3). J Cell Biochem. 2010;110:1195-1207
17. Sun Z, Klein AS, Radaeva S, Hong F, El-Assal O, Pan HN, Jaruga B. et al. In vitro interleukin-6 treatment prevents mortality associated with fatty liver transplants in rats. Gastroenterology. 2003;125:202-215
18. Gao B. Therapeutic potential of interleukin-6 in preventing obesity- and alcohol-associated fatty liver transplant failure. Alcohol. 2004;34:59-65
19. Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 2010;51:1820-1832
20. Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology. 2010;51:679-689
21. Vuppalanchi R, Chalasani N. Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: Selected practical issues in their evaluation and management. Hepatology. 2009;49:306-317
22. Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52:774-788
23. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836-1846
24. Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM. et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. 2010;139:323-334
25. Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JE, van Rooijen N, Staels B. et al. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology. 2010;51:511-522
26. Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, Thurman RG. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117:942-952
27. El-Assal O, Hong F, Kim WH, Radaeva S, Gao B. IL-6-deficient mice are susceptible to ethanol-induced hepatic steatosis: IL-6 protects against ethanol-induced oxidative stress and mitochondrial permeability transition in the liver. Cell Mol Immunol. 2004;1:205-211
28. Zhang X, Tachibana S, Wang H, Hisada M, Williams GM, Gao B, Sun Z. Interleukin-6 is an important mediator for mitochondrial DNA repair after alcoholic liver injury in mice. Hepatology. 2010;52:2137-2147
29. Hong F, Radaeva S, Pan HN, Tian Z, Veech R, Gao B. Interleukin 6 alleviates hepatic steatosis and ischemia/reperfusion injury in mice with fatty liver disease. Hepatology. 2004;40:933-941
30. Ki SH, Park O, Zheng M, Morales-Ibanez O, Kolls JK, Bataller R, Gao B. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatology. 2010;52:1291-1300
31. Yang L, Zhang Y, Wang L, Fan F, Zhu L, Li Z, Ruan X. et al. Amelioration of high fat diet induced liver lipogenesis and hepatic steatosis by interleukin-22. J Hepatol. 2010;53:339-347
32. Kroy DC, Beraza N, Tschaharganeh DF, Sander LE, Erschfeld S, Giebeler A, Liedtke C. et al. Lack of interleukin-6/glycoprotein 130/signal transducers and activators of transcription-3 signaling in hepatocytes predisposes to liver steatosis and injury in mice. Hepatology. 2010;51:463-473
33. Horiguchi N, Wang L, Mukhopadhyay P, Park O, Jeong WI, Lafdil F, Osei-Hyiaman D. et al. Cell type-dependent pro- and anti-inflammatory role of signal transducer and activator of transcription 3 in alcoholic liver injury. Gastroenterology. 2008;134:1148-1158
34. Inoue H, Ogawa W, Ozaki M, Haga S, Matsumoto M, Furukawa K, Hashimoto N. et al. Role of STAT-3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism in vivo. Nat Med. 2004;10:168-174
35. Ramadoss P, Unger-Smith NE, Lam FS, Hollenberg AN. STAT3 targets the regulatory regions of gluconeogenic genes in vivo. Mol Endocrinol. 2009;23:827-837
36. Bustos M, Beraza N, Lasarte JJ, Baixeras E, Alzuguren P, Bordet T, Prieto J. Protection against liver damage by cardiotrophin-1: a hepatocyte survival factor up-regulated in the regenerating liver in rats. Gastroenterology. 2003;125:192-201
37. Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology. 2004;39:1332-1342
38. Sun R, Tian Z, Kulkarni S, Gao B. IL-6 prevents T cell-mediated hepatitis via inhibition of NKT cells in CD4+ T cell- and STAT3-dependent manners. J Immunol. 2004;172:5648-5655
39. Bozza M, Bliss JL, Maylor R, Erickson J, Donnelly L, Bouchard P, Dorner AJ. et al. Interleukin-11 reduces T-cell-dependent experimental liver injury in mice. Hepatology. 1999;30:1441-1447
40. Wahl C, Wegenka UM, Leithauser F, Schirmbeck R, Reimann J. IL-22-dependent attenuation of T cell-dependent (ConA) hepatitis in herpes virus entry mediator deficiency. J Immunol. 2009;182:4521-4528
41. Pellegrini M, Calzascia T, Toe JG, Preston SP, Lin AE, Elford AR, Shahinian A. et al. IL-7 engages multiple mechanisms to overcome chronic viral infection and limit organ pathology. Cell. 2011;144:601-613
42. Park O, Wang H, Weng H, Feigenbaum L, Li H, Yin S, Ki SH, Yoo SH, Dooley S, Wang FS, Young HA, Gao B. In vivo consequences of liver-specific interleukin-22 expression: implications for human liver disease progression. Hepatology. 2011 DOI: 10.1002/hep.24339
43. Zhang W, Tsuda M, Yang GX, Tsuneyama K, Rong G, Ridgway WM, Ansari AA. et al. Deletion of interleukin-6 in mice with the dominant negative form of transforming growth factor beta receptor II improves colitis but exacerbates autoimmune cholangitis. Hepatology. 2010;52:215-222
44. Kovalovich K, Li W, DeAngelis R, Greenbaum LE, Ciliberto G, Taub R. Interleukin-6 protects against Fas-mediated death by establishing a critical level of anti-apoptotic hepatic proteins FLIP, Bcl-2, and Bcl- xL. J Biol Chem. 2001;276:26605-26613
45. Streetz KL, Tacke F, Leifeld L, Wustefeld T, Graw A, Klein C, Kamino K. et al. Interleukin 6/gp130-dependent pathways are protective during chronic liver diseases. Hepatology. 2003;38:218-229
46. Streetz KL, Wustefeld T, Klein C, Kallen KJ, Tronche F, Betz UA, Schutz G. et al. Lack of gp130 expression in hepatocytes promotes liver injury. Gastroenterology. 2003;125:532-543
47. Nakamura K, Nonaka H, Saito H, Tanaka M, Miyajima A. Hepatocyte proliferation and tissue remodeling is impaired after liver injury in oncostatin M receptor knockout mice. Hepatology. 2004;39:635-644
48. Iniguez M, Berasain C, Martinez-Anso E, Bustos M, Fortes P, Pennica D, Avila MA. et al. Cardiotrophin-1 defends the liver against ischemia-reperfusion injury and mediates the protective effect of ischemic preconditioning. J Exp Med. 2006;203:2809-2815
49. Marques JM, Belza I, Holtmann B, Pennica D, Prieto J, Bustos M. Cardiotrophin-1 is an essential factor in the natural defense of the liver against apoptosis. Hepatology. 2007;45:639-648
50. Horiguchi N, Lafdil F, Miller AM, Park O, Wang H, Rajesh M, Mukhopadhyay P. et al. Dissociation between liver inflammation and hepatocellular damage induced by carbon tetrachloride in myeloid cell-specific signal transducer and activator of transcription 3 gene knockout mice. Hepatology. 2010;51:1724-1734
51. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity. 2007;27:647-659
52. Haga S, Terui K, Zhang HQ, Enosawa S, Ogawa W, Inoue H, Okuyama T. et al. Stat3 protects against Fas-induced liver injury by redox-dependent and -independent mechanisms. J Clin Invest. 2003;112:989-998
53. Mair M, Zollner G, Schneller D, Musteanu M, Fickert P, Gumhold J, Schuster C. et al. Signal transducer and activator of transcription 3 protects from liver injury and fibrosis in a mouse model of sclerosing cholangitis. Gastroenterology. 2010;138:2499-2508
54. Sakamori R, Takehara T, Ohnishi C, Tatsumi T, Ohkawa K, Takeda K, Akira S. et al. Signal transducer and activator of transcription 3 signaling within hepatocytes attenuates systemic inflammatory response and lethality in septic mice. Hepatology. 2007;46:1564-1573
55. Jaruga B, Hong F, Kim WH, Gao B. IFN-{gamma}/STAT1 acts as a proinflammatory signal in T cell-mediated hepatitis via induction of multiple chemokines and adhesion molecules: a critical role of IRF-1. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1044-G1052
56. Hong F, Jaruga B, Kim WH, Radaeva S, El-Assal ON, Tian Z, Nguyen VA. et al. Opposing roles of STAT1 and STAT3 in T cell-mediated hepatitis: regulation by SOCS. J Clin Invest. 2002;110:1503-1513
57. Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39-49
58. Lafdil F, Wang H, Park O, Zhang W, Moritoki Y, Yin S, Fu XY. et al. Myeloid STAT3 inhibits T cell-mediated hepatitis by regulating T helper 1 cytokine and interleukin-17 production. Gastroenterology. 2009;137:2125-2135
59. Wang H, Park O, Lafdil F, Shen K, Horiguchi N, Yin S, Fu XY. et al. Interplay of hepatic and myeloid signal transducer and activator of transcription 3 in facilitating liver regeneration via tempering innate immunity. Hepatology. 2010;51:1354-1362
60. Carrithers M, Tandon S, Canosa S, Michaud M, Graesser D, Madri JA. Enhanced susceptibility to endotoxic shock and impaired STAT3 signaling in CD31-deficient mice. Am J Pathol. 2005;166:185-196
61. Wang M, Zhang W, Crisostomo P, Markel T, Meldrum KK, Fu XY, Meldrum DR. Endothelial STAT3 plays a critical role in generalized myocardial proinflammatory and proapoptotic signaling. Am J Physiol Heart Circ Physiol. 2007;293:H2101-2108
62. Kano A, Wolfgang MJ, Gao Q, Jacoby J, Chai GX, Hansen W, Iwamoto Y. et al. Endothelial cells require STAT3 for protection against endotoxin-induced inflammation. J Exp Med. 2003;198:1517-1525
63. Miller AM, Wang H, Park O, Horiguchi N, Lafdil F, Mukhopadhyay P, Moh A. et al. Anti-inflammatory and anti-apoptotic roles of endothelial cell STAT3 in alcoholic liver injury. Alcohol Clin Exp Res. 2010;34:719-725
64. Michalopoulos GK. Liver regeneration. J Cell Physiol. 2007;213:286-300
65. Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006;43:S45-53
66. Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol. 2004;5:836-847
67. Beraza N, Marques JM, Martinez-Anso E, Iniguez M, Prieto J, Bustos M. Interplay among cardiotrophin-1, prostaglandins, and vascular endothelial growth factor in rat liver regeneration. Hepatology. 2005;41:460-469
68. Ren X HB, Colletti LM. IL-22 is involved in liver regeneration after hepatectomy. Am J Physiol Gastrointest Liver Physiol. 2010;298:G74-G80
69. Yin S, Wang H, Park O, Wei W, Shen J, Gao B. Enhanced Liver Regeneration in IL-10-Deficient Mice after Partial Hepatectomy via Stimulating Inflammatory Response and Activating Hepatocyte STAT3. Am J Pathol. 2011;178:1614-1621
70. Cornell RP. Gut-derived endotoxin elicits hepatotrophic factor secretion for liver regeneration. Am J Physiol. 1985;249:R551-562
71. Xu CP, Liu J, Liu JC, Han DW, Zhang Y, Zhao YC. Dynamic changes and mechanism of intestinal endotoxemia in partially hepatectomized rats. World J Gastroenterol. 2007;13:3592-3597
72. Yamada Y, Kirillova I, Peschon JJ, Fausto N. Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc Natl Acad Sci U S A. 1997;94:1441-1446
73. Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V, Taub R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 1996;274:1379-1383
74. Sakamoto T, Liu Z, Murase N, Ezure T, Yokomuro S, Poli V, Demetris AJ. Mitosis and apoptosis in the liver of interleukin-6-deficient mice after partial hepatectomy. Hepatology. 1999;29:403-411
75. Blindenbacher A, Wang X, Langer I, Savino R, Terracciano L, Heim MH. Interleukin 6 is important for survival after partial hepatectomy in mice. Hepatology. 2003;38:674-682
76. Sun R, Jaruga B, Kulkarni S, Sun H, Gao B. IL-6 modulates hepatocyte proliferation via induction of HGF/p21cip1: regulation by SOCS3. Biochem Biophys Res Commun. 2005;338:1943-1949
77. Wuestefeld T, Klein C, Streetz KL, Betz U, Lauber J, Buer J, Manns MP. et al. Interleukin-6/glycoprotein 130-dependent pathways are protective during liver regeneration. J Biol Chem. 2003;278:11281-11288
78. Zimmers TA, McKillop IH, Pierce RH, Yoo JY, Koniaris LG. Massive liver growth in mice induced by systemic interleukin 6 administration. Hepatology. 2003;38:326-334
79. Wustefeld T, Rakemann T, Kubicka S, Manns MP, Trautwein C. Hyperstimulation with interleukin 6 inhibits cell cycle progression after hepatectomy in mice. Hepatology. 2000;32:514-522
80. Teixeira-Clerc F, Belot MP, Manin S, Deveaux V, Cadoudal T, Chobert MN, Louvet A. et al. Beneficial paracrine effects of cannabinoid receptor 2 on liver injury and regeneration. Hepatology. 2010;52:1046-1059
81. Tumanov AV, Koroleva EP, Christiansen PA, Khan MA, Ruddy MJ, Burnette B, Papa S. et al. T cell-derived lymphotoxin regulates liver regeneration. Gastroenterology. 2009;136:694-704
82. Zorde-Khvalevsky E, Abramovitch R, Barash H, Spivak-Pohis I, Rivkin L, Rachmilewitz J, Galun E. et al. Toll-like receptor 3 signaling attenuates liver regeneration. Hepatology. 2009;50:198-206
83. Ren X, Hu B, Colletti LM. IL-22 is involved in liver regeneration after hepatectomy. Am J Physiol Gastrointest Liver Physiol. 2010;298:G74-80
84. Campbell JS, Prichard L, Schaper F, Schmitz J, Stephenson-Famy A, Rosenfeld ME, Argast GM. et al. Expression of suppressors of cytokine signaling during liver regeneration. J Clin Invest. 2001;107:1285-1292
85. Li W, Liang X, Kellendonk C, Poli V, Taub R. STAT3 contributes to the mitogenic response of hepatocytes during liver regeneration. J Biol Chem. 2002;277:28411-28417
86. Sakuda S, Tamura S, Yamada A, Miyagawa J, Yamamoto K, Kiso S, Ito N. et al. Activation of signal transducer and activator transcription 3 and expression of suppressor of cytokine signal 1 during liver regeneration in rats. J Hepatol. 2002;36:378-384
87. Moh A, Iwamoto Y, Chai GX, Zhang SS, Kano A, Yang DD, Zhang W. et al. Role of STAT3 in liver regeneration: survival, DNA synthesis, inflammatory reaction and liver mass recovery. Lab Invest. 2007;87:1018-1028
88. Riehle KJ, Campbell JS, McMahan RS, Johnson MM, Beyer RP, Bammler TK, Fausto N. Regulation of liver regeneration and hepatocarcinogenesis by suppressor of cytokine signaling 3. J Exp Med. 2008;205:91-103
89. Haga S, Ogawa W, Inoue H, Terui K, Ogino T, Igarashi R, Takeda K. et al. Compensatory recovery of liver mass by Akt-mediated hepatocellular hypertrophy in liver-specific STAT3-deficient mice. J Hepatol. 2005;43:799-807
90. Haga S, Ozaki M, Inoue H, Okamoto Y, Ogawa W, Takeda K, Akira S. et al. The survival pathways phosphatidylinositol-3 kinase (PI3-K)/phosphoinositide-dependent protein kinase 1 (PDK1)/Akt modulate liver regeneration through hepatocyte size rather than proliferation. Hepatology. 2009;49:204-214
91. Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655-1669
92. Dranoff JA, Wells RG. Portal fibroblasts: Underappreciated mediators of biliary fibrosis. Hepatology. 2010;51:1438-1444
93. Kovalovich K, DeAngelis RA, Li W, Furth EE, Ciliberto G, Taub R. Increased toxin-induced liver injury and fibrosis in interleukin-6-deficient mice. Hepatology. 2000;31:149-159
94. Natsume M, Tsuji H, Harada A, Akiyama M, Yano T, Ishikura H, Nakanishi I. et al. Attenuated liver fibrosis and depressed serum albumin levels in carbon tetrachloride-treated IL-6-deficient mice. J Leukoc Biol. 1999;66:601-608
95. Rio A, Gassull MA, Aldeguer X, Ojanguren I, Cabre E, Fernandez E. Reduced liver injury in the interleukin-6 knockout mice by chronic carbon tetrachloride administration. Eur J Clin Invest. 2008;38:306-316
96. Sripa B, Mairiang E, Thinkhamrop B, Laha T, Kaewkes S, Sithithaworn P, Tessana S. et al. Advanced periductal fibrosis from infection with the carcinogenic human liver fluke Opisthorchis viverrini correlates with elevated levels of interleukin-6. Hepatology. 2009;50:1273-1281
97. Wieckowska A, Papouchado BG, Li Z, Lopez R, Zein NN, Feldstein AE. Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am J Gastroenterol. 2008;103:1372-1379
98. Falleti E, Fabris C, Vandelli C, Colletta C, Cussigh A, Smirne C, Fontanini E. et al. Genetic polymorphisms of interleukin-6 modulate fibrosis progression in mild chronic hepatitis C. Hum Immunol. 2010;71:999-1004
99. Starkel P, De Saeger C, Leclercq I, Strain A, Horsmans Y. Deficient Stat3 DNA-binding is associated with high Pias3 expression and a positive anti-apoptotic balance in human end-stage alcoholic and hepatitis C cirrhosis. J Hepatol. 2005;43:687-695
100. Starkel P, Saeger CD, Leclercq I, Horsmans Y. Role of signal transducer and activator of transcription 3 in liver fibrosis progression in chronic hepatitis C-infected patients. Lab Invest. 2007;87:173-181
101. Plum W, Tschaharganeh DF, Kroy DC, Corsten E, Erschfeld S, Dierssen U, Wasmuth H. et al. Lack of glycoprotein 130/signal transducer and activator of transcription 3-mediated signaling in hepatocytes enhances chronic liver injury and fibrosis progression in a model of sclerosing cholangitis. Am J Pathol. 2010;176:2236-2246
102. Ogata H, Chinen T, Yoshida T, Kinjyo I, Takaesu G, Shiraishi H, Iida M. et al. Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-beta1 production. Oncogene. 2006;25:2520-2530
103. Ikejima K, Takei Y, Honda H, Hirose M, Yoshikawa M, Zhang YJ, Lang T. et al. Leptin receptor-mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology. 2002;122:1399-1410
104. Saxena NK, Ikeda K, Rockey DC, Friedman SL, Anania FA. Leptin in hepatic fibrosis: evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology. 2002;35:762-771
105. Lakner AM, Moore CC, Gulledge AA, Schrum LW. Daily genetic profiling indicates JAK/STAT signaling promotes early hepatic stellate cell transdifferentiation. World J Gastroenterol. 2010;16:5047-5056
106. Cao Q, Mak KM, Ren C, Lieber CS. Leptin stimulates tissue inhibitor of metalloproteinase-1 in human hepatic stellate cells: respective roles of the JAK/STAT and JAK-mediated H2O2-dependant MAPK pathways. J Biol Chem. 2004;279:4292-4304
107. Jiang JX, Mikami K, Venugopal S, Li Y, Torok NJ. Apoptotic body engulfment by hepatic stellate cells promotes their survival by the JAK/STAT and Akt/NF-kappaB-dependent pathways. J Hepatol. 2009;51:139-148
108. Wang J, Leclercq I, Brymora JM, Xu N, Ramezani-Moghadam M, London RM, Brigstock D. et al. Kupffer cells mediate leptin-induced liver fibrosis. Gastroenterology. 2009;137:713-723
109. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41-51
110. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798-809
111. Calvisi DF, Ladu S, Gorden A, Farina M, Conner EA, Lee JS, Factor VM. et al. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology. 2006;130:1117-1128
112. Rebouissou S, Amessou M, Couchy G, Poussin K, Imbeaud S, Pilati C, Izard T. et al. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature. 2009;457:200-204
113. Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL, Travis WD. et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest. 2007;117:3846-3856
114. Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, Nebelsiek T. et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91-102
115. Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J. et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103-113
116. Levy DE, Darnell JEJr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651-662
117. Chan KS, Sano S, Kiguchi K, Anders J, Komazawa N, Takeda J, DiGiovanni J. Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J Clin Invest. 2004;114:720-728
118. Tebbutt NC, Giraud AS, Inglese M, Jenkins B, Waring P, Clay FJ, Malki S. et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat Med. 2002;8:1089-1097
119. Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, Lehr HA. et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465-1472
120. Waldner MJ, Wirtz S, Jefremow A, Warntjen M, Neufert C, Atreya R, Becker C. et al. VEGF receptor signaling links inflammation and tumorigenesis in colitis-associated cancer. J Exp Med. 2010;207(13):2855-68
121. Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest. 2008;118:3367-3377
122. Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, Zhang S. et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21:2000-2008
123. Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G. et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11:1314-1321
124. Lee H, Deng J, Kujawski M, Yang C, Liu Y, Herrmann A, Kortylewski M. et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med. 2010;16:1421-1428
125. Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009;69:2506-2513
126. Ferenci P, Fried M, Labrecque D, Bruix J, Sherman M, Omata M, Heathcote J. et al. World Gastroenterology Organisation Guideline. Hepatocellular carcinoma (HCC): a global perspective. J Gastrointestin Liver Dis. 2010;19:311-317
127. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557-2576
128. Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312-1327
129. Asahina Y, Tsuchiya K, Tamaki N, Hirayama I, Tanaka T, Sato M, Yasui Y. et al. Effect of aging on risk for hepatocellular carcinoma in chronic hepatitis C virus infection. Hepatology. 2010;52:518-527
130. Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121-124
131. Abiru S, Migita K, Maeda Y, Daikoku M, Ito M, Ohata K, Nagaoka S. et al. Serum cytokine and soluble cytokine receptor levels in patients with non-alcoholic steatohepatitis. Liver Int. 2006;26:39-45
132. Bosch FX, Ribes J, Diaz M, Cleries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127:S5-S16
133. Ramani K, Yang H, Xia M, Ara AI, Mato JM, Lu SC. Leptin's mitogenic effect in human liver cancer cells requires induction of both methionine adenosyltransferase 2A and 2beta. Hepatology. 2008;47:521-531
134. Saxena NK, Sharma D, Ding X, Lin S, Marra F, Merlin D, Anania FA. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 2007;67:2497-2507
135. Sharma D, Wang J, Fu PP, Sharma S, Nagalingam A, Mells J, Handy J. et al. Adiponectin antagonizes the oncogenic actions of leptin in hepatocellular carcinogenesis. Hepatology. 2010;52:1713-1722
136. Lin L, Amin R, Gallicano GI, Glasgow E, Jogunoori W, Jessup JM, Zasloff M. et al. The STAT3 inhibitor NSC 74859 is effective in hepatocellular cancers with disrupted TGF-beta signaling. Oncogene. 2009;28:961-972
137. Kusaba M, Nakao K, Goto T, Nishimura D, Kawashimo H, Shibata H, Motoyoshi Y. et al. Abrogation of constitutive STAT3 activity sensitizes human hepatoma cells to TRAIL-mediated apoptosis. J Hepatol. 2007;47:546-555
138. Liu Y, Li PK, Li C, Lin J. Inhibition of STAT3 signaling blocks the anti-apoptotic activity of IL-6 in human liver cancer cells. J Biol Chem. 2010;285:27429-27439
139. Yang SF, Wang SN, Wu CF, Yeh YT, Chai CY, Chunag SC, Sheen MC. et al. Altered p-STAT3 (tyr705) expression is associated with histological grading and intratumour microvessel density in hepatocellular carcinoma. J Clin Pathol. 2007;60:642-648
140. Niwa Y, Kanda H, Shikauchi Y, Saiura A, Matsubara K, Kitagawa T, Yamamoto J. et al. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene. 2005;24:6406-6417
141. Cui Q, Jiang W, Wang Y, Lv C, Luo J, Zhang W, Lin F. et al. Transfer of suppressor of cytokine signaling 3 by an oncolytic adenovirus induces potential antitumor activities in hepatocellular carcinoma. Hepatology. 2008;47:105-112
142. He G, Yu GY, Temkin V, Ogata H, Kuntzen C, Sakurai T, Sieghart W. et al. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell. 2010;17:286-297
143. Machida K, Tsukamoto H, Liu JC, Han YP, Govindarajan S, Lai MM, Akira S. et al. c-Jun mediates hepatitis C virus hepatocarcinogenesis through signal transducer and activator of transcription 3 and nitric oxide-dependent impairment of oxidative DNA repair. Hepatology. 2010;52:480-492
144. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH. et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197-208
145. Tang Y, Kitisin K, Jogunoori W, Li C, Deng CX, Mueller SC, Ressom HW. et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc Natl Acad Sci U S A. 2008;105:2445-2450
146. Bertola A, Bonnafous S, Anty R, Patouraux S, Saint-Paul MC, Iannelli A, Gugenheim J. et al. Hepatic expression patterns of inflammatory and immune response genes associated with obesity and NASH in morbidly obese patients. PLoS One. 2010;5:e13577
147. Hill DB, Marsano L, Cohen D, Allen J, Shedlofsky S, McClain CJ. Increased plasma interleukin-6 concentrations in alcoholic hepatitis. J Lab Clin Med. 1992;119:547-552
148. Selzner M, Graf R, Clavien PA. IL-6: a magic potion for liver transplantation? Gastroenterology. 2003;125:256-259
149. Wolk K, Witte E, Witte K, Warszawska K, Sabat R. Biology of interleukin-22. Semin Immunopathol. 2010;32:17-31
150. Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N Engl J Med. 2009;360:2758-2769
151. Blechacz BR, Smoot RL, Bronk SF, Werneburg NW, Sirica AE, Gores GJ. Sorafenib inhibits signal transducer and activator of transcription-3 signaling in cholangiocarcinoma cells by activating the phosphatase shatterproof 2. Hepatology. 2009;50:1861-1870
152. Page BD, Ball DP, Gunning PT. Signal transducer and activator of transcription 3 inhibitors: a patent review. Expert Opin Ther Pat. 2011;21:65-83
Author contact
![]() Corresponding author: Bin Gao, M.D., Ph.D., Laboratory of Liver Diseases, NIAAA/NIH, 5625 Fishers Lane, Rm 2S-33, Bethesda, MD 20892. Email: bgaonih.gov
Corresponding author: Bin Gao, M.D., Ph.D., Laboratory of Liver Diseases, NIAAA/NIH, 5625 Fishers Lane, Rm 2S-33, Bethesda, MD 20892. Email: bgaonih.gov