Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(5):645-650. doi:10.7150/ijbs.7.645 This issue Cite
Short Research Communication
Mechanism of AMPK Suppression of LXR-dependent Srebp-1c Transcription
Fuichi Yap, Lauren Craddock, Jian Yang ![]()
Department of Physiology, University of South Alabama College of Medicine, Mobile, Alabama 36688.
Received 2011-3-4; Accepted 2011-5-13; Published 2011-5-19
Abstract
Activation of AMP-activated protein kinase (AMPK) inhibits hepatic fatty acid synthesis by suppressing sterol regulatory element-binding protein (SREBP)-1c, a master regulator of hepatic lipogenic gene expression. Using a model cell line rat hepatoma McA-RH7777 (CRL-1601) that mimics the behavior of the intact liver by producing high levels of SREPB-1c mRNA and protein, we previously showed that AMPK suppresses hepatic Srebp-1c transcription by inhibiting endogenous liver X receptor (LXR) ligand production and SREBP-1c processing. However, whether AMPK directly inhibits ligand-induced LXR activity remained undetermined. In this study we used a series of mutant Srebp-1c promoter linked to a luciferase reporter to determine the inhibitory mechanism in rat hepatoma McA-RH7777 cells. AMPK activation by either AICAR or metformin decreases Srebp-1c promoter activity by about 75%. Normally, the synthetic LXR ligand T0901317 compound increases the wild-type Srebp-1c promoter activity by about 3-fold, which is similar to that observed in the presence of AICAR or metformin. When endogenous LXR ligand production was blocked by the potent HMG CoA reductase inhibitor compactin, T0901317-induced Srebp-1c promoter activity was decreased by AICAR or metformin treatment. In the mutant Srebp-1c promoter in which two LXR elements are intact but the sterol regulatory element (SRE) is disrupted, the fold inductions of the promoter activity by T0901317 without AMPK activators are significantly higher than those with AMPK activators. Furthermore, AMPK activation attenuates induction of endogenous SREBP-1c mRNA by T0901317. These results indicate that AMPK directly inhibits ligand-induced LXR activity in addition to blocking production of endogenous LXR ligands.
Keywords: AMPK, SREBP-1c, LXR, McA-RH7777 cells, Liver gene expression
Introduction
The 5' adenosine monophosphate (AMP)-activated protein kinase (AMPK) is a heterotrimeric enzyme complex consisting of a catalytic subunit (α) and two regulatory subunits (β and γ) [1]. Activation of AMPK requires phosphorylation of threonine 172 (T172) in the catalytic (α) subunit by one of the three upstream kinases, liver kinase B1 (LKB1) [2-4], calmodulin-dependent protein kinase kinase (CaMKK)-β [5-7], or transforming growth factor-β (TGFβ)-activated kinase-1 (TAK1) [8]. AMPK inhibits hepatic fatty acid synthesis [9]. One of the mechanisms is suppression of sterol regulatory element-binding protein 1c (SREBP-1c) [10-14], a master regulator of hepatic lipogenic gene expression [15].
SREBP-1c is regulated at both transcriptional and posttranslational levels. At transcriptional level, Srebp-1c promoter contains at least four DNA elements (LXR responsive element or LXRE, NFY, SRE, and Sp1 element), which bind to liver X receptors (LXR), nuclear factor-Y (NF-Y), SREBP, and Sp1, respectively [16,17]. Srebp-1c transcription is greatly stimulated by insulin [18]. Insulin activates Srebp-1c promoter primarily by increasing the activity of LXR, whereas SREBPs and NFY play permissive roles [17].
In a recent study [19], we demonstrate that AMPK suppresses SREBP-1c mRNA expression by inhibiting LXR-dependent Srebp-1c transcription via inhibition of endogenous LXR ligand production and by inhibiting SREBP-1c processing in a model cell line - rat hepatoma McA-RH7777 (ATCC Number CRL-1601) that mimics the behavior of the intact liver by producing high levels of SREBP-1c mRNA and protein [20]. However, whether AMPK directly inhibits ligand-induced LXR activity remained unresolved. In this study we used a series of mutant Srebp-1c promoter linked to a luciferase reporter to test this hypothesis in rat hepatoma McA-RH7777 cells.
Materials and Methods
Materials. McA-RH7777 cell line (ATCC#CRL-1601) was purchased from American Type Culture Collection (ATCC, Manassas, VA). Fao cell line (Cat# 89042701) was purchased from Sigma. Fetal bovine serum (FBS, Cat.#16000-044) and first-strand cDNA synthesis kit were obtained from Invitrogen (Carlsbad, CA), delipidated FBS (Cat.#55-0116) from Cocalico Biologicals (Reamstown, PA), Dual Luciferase Reporter Assay System (E1960) from Promega (Madison, WI), 5'-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR, Cat.#A611700) from Toronto Research Chemicals, metformin (Cat.#D5035), compactin (Cat.#M4667), and 1x PBS/Tween 20 (PBST) from Sigma (St. Louis, MO), the synthetic LXR ligand T0901317 (Cat.#71810) from Cayman Chemical (Ann Arbor, MI), RNA STAT-60 from TEL-TEST (Friendswood, TX), DNA-freeTM kit from Ambion, 2x SYBR Green Supermix (Cat.#1708880) from Bio-Rad, BCA assay kit from Fisher (Pittsburgh, PA), and FuGENE 6 transfection reagent from Roche (Indianapolis, IN). Galacto-Light Plus chemiluminescent reporter gene assay system (Cat.#BL300) was from Applied Biosystems (Bedford, Massachusetts).
Plasmids. The plasmid pD containing the wild-type mouse Srebp-1c promoter linked to firefly luciferase reporter gene and the mutant constructs pM24 (-SRE), pM31 (-LXRE1, 2), and pM34 (-LXRE1,2/SRE) were previously described [17,21] and kindly provided by Dr. Guoxun Chen (University of Tennessee at Knoxville). The plasmid pCMV-β-Gal containing the beta-galactosidase reporter gene was from Invitrogen (Carlsbad, CA).
Cell culture. McA-RH7777 cells were cultured and maintained in Medium A (DMEM + 100 units/ml of penicillin and 100 µg/ml streptomycin) supplemented with 10% FBS at 37°C in a 5% CO2 incubator [19,20]. Fao cells were cultured and maintained in Medium B (Kaighn's Modified Ham's F-12K medium + 100 units/ml of penicillin and 100 µg/ml streptomycin) supplemented with 10% FBS at 37°C in a 5% CO2 incubator according to manufacturer's instructions.
Transfection of McA-RH7777 cells and luciferase reporter assay. On Day 0, McA-RH7777 cells were plated at density of 5 x 104 cells per well in 24-well plates in Medium A supplemented with 10% FBS and incubated at 37°C in a 5% CO2 incubator. On Day 1, cells were washed with 0.5 ml PBS and 0.5 ml fresh Medium A was added to each well before transfection. Cells were cotransfected with 0.25 μg plasmids containing wild-type or mutant Srebp-1c promoter linked to the firefly luciferase reporter gene and 10 ng control plasmid pCMV-β-Gal containing the beta-galactosidase reporter gene by using FuGENE 6 transfection reagent. Each transfection was done in triplicate wells. Six hours after transfection cells were washed with 0.5 ml PBS, switched to Medium A supplemented with 10% delipidated FBS and various compounds as indicated in the figure legends, and incubated for 16 hours at 37°C and 5% CO2. On Day 2, cells were washed with 0.5 ml PBS and firefly luciferase activities were measured using the Luciferase Reporter Assay System and Turner Designs TD-20/20 Luminometer. Beta-galactosidae activity was measured by Galacto-Light Plus chemiluminescent reporter gene assay system. Firefly luciferase activities in the transfected lysates were normalized by beta-galactosidase activity or protein concentration from the same tube.
Preparation of total cellular RNA. On Day 0, McA-RH7777 cells were plated at the density of 7 x 105 cells per 100-mm dish and cultured in Medium A supplemented with 10% FBS at 37°C in a 5% CO2 incubator as previously described. On Day 2, cells were washed with 1x phosphate-buffered saline (PBS) and cultured for 16 h in Medium A supplemented with 10% delipidated FBS and various compounds as indicated in the figure legends. Each treatment was performed in duplicate dishes. On Day 3, the medium was aspirated, the cells in duplicate dishes were harvested and combined, and total cellular RNA was prepared with RNA STAT-60 according to manufacturer's instructions.
Real-time PCR. Real-time PCR was carried out as previously described [19]. Briefly, total cellular RNAs were prepared with RNA STAT-60, and treated with DNase I before cDNA synthesis. First strand cDNA was synthesized from 5 μg of DNase I-treated total RNA with random hexamer primers by using the first-strand cDNA synthesis system. Real-time PCR of samples was prepared in a final volume of 25 μl containing 50 ng of reverse transcribed total RNA, 167 nM forward and reverse primers, 12.5 μl of 2x SYBR Green Supermix. Real-time PCR was carried out in a 96-well plate on the iCycler iQ Real-Time Detection System (Bio-Rad). All reactions were done in triplicate. The relative amount of all mRNAs was calculated using the comparative CT method (Applied Biosystem 2001. User Bulletin No.2, Applied Biosystems, Forster City, CA). Ribosomal phosphoprotein 36B4 mRNA was used as the invariant control.
Statistics. Experimental data were statistically analyzed using a two-tailed Student's t test between two means. Results were expressed as mean ± SD. The difference between two groups with P<0.05 was considered to be statistically significant.
Results and Discussion
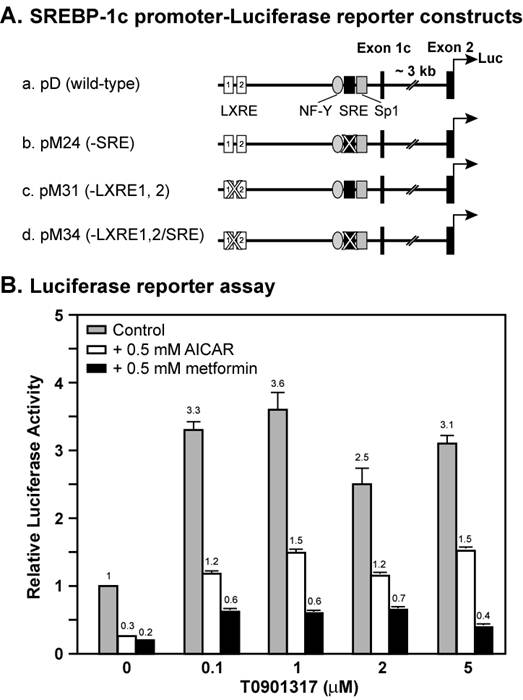
Figure 1 shows a dose curve of the synthetic LXR ligand T0901317 compound on the induction of wild-type Srebp-1c promoter linked to a luciferase reporter in the absence or presence of AMPK activators AICAR and metformin. In the absence of AMPK activators, the luciferase reporter activity was increased by 3.3-fold by 0.1 μM T0901317 compound, and 3.6-fold by 1 μM. Further increase of T0901317 concentration did not enhance the promoter activity, suggesting that 1 μM reached a saturating concentration. Activation of AMPK by 0.5 mM either AICAR or metformin inhibited the Srebp-1c promoter activity by 70% and 80%, respectively, consistent with our previous results [19]. In the presence of AICAR, T0901317 compound induced Srebp-1c promoter activity by 4-fold at 0.1 μM and 5-fold at 1 μM, whereas in the presence of metformin, T0901317 compound induced Srebp-1c promoter activity by 3-fold at 0.1 μM and 3-fold at 1 μM. These data indicate that the LXR ligand T0901317 induces Srebp-1c promoter activity to the same extent with or without activation of AMPK under normal conditions.
AMPK activation suppresses LXR ligand-mediated induction of Srebp-1c promoter activity in McA-RH7777 cells. A, Diagram of plasmids containing the wild-type or mutant mouse Srebp-1c promoter linked to the firefly luciferase reporter gene. B, Luciferase reporter assay. On Day 0, McA-RH7777 cells were plated at density of 5 x 104 cells per well in 24-well plates in Medium A supplemented with 10% FBS and incubated at 37°C in a 5% CO2 incubator. On Day 1, cells were washed with 0.5 ml PBS and 0.5 ml fresh Medium A was added to each well before transfection. Cells were cotransfected with plasmid D and a control plasmid as described in Materials and Methods. Six hours after transfection, cells were washed with 0.5 ml PBS, switched to Medium A supplemented with 10% delipidated FBS and 0.5 mM AICAR or metformin or various amounts of T0901317, and incubated for 16 hours at 37°C and 5% CO2. On Day 2, cells were washed with 0.5 ml PBS, and luciferase and beta-gal activities were measured as described in the Materials and Methods. Normalized firefly luciferase activity without treatment was arbitrarily set as 1.
Activation of AMPK is known to inhibit endogenous LXR ligand production by inhibiting HMG CoA reductase activity, thereby decreasing Srebp-1c promoter activity and SREBP-1c mRNA expression [19,20]. There is minimal SREBP-1c mRNA expression (<10% compared with control) in the McA-RH7777 cells treated with compactin, a potent inhibitor of HMG CoA reductase [19]. We carried out luciferase reporter assays in the presence of compactin (Fig. 2). There was negligible but detectable basal luciferase reporter activity in the presence of compactin (Fig. 2A). Activation of AMPK by AICAR and metformin further reduced basal SREBP-1c promoter activity by 50% and 40%, respectively. In the absence of AMPK activators, T0901317 compound (1 μM) increased Srebp-1c promoter activity by 10.2-fold. The fold induction by T0901317 compound was reduced to 6.6 in the presence of AICAR, and to 7 in the presence of metformin.
Activation of AMPK decreases the fold induction of wild-type and mutant Srebp-1c promoter activities by LXR ligand T0901317 in McA-RH7777 cells treated with compactin. McA-RH7777 cells were cultured, transfected, and treated with AICAR (0.5 mM) or metformin (0.5 mM) with or without T0901317 (1 μM) in the presence of compactin (50 μM compactin + 50 μM sodium mevalonate) as in Fig. 1. Cells were lysed and luciferase reporter assays were performed as in Fig. 1 with (A) wild-type Srebp-1c promoter construct pD, (B) pM31 (-LXRE1, 2), (C) pM24 (-SRE), and (D) pM34 (-LXRE1, 2/SRE). The value on each column represents the fold change (the average of three assays) as compared with control (-AICAR/Metformin/T0901317). White bar, - T0901317; grey bar, + T0901317. *, P<0.05 as compared with control (+ T0901317) without AICAR/metformin treatment for each promoter construct.
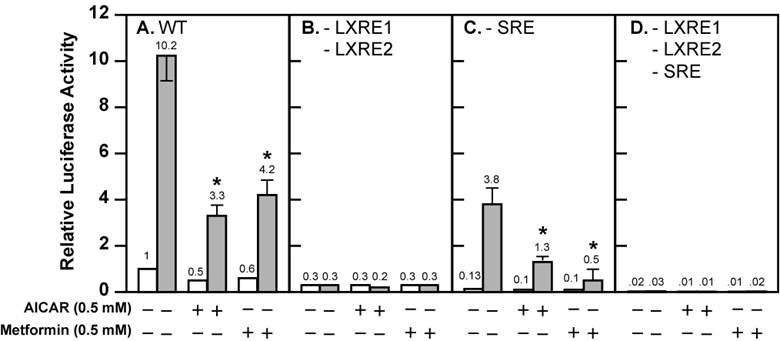
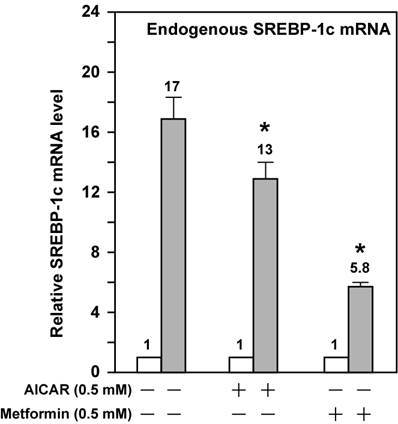
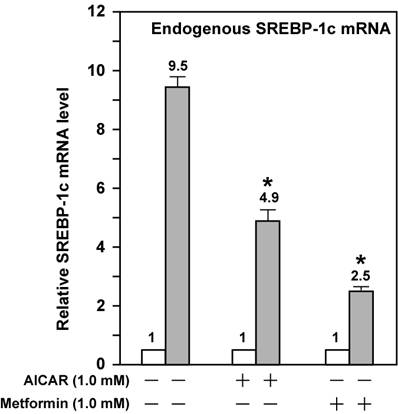
These data suggest that activation of AMPK directly effects ligand-induced LXR activity. However, SREBP-1c regulates its own transcription due to the presence of the SRE in its promoter; this leads to amplification of the LXR ligand-induced Srebp-1c promoter activity [17,22]. To bypass the potential complication by this autoregulation, we assayed three mutant Srebp-1c promoter constructs, pM24 with mutation in the SRE, pM31 with double mutations in LXRE1 and LXRE2, and pM34 with triple mutations in LXRE1, LXRE2, and SRE (Fig. 1A). Fig. 2 shows that pM31 (-LXRE1, 2) had about 30% wild-type Srebp-1c promoter activity, pM24 (-SRE) had less than 15% activity, and pM34 (-LXRE1, 2/SRE) virtually had no activity, consistent with the published data [17]. For the mutant pM31 (-LXRE1, 2), addition of T0901317 compound (1 μM) did not induce the promoter activity and AMPK activation did not further decrease the mutant promoter activity in the absence or presence of T0901317 compound (1 μM). For the mutant pM24 (-SRE), the fold induction by T0901317 compound was 28.4 in the absence of AMPK activators, but was significantly reduced to 13 and 5 in the presence of AICAR and metformin, respectively. It is noted that activation of AMPK consistently reduced the absolute amount of T0901317-induced Srebp-1c promoter activity with SRE (pD) or without SRE (pM24) (Fig. 2). Fig. 3 shows that activation of AMPK by either AICAR or metformin attenuated the fold induction of SREBP-1c mRNA by T0901317 in McA-RH7777 cells treated with compactin, in agreement with the results from luciferase reporter assays. Similar results were obtained from rat hepatoma Fao cells (Fig. 4). These data indicate that activation of AMPK reduces LXR ligand-induced Srebp-1c promoter activity when the endogenous LXR ligand is blocked, suggesting that AMPK directly inhibits ligand-induced LXR activity. However, because LXR has to form heterodimers with retinoid X receptor (RXR) in order to be functional [23], our data do not rule out the possibility that AMPK activation inhibits RXR as well.
AMPK activation attenuates the fold induction of endogenous SREBP-1c mRNA by LXR ligand T0901317 in McA-RH7777 cells treated with compactin. On Day 0, McA-RH7777 cells were plated at the density of 7 x 105 cells per 100-mm dish and cultured in Medium A supplemented with 10% FBS at 37°C in a 5% CO2 incubator. On Day 2, cells were washed with 1x PBS and cultured for 16 h in Medium A supplemented with 10% delipidated FBS and compactin (50 μM compactin + 50 μM sodium mevalonate) in the absence or presence of additional compounds (AICAR, 0.5 mM; metformin, 0.5 mM; and T0901317, 1 μM). Each treatment was performed in duplicate dishes. On Day 3, cells from duplicate dishes were harvested and combined, total RNA was prepared and reverse transcribed, and endogenous SREBP-1c mRNA levels were measured by real-time PCR as described in Materials and Methods. The value on each column represents the fold change (the average of three assays) as compared with control (-T0901317). White bar, - T0901317; grey bar, + T0901317. *, P<0.05 as compared with control (+ T0901317) without AICAR/metformin treatment.
AMPK activation attenuates the fold induction of endogenous SREBP-1c mRNA by LXR ligand T0901317 in Fao cells. Fao cells were cultured and treated as McA-RH7777 cells in Fig. 3 except that Medium A was replaced with Medium B and the concentration of both AICAR and metformin was 1 mM. Cells from duplicate dishes were harvested and combined, total RNA was prepared and reverse transcribed, and endogenous SREBP-1c mRNA levels were measured by real-time PCR as described in Materials and Methods. The value on each column represents the fold change (the average of three assays) as compared with control (-T0901317). White bar, - T0901317; grey bar, + T0901317. *, P<0.05 as compared with control (+ T0901317) without AICAR/metformin treatment.
In conclusion, the data from this study support the hypothesis that AMPK inhibits ligand-induced LXR activity on Srebp-1c promoter. Further work should be conducted to determine whether AMPK directly phosphorylates LXR or RXR and inhibits their transcriptional activities.
Abbreviations
AMPK: AMP-activated protein kinase; AICAR: 5'-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside; beta-gal: beta-galactosidase; HMG CoA: 3-hydroxy-3-methylglutaryl coenzyme A; LXR: liver X receptor; NFY: Nuclear factor Y; RXR: retinoid X receptor; SREBP: sterol regulatory element-binding protein.
Acknowledgements
We thank Dr. Guoxun Chen (University of Tennessee at Knoxville) for the Srebp-1c promoter constructs. This work was supported by National Scientist Development Grant #0635341N from American Heart Association National Center (JY).
Conflict of Interests
No conflict of interests exists.
References
1. Hardie D.G. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774-785
2. Hawley S.A, Boudeau J, Reid J.L. et al. Complexes between the LKB1 tumor suppressor, STRADalpha/beta and MO25alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28
3. Shaw R.J, Kosmatka M, Bardeesy N. et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329-3335
4. Woods A, Johnstone S.R, Dickerson K. et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004-2008
5. Woods A, Dickerson K, Heath R. et al. C(Ca2+)/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21-33
6. Hawley S.A, Pan D.A, Mustard K.J. et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9-19
7. Hurley R.L, Anderson K.A, Franzone J.M, Kemp B.E, Means A.R, Witters L.A. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060-29066
8. Momcilovic M, Hong S.P, Carlson M. Mammalian TAK1 Activates Snf1 Protein Kinase in Yeast and Phosphorylates AMP-activated Protein Kinase in Vitro. J Biol Chem. 2006;281:25336-25343
9. Viollet B, Foretz M, Guigas B. et al. Activation of AMP-activated protein kinase in the liver: a new strategy for the management of metabolic hepatic disorders. J Physiol. 2006;574:41-53
10. Zhou G, Myers R, Li Y. et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167-1174
11. Lin H.Z, Yang S.Q, Chuckaree C, Kuhajda F, Ronnet G, Diehl A.M. Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nat Med. 2000;6:998-1003
12. Foretz M, Pacot C, Dugail I. et al. ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Mol Cell Biol. 1999;19:3760-3768
13. Shaw R.J, Lamia K.A, Vasquez D. et al. The Kinase LKB1 Mediates Glucose Homeostasis in Liver and Therapeutic Effects of Metformin. Science. 2005;310:1642-1646
14. Yang J, Maika S, Craddock L, King J.A, Liu Z.M. Chronic activation of AMP-activated protein kinase-alpha1 in liver leads to decreased adiposity in mice. Biochem Biophys Res Commun. 2008;370:248-253
15. Horton J.D, Goldstein J.L, Brown M.S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125-1131
16. Cagen L.M, Deng X, Wilcox H.G, Park E.A, Raghow R, Elam M.B. Insulin activates the rat sterol-regulatory-element-binding protein 1c (SREBP-1c) promoter through the combinatorial actions of SREBP, LXR, Sp-1 and NF-Y cis-acting elements. Biochem J. 2005;385:207-216
17. Chen G, Liang G, Ou J, Goldstein J.L, Brown M.S. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc Natl Acad Sci U S A. 2004;101:11245-11250
18. Horton J.D, Bashmakov Y, Shimomura I, Shimano H. Regulation of sterol regulatory element binding proteins in livers of fasted and refed mice. Proc Natl Acad Sci U S A. 1998;95:5987-5992
19. Yang J, Craddock L, Hong S, Liu Z.M. AMP-activated protein kinase suppresses LXR-dependent sterol regulatory element-binding protein-1c transcription in rat hepatoma McA-RH7777 cells. J Cell Biochem. 2009;106:414-426
20. DeBose-Boyd R.A, Ou J, Goldstein J.L, Brown M.S. Expression of sterol regulatory element-binding protein 1c (SREBP-1c) mRNA in rat hepatoma cells requires endogenous LXR ligands. Proc Natl Acad Sci U S A. 2001;98:1477-1482
21. Li R, Chen W, Li Y, Zhang Y, Chen G. Retinoids synergized with insulin to induce Srebp-1c expression and activated its promoter via the two liver X receptor binding sites that mediate insulin action. Biochem Biophys Res Commun. 2011;406:268-272
22. Amemiya-Kudo M, Shimano H, Yoshikawa T. et al. Promoter analysis of the mouse sterol regulatory element-binding protein-1c gene. J Biol Chem. 2000;275:31078-31085
23. Repa J.J, Mangelsdorf D.J. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu Rev Cell Dev Biol. 2000;16:459-481
Author contact
![]() Corresponding author: Jian Yang, Ph.D., Department of Physiology, MSB 3074, 307 University Boulevard, Mobile, Alabama 36688-0002. Phone: (251) 460-6814; Fax: (251) 460-6386; E-mail: jyangedu
Corresponding author: Jian Yang, Ph.D., Department of Physiology, MSB 3074, 307 University Boulevard, Mobile, Alabama 36688-0002. Phone: (251) 460-6814; Fax: (251) 460-6386; E-mail: jyangedu