Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Inflammation and cancer
Immune cells within the tumor...
Immunosuppressive cytokines...
Concluding Remarks
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(5):651-658. doi:10.7150/ijbs.7.651 This issue Cite
Review
Dual Roles of Immune Cells and Their Factors in Cancer Development and Progression
Brian F. Zamarron, WanJun Chen ![]()
Mucosal Immunology Unit, Oral Infection and Immunity Branch, National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD 20892, USA
Received 2011-3-3; Accepted 2011-4-12; Published 2011-5-21
Abstract
Traditional wisdom holds that intact immune responses, such as immune surveillance or immunoediting, are required for preventing and inhibiting tumor development; but recent evidence has also indicated that unresolved immune responses, such as chronic inflammation, can promote the growth and progression of cancer. Within the immune system, cytotoxic CD8+ and CD4+ Th1 T cells, along with their characteristically produced cytokine IFN-γ, function as the major anti-tumor immune effector cells, whereas tumor associated macrophages (TAM) or myeloid-derived suppressive cells (MDSC) and their derived cytokines IL-6, TNF, IL-1β and IL-23 are generally recognized as dominant tumor-promoting forces. However, the roles played by Th17 cells, CD4+ CD25+ Foxp3+ regulatory T lymphocytes and immunoregulatory cytokines such as TGF-β in tumor development and survival remain elusive. These immune cells and the cellular factors produced from them, including both immunosuppressive and inflammatory cytokines, play dual roles in promoting or discouraging cancer development, and their ultimate role in cancer progression may rely heavily on the tumor microenvironment and the events leading to initial propagation of carcinogenesis.
Keywords: TGF-β, Tumor Associated Macrophages, Hepatocellular Carcinoma, Treg
Introduction
Cancer cell development and survival is a multifactor process, involving genetic mutation of normal cells as well as physiological changes within both cancer cells and also the body's defense mechanisms1, 2. Cellular changes, such as gain of function mutations of oncogenes and loss of function mutations in tumor suppressor genes, can lead to relative cellular immortality, proliferation, and carcinogenesis. In addition to cellular changes, the body's normal defense mechanisms can play a key role in encouraging or combating carcinogenesis and tumor spread. Of particular importance is the immune response to cancer cell development and progression, as well as the potential role the immune response might play in initial tumor formation. In this regard, compelling evidence has been documented, both in animal tumor models and in human cancers, that chronic inflammation plays a decisive role in the development of certain cancers such as hepatocellular carcinoma (HCC) and colon cancer. However, cells of the immune system can also inhibit tumor growth and progression through the recognition and rejection of malignant cells, a process referred to as immunosurveillance or immunoediting 3, 4. In this respect, immunodeficiency can predispose an individual to the development of both spontaneous and virally induced cancers, and established tumors often generate immunosuppressive microenvironments that can block productive antitumor immunity. Of the plethora of cells, soluble factors and cytokines involved, tumor-associated macrophages (TAM) or myeloid-derived suppressive cells (MDSC) and their secreted cytokines IL-6, TNF, and IL-1β may be key in promoting the progress of tumor development. However, the cellular and molecular mechanisms that positively and negatively regulate the function and phenotype of TAM remain largely unknown. Elucidation of these mechanisms would have a significant impact on understanding the pathogenesis of inflammation-associated cancers, and could aid progress in the development of more effective cancer therapies. This review will explore the dual-roles played by key T lymphocyte subsets in cancer development and progression, as well as discussing the potential link between TAM cells and T lymphocytes as they relate to tumor formation, growth and survival.
Inflammation and cancer
A causal association between viral infection and tumor development has been well established and supported in animal studies as well as epidemiological studies. For example, the risk of developing HCC appears to be intimately linked to the duration of Hepatitis B and C viral infection-induced inflammatory processes 1, 5-7. Normally, inflammation serves to protect a defined region of infected or damaged tissue by recruiting cells necessary to resolve the insult while also isolating the area to prevent the spread of infection and, once resolved, normal tissue function is typically restored. However, inflammation sometimes fails to subside and this unresolved inflammation can promote tumor cell growth, survival and angiogenesis 1. Intriguingly, evidence continues to accumulate indicating that unresolved chronic inflammation plays a critical role in the initiation, promotion, malignant conversion, and metastasis of several human cancers 1. Additionally, inflammation associated with the hepatitis B and C viruses is the primary cause of liver cancer. Infection with the bacterium Helicobacter pylori plays a central role in the development of most cancers of the stomach 8. Beyond infections, many autoimmune diseases are associated with an increased risk of lymphoma, and inappropriate immune responses to commensal flora in ulcerative colitis are strongly linked to colon cancer 1, 8-10. Evidence now links inflammation to tumor development in both genetic tumor syndromes and in the context of chronic carcinogen exposure. Administration of non-steroidal anti-inflammatory drugs (NSAIDs) in randomized studies have shown reduced incidence of colon cancer in patients with familial adenomatous polyposis and, perhaps more remarkably, reduces the incidence of lung cancer in smokers 10-13.
Immune cells within the tumor microenvironment
Tumor-associated macrophages and myeloid-derived suppressive cells
Within the microenvironment, TAM −isolated from established metastatic tumors in both mice and humans− play a vital role in tumor cell development, survival and growth. TAM are a heterogeneous cell population according to oxygen availability (hypoxia vs. normoxia) and to tumor progression (small vs. advanced tumors) 14, 15. In early stage tumor development Type 1 macrophages (M1) may infiltrate, activated in response to inflammatory mediators, and release pro-inflammatory cytokines and chemokines, such as CXCL19 and CXCL10, in order to attract and encourage Th1, Th17 and NK cell development and differentiation. M1 macrophages are activated by signals such as bacteria and LPS, however the cellular factors GM-CSF and IFN-γ produced at sites of inflammation will also tend to polarize towards the M1 state. It has been shown that these conditions promote IRF5 expression in macrophages, leading to production and maintenance of M1 phenotypic markers and encouraging lymphocyte proliferation and Th1/Th17 cellular responses by directly up-regulating the transcription of IL-12p35, IL-12p40, IL-23 and TNF-α 16. In contrast, in more advanced tumors or in hypoxic regions of the tumor microenvironment TAM polarize to a more type 2 macrophage (M2) related cell, releasing factors to encourage Th2 differentiation and recruitment 15. M2-like TAM express a distinct set of cytokines and chemokines including CCL17, CCL22 and CCL24 favoring regulatory T cell (Treg) recruitment and development. Moreover, M2 macrophages support tissue repair and remodeling, as well as angiogenesis through the production of VEGF or EGF 14. Overall, these TAM cells can be pro-inflammatory with M1-type cells promoting cell growth and recruitment through the production of chemokines and cytokines such as IL-6, TNF-α, IL-23 and IL-12 1, 5, 17, 18, and may also serve to promote tumor development and immunoregulation with M2-like TAM inhibiting anti-cancer immunity through the production of transforming growth factor β1 (TGF-β) and IL-10 14, 18, 19.
Myeloid-derived suppressive cells represent a heterogeneous population of immature myeloid cells not already committed into macrophages, dendritic cells or granulocytes. These MDSC exert suppressive functions, regulating T cell responses through nitric oxide, reactive oxygen species and TGF-β secretion while also promoting Treg induction and favoring anti-inflammatory responses 19, 20. These cells are found within both human and mouse tumors. MDSC produce high levels of IL-6 and are responsive to IL-6 but also IL-1β and IFN-γ. Moreover, expansion of MDSC in response to macrophage-derived factors, such as VEGF, has been reported 19, 20. Whereas MDSC would normally differentiate after migration, the cytokines and cellular factors found within the tumor microenvironment prevent differentiation and instead encourage expansion and activation of the immature myeloid cell population 19, and may result in the suppression of tumor immunity through a variety of mechanisms.
IL-6 is a key cytokine amongst these cellular factors, encouraging cancer cell proliferation while also inhibiting their apoptosis through activation of signal transducer and activator of transcription 3 (Stat3) 1, 21. IL-6 signaling can also influence T cell subset differentiation, particularly in the presence of other cytokines such as TGF-β 22. IL-6 has also been shown to play an important role in carcinogen-driven liver cancer development 7, 23. Additionally, IL-6 acts as an angiogenic factor, and has been implicated in many of the same processes as TNF. TNF itself also plays an essential role in several models of cancer, and is a critical inflammatory mediator in many autoimmune diseases of both mice and humans primarily acting via induction of NF-κB 5, 6. TNF plays an indispensible role in the hepatic procarcinogen diethylnitrosamine-induced experimental hepatocellular carcinoma, as ablation of TNF receptor I in mice almost completely abolished obesity-enhanced HCC development 7. IL-1β can activate NF-κB in a manner similar to TNF, and polymorphisms in IL-1β have been linked to gastric cancers 24. Notably, during the cross-talk between cancer and inflammatory cells, Stat3 and NF-κB seem to be key transcription factors linking a mutual positive feedback loop and promoting cancer progression 5, 21. In an experimental model of Src oncogene-dependent cancer transformation, induction of inflammation was critical for oncogenesis and Src-induced inflammation was mediated by NF-κB leading to increased IL-6 production, which then in turn increased Stat3 activation. The IL-6 in this tumor model served to further activate NF-κB, causing a positive feedback loop of inflammation and tumorigenesis 25.
CD4+ Th1 and CD8+ T cells
It is generally recognized that IFN-γ producing CD4+ Th1 cells and CD8+ T cells play an important role in inhibiting and killing tumor cells and impeding tumor growth. Spontaneous lymphocytic infiltrates can be consistently observed in a variety of human cancers, and in some cases these infiltrating lymphocytes correlate with a favorable prognosis 3. Anti-tumor specific effector immune cells develop within the draining lymph nodes, but have also been found to proliferate within the tumor microenvironment despite potential immunosuppressive factors 26, however the extent to which these cells are able to contribute to anti-cancer immunity is unclear; particularly with respect to the immunosuppressive conditions present within the tumor. Amongst the many factors Th1 and CD8+ T cells produce, IFN-γ seems to be one of most significant cytokines preventing and suppressing the development of cancers. In addition, the cytotoxic effects of CD8+ T cells may also directly mediate the death of tumor cells. Given the recognized tumor promoting function of macrophages and their inflammatory cytokines, the potential effects of these anti-tumor immune Th1 and CD8+ T cells on TAM over the course of HCC tumor development and progression are desperately needed.
CD4+ CD25+ Foxp3+ regulatory T cells
Not all T cells are anti-tumor effector immune cells; a subpopulation of CD4+ T cells expressing CD25 and the master transcription factor Foxp3 (CD4+CD25+Foxp3+), termed regulatory T cells (Tregs), play a role in promoting tumor growth and progress by inhibiting the immune response against cancer 27-29. A critical function of these cells is to suppress the activation of effector immune cells that are specific for self-antigens, limiting autoimmunity and inflammation under physiologically normal conditions. Loss of function mutations of the Foxp3 gene in both mice and humans results in fatal autoimmunity affecting multiple organ systems, and depletion of the Foxp3+ Treg population in adult mice was also found to result in severe and fatal full-body autoimmunity 30-32. Tregs inhibit the activation of both CD4+ and CD8+ T cells, and within the tumor microenvironment may serve to suppress anti-cancer cell immunity. Studies of murine tumors indicate that Tregs inhibit the immune response to tumors, and depletion of these cells promotes rejection of several murine tumor cell lines including melanoma, fibrosarcoma, leukemia and myeloma. In some murine tumors (melanoma and fibrosarcoma), both CD4+ and CD8+ T cells were found to be required for tumor rejection, while only CD8+ T cells were required for the rejection of murine myeloma 33. The role of Tregs in carcinogenesis and metastasis in humans is just beginning to emerge. In pancreatic and breast cancer patients, the prevalence of CD25+ regulatory T cells in the blood has been shown to be much higher than that found in normal donors. In cervical and endometrial cancer patients, functioning CD25+ regulatory T cells have been identified within the tumor draining lymph nodes. In non-small cell lung cancer and ovarian cancer, tumor associated T cells contained increased proportions of CD25+ regulatory T cells and it was further shown that these cells secrete the immunosuppressive cytokine TGF-β 34. The role of TGF-β mediated immunosuppression by CD25+ regulatory T cells in colorectal cancer has also been documented 35. The role of Tregs in the development of liver carcinomas, however, remains largely unknown. It has been reported that in both experimental murine models and human patients with HCC, the frequency of CD4+ Foxp3+ Tregs was increased 36. However, the impact of Tregs in the development of HCC, initiated by TAM-mediated inflammation, remains elusive. In particular, whether and how CD4+ Tregs affect the behavior and function of TAM is unknown. Conversely, it is also unclear how TAM and TGF-β regulate the generation and function of Tregs in the developing and established solid tumor microenvironment.
Contrasted with the possible tumor-promoting role of these cells in the progression of established tumors, the role of CD4+ Foxp3+ Tregs in initial tumor transformation caused by chronic inflammation remains elusive. Based on the potent immunoregulatory function these cells have on immune responses and inflammation, it is reasonable to conceive that Tregs may in fact help prevent and/or delay inflammation-mediated tumor development. In this regard, the CD4+ Tregs in the gut may prevent chronic colitis-triggered colon cancer by inhibiting inflammation within the colon. However, the experimental evidence supporting this notion awaits further investigation.
Th17 cells
Similar to Tregs, the role of IL-17 producing CD4+ cells (Th17) in the growth, survival, and progression of HCC also remain unknown. Th17 cells are involved in several autoimmune diseases and chronic inflammatory syndromes. Naive CD4+ T cells preferentially differentiate into the Th17 T cell subset in response to the combined signals of TGF-β and IL-6, mediated through Stat3 activation. In addition, IL-23 is a key factor for maintaining and expanding Th17 inflammatory cell populations 37. IL-23 is another recently identified pro-carcinogenic cytokine, promoting inflammation and angiogenesis within the tumor microenvironment while reducing CD8+ T cell infiltration 38. The presence of these Th17 cells within the tumor microenvironment could antagonize and counter the tumor-suppressive IFN-γ producing CD4+ Th1 cells 22, 24. In addition, a study of murine tumor formation, using the B16 melanoma cell line, found that IL-17 signaling was critical for tumor development, with direct effects on tumor and stromal cells inducing IL-6 production which then led to activation of Stat3 39. Th17 cells antagonize the differentiation and function of IFN-γ producing Th1 cells, and are thereby likely involved the promotion of tumor growth. However, another study found a potential anti-tumor role for adoptively transferred tumor-specific Th17 cells by promoting tumor-specific CD8+ T cell activation in a model of lung melanoma in IL-17A deficient mice 40. Since HCC cells, TAM and MDSC are able to produce large amounts of IL-6 and TGF-β, it is tempting to envision that Th17 cells could be preferentially differentiated in the tumor microenvironment of HCC, particularly in established tumor tissues. Thus, the combined cellular products from both Th17 and myeloid cells infiltrating the tumor, and the interaction between these cell types, may play a critical role in the balance of anti- and pro-tumor immune responses within the HCC microenvironment.
Immunosuppressive cytokines within the tumor microenvironment
TGF-β
As mentioned, TAM also produce immunosuppressive cytokines that inhibit anti-tumor immune cells such as CD8+ and Th1 cells in established cancer tissues, further facilitating cancer progression. TGF-β and IL-10 are two such immunoregulatory cytokines identified and produced within the tumor microenvironment. TGF-β plays an indispensible, yet complex, role in carcinogenesis and the progression of tumors 41, 42. With regard to immune cells, it is recognized that TGF-β is largely an immunosuppressive cytokine, generally inhibiting immune responses including anti-cancer immunity. In addition, TGF-β plays a central role in the generation and function of CD4+ CD25+ Tregs 27. However, the ultimate effect of TGF-β on immune cells is heavily dependent on the microenvironment and the presence of other cytokines and factors 43. For example, TGF-β suppresses IFN-γ production by Th1 and CD8+ T cells while also promoting the generation of Foxp3+ Tregs and differentiation of Th17 cells, which, when taken together, likely favors the growth and progression of cancer. However, TGF-β is also a potent inhibitor of macrophage activation and decreases their production of inflammatory cytokines such as IL-6, TNF and IL-1β, which may impede inflammation-associated cancer development. It will be critical to understand what role, if any, TGF-β has at the interface between T cells and macrophages within the tumor microenvironment.
IL-10
IL-10 may play a similarly complex role in the development and survival of cancer cells. It has been shown that IL-10 production from CD25+ Tregs aids in regulating immune responses, and, in certain cases of chronic infection, IL-10 is essential for proper downregulation of inflammation and prevention of eventual carcinogenesis in microbial-induced colitis mice models 44-46. IL-10 can also inhibit NF-κB activation and may hinder angiogenesis within the tumor microenvironment. Despite this, however, IL-10 can also activate STAT3 and may thereby help to promote tumor development, playing a part in cell proliferation and survival 24, 47. The role of IL-10 in cancer development being either primarily anti-tumor, preventing inflammation and tumor angiogenesis, or primarily pro-tumor, encouraging cell survival and suppressing effector T cells, may depend upon the conditions of initial carcinogenesis as well as the presence of other cells such as Tregs.
Concluding Remarks
Accumulated evidence has indicated that chronic and unresolved inflammation may play a critical role in the initiation, development, growth, and metastasis of cancer. The net outcome of the battle between the anti-tumor and the tumor promoting immune cells and their associated cytokines within the tumor environment will determine the ultimate fate of the affected tumors. Thus, understanding the interactions and mutual regulation of these complex immune networks within the tumor environment, rather than the role of individual components, is much more likely to guide us towards developing effective and specific cancer therapeutics. It is generally believed that CD4+ Th1 and CD8+ T cells comprise the primary force of immune cells responsible for inhibiting tumor growth and development, while TAM, MDSC, Cd4+Foxp3+Treg cells and Th17 cells and their associated cytokines IL-6, TNF, IL-1β, IL-23 and TGF-β may play important roles in promoting the growth and survival of cancer. However, it is difficult to draw a distinction and establish clear “black and white” classifications for which cell types are always pro-tumor or always anti-tumor.
Of particularly complexity is the immunoregulatory cytokine TGF-β. It has been long known that most tumor cells produce large amounts of TGF-β. Macrophages are a major cellular source of TGF-β during normal immune system responses. However, surprisingly, it is less known whether and how TGF-β functions in the cross-talk between TAM and cancer cells. In terms of the direct relationship between tumor cells and TGF-β, as well as the potential role TGF-β plays in determining T cell differentiation fate within developing and established tumors, many outstanding questions remain to be answered. For example, what is the effect of TGF-β on TAM within the tumor environment? Does TGF-β influence the production of proinflammatory cytokines IL-6, TNF and TGF-β in TAM? Of further importance is understanding whether TGF-β production, by either TAM, MDSC or cancer cells, preferentially induces CD4+Foxp3+ regulatory T cells, or instead promotes the development of Th17 cells within the microenvironment of tumors. The ultimate outcome of TGF-β's effects on tumor development and survival will most probably depend upon the balance of its activity at the interface between the cancer cells and the tumor associated immune cells. Further research into better understanding this balance at all stages of carcinogenesis is essential for the development of effective cancer therapies that target or utilize immunological mechanisms.
A similar paradox exists in determining the role of CD4+ Foxp3+ Tregs in tumor development, which is further complicated and particularly important given the control TGF-β has over Treg induction. With chronic infection or autoimmunity, CD4+ Treg cells help to limit inflammation and may hinder carcinogenesis by limiting cellular damage and inhibiting cycles of proliferation. Once a tumor is established, however, Tregs and their produced cytokines may instead play a role in promoting tumor survival by inhibiting cancer cell destruction. It is reasonable to assume that effector immune T cells could infiltrate into tumors and counter the growth and progression of cancer cells. However, to what extent Tregs may influence the effector T cell population within the tumor microenvironment remains unclear. Further unknown is what role Th1 and CD8+ T cells have on influencing both the local cytokine profile within the microenvironment as well as what influence these effector T cells have on TAM activity in either developing or established solid tumors. Given the recognized tumor promoting function of macrophages and their inflammatory cytokines, the potential effects of these anti-tumor immune Th1 and CD8+ T cells on TAM over the course of tumor development are desperately needed.
Our belief is that the tumor-associated macrophages and oncogenic inflammation are regulated at multiple levels by T cell subsets, immunoregulatory cytokines, and also intracellular transcription factors such as NF-κB and STAT3. The net outcome of these complex regulations determines whether or not tumor cells grow, recur, and intrude, and thereby have an impact on the eventual survival of patients with cancer. While Treg cells and their cytokines likely serve to promote cancer cell growth and survival in established tumors, these same immunosuppressive properties also prevent autoimmunity and early stage cancer development. Even within established tumors immunoregulatory cytokines, such as TGF-β and IL-10, play potentially opposing roles in both promoting and discouraging cancer cell survival and a more thorough understanding of their effect in relation to the plethora of other cytokines and immune cells within the tumor microenvironment is needed. Th17 cells may play a key role in providing a constant proinflammatory environment within established tumors while also influencing cell fate decisions and activation. Chronic infection and inflammation could provide the priming to encourage initial carcinogenesis, and, whilst establishing, TAM and MDSC cells may heavily encourage Th17 cell development and proliferation through the production of TGF-β and IL-6 cytokines. A proposed model outlining how this cycle of inflammation may perpetuate and lead to cancer development and survival can be found in Figure 1.
A proposed model of chronic inflammation progressing to tumor formation. A chronic insult such as autoimmunity or infection leads to a steady inflamed tissue state. Tumor-associated macrophages (TAM) of the M1 variety begin to produce cytokines that promote Th17 cell differentiation from naïve CD4+ T lymphocytes such as IL-6, IL-23, IL-1β and TNF-α. One of the key cytokines produced by M2 TAM is TGF-β, which may further encourage Th17 development. Myeloid-derived suppressor cells serve an immunosuppressive role within the microenvironment by producing TGF-β as well as other regulatory factors, but may also encourage further Th17 differentiation by secreting IL-6. The production of TGF-β would likely also have the effect of inducing CD4+Foxp3+Tregs within the tumor microenvironment while also suppressing inflammatory cytokine production from the TAM themselves. However, these combined suppressive signals may not be sufficient to overcome the self-perpetuating cycle of activation established between Th17 cells and TAM. At the same time, this suppressive environment may greatly hamper, or even eliminate, the possibility for Th1 and CD8+ T cells to mount an immune response. The net effect of these cellular interactions within the tumor microenvironment is to limit anti-tumor immunity from Th1 and CD8 T cells while continuing to encourage chronic inflammation, angiogenesis, and overall cancer cell survival and proliferation.
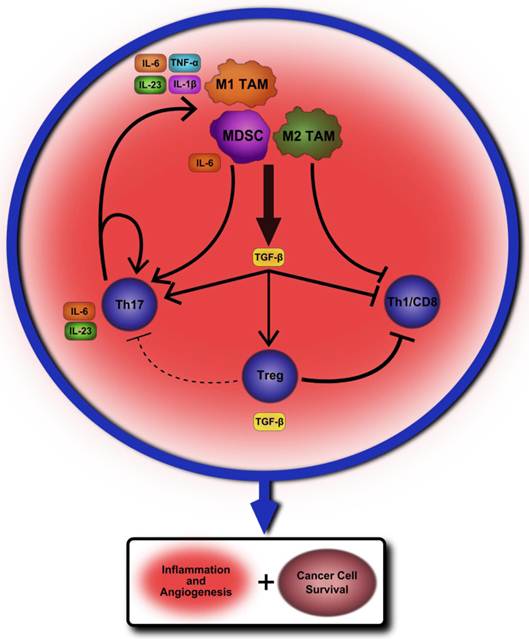
Once initially established, Th17 cells may then create a chronic environment of inflammatory factors that further activates TAM and self-propagates, encouraging angiogenesis and cancer cell survival through STAT3 activation and anti-apoptotic protein production. The immunosuppressive effects of Tregs within the tumor microenvironment may prevent initiation of antitumor immune responses by IFN-γ producing CD4+ Th1 cells and CD8+ T cells, but may not be sufficient to overcome the already established cycle of IL-6 and STAT3-mediated inflammation. There remains much to be learned of the interactions between these various cell types within the tumor microenvironment. However, teasing apart and understanding these mechanisms is essential to finding an effective therapeutic approach for altering pro-tumor immune signaling while also limiting potential side-effects for the patient.
Acknowledgements
We thank Drs. Joanne Konkel and Takashi Maruyama, OIIB, NIDCR, NIH for critically reading this manuscript. This research was supported by the Intramural Research Program of the NIH, NIDCR.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883-99
2. Raza SA, Clifford GM, Franceschi S. Worldwide variation in the relative importance of hepatitis B and hepatitis C viruses in hepatocellular carcinoma: a systematic review. Br J Cancer. 2007;96(7):1127-34
3. Bui JD, Schreiber RD. Cancer immunosurveillance, immunoediting and inflammation: independent or interdependent processes? Curr Opin Immunol. 2007;19(2):203-8
4. Dunn GP, Ikeda H, Bruce AT, Koebel C, Uppaluri R, Bui J, Chan R, Diamond M, White JM, Sheehan KC, Schreiber RD. Interferon-gamma and cancer immunoediting. Immunologic research. 2005;32(1-3):231-45
5. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092):431-6
6. Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124(4):823-35
7. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140(2):197-208
8. de Martel C, Franceschi S. Infections and cancer: established associations and new hypotheses. Crit Rev Oncol Hematol. 2009;70(3):183-94
9. Coussens LM, Werb Z. Inflammatory cells and cancer: think different!. J Exp Med. 2001;193(6):F23-6
10. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860-7
11. Giardiello FM, Hamilton SR, Krush AJ, Piantadosi S, Hylind LM, Celano P, Booker SV, Robinson CR, Offerhaus GJA. Treatment of Colonic and Rectal Adenomas with Sulindac in Familial Adenomatous Polyposis. New Eng J Med. 1993;328:1313-6
12. Labayle D, Fischer D, Vielh P, Drouhin F, Pariente A, Bories C, Duhamel O, Trousset M, Attali P. Sulindac causes regression of rectal polyps in familial adenomatous polyposis. Gastroenterology. 1991;101:5
13. Phillips RKS, Wallace MH, Lynch PM, Hawk E, Gordon GB, Saunders BP, Wakabayashi N, Shen Y, Zimmerman S, Godio L, Rodrigues-Bigas M, Su LK, Sherman J, Kelloff G, Levin B, Steinbach G. A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut. 2002;50:857-60
14. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889-96
15. Movahedi K, Laoui D, Gysemans C, Baeten M, Stangà G, Van den Bossche J, Mack M, Pipeleers D, In't Veld P, De Baetselier P, Van Ginderachter JA. Different Tumor Microenvironments Contain Functionally Distinct Subsets of Macrophages Derived from Ly6C(high) Monocytes. Cancer Res. 2010;70:5728-39
16. Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, Hussell T, Feldmann M, Udalova IA. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. 2011;12(3):231-8
17. Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells in human cancer. Cancer journal. 2010;16(4):348-53
18. Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, Carbone DP, Matrisian LM, Richmond A, Lin PC, Moses HL. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13(1):23-35
19. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162-74
20. Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends in immunology. 2011;32(1):19-25
21. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798-809
22. Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell. 2010;140(6):845-58
23. Grivennikov S, Karin M. Autocrine IL-6 signaling: a key event in tumorigenesis? Cancer Cell. 2008;13(1):7-9
24. Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. The Journal of clinical investigation. 2007;117(5):1175-83
25. Iliopoulos D, Hirsch HA, Struhl K. An Epigenetic Switch Involving NF-κB, Lin28, Let-7 MicroRNA, and IL6 Links Inflammation to Cell Transformation. Cell. 2009;139(4):693-706
26. Thompson ED, Enriquez HL, Fu Y-X, Engelhard VH. Tumor masses support naive T cell infiltration, activation, and differentiation into effectors. J Exp Med. 2010;207:1791-804
27. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198(12):1875-86
28. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942-9
29. Wei S, Kryczek I, Zou W. Regulatory T-cell compartmentalization and trafficking. Blood. 2006;108(2):426-31
30. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20-1
31. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko S-A, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27(1):68-73
32. Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8(2):191-7
33. Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 1999;59(13):3128-33
34. Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, Rubin SC, Kaiser LR, June CH. Regulatory CD4+CD25+ T Cells in Tumors from Patients with Early-Stage Non-Small Cell Lung Cancer and Late-Stage Ovarian Cancer. Cancer Res. 2001;61:4766-72
35. Somasundaram R, Jacob L, Swoboda R, Caputo L, Song H, Basak S, Monos D, Peritt D, Marincola F, Cai D, Birebent B, Bloome E, Kim J, Berencsi K, Mastrangelo M, Herlyn D. Inhibition of Cytolytic T Lymphocyte Proliferation by Autologous CD4+/CD25+ Regulatory T Cells in a Colorectal Carcinoma Patient Is Mediated by Transforming Growth Factor-β. Cancer Res. 2002;62:5267-72
36. Fu J, Xu D, Liu Z, Shi M, Zhao P, Fu B, Zhang Z, Yang H, Zhang H, Zhou C, Yao J, Jin L, Wang H, Yang Y, Fu YX, Wang FS. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology. 2007;132(7):2328-39
37. Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T, Drake C, Pardoll D, Yu H. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell. 2009;15(2):114-23
38. Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, Basham B, McClanahan T, Kastelein RA, Oft M. IL-23 promotes tumour incidence and growth. Nature. 2006;442(7101):461-5
39. Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6--Stat3 signaling pathway. JEM. 2009;206:1457-64
40. Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, Hwu P, Restifo NP, Overwijk WW, Dong C. T Helper 17 Cells Promote Cytotoxic T Cell Activation in Tumor Immunity. Immunity. 2009;31(5):787-98
41. Massague J. TGFbeta in Cancer. Cell. 2008;134(2):215-30
42. Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103(2):295-309
43. Chen C-J, Yang H-I, Su J, Jen C-L, You S-L, Lu S-N, Huang G-T, Iloeje UH, for the R-HBVSG. Risk of Hepatocellular Carcinoma Across a Biological Gradient of Serum Hepatitis B Virus DNA Level. JAMA. 2006;295:65-73
44. Erdman SE, Poutahidis T, Tomczak M, Rogers AB, Cormier K, Plank B, Horwitz BH, Fox JG. CD4+ CD25+ Regulatory T Lymphocytes Inhibit Microbially Induced Colon Cancer in Rag2-Deficient Mice. Am J Pathol. 2003;162:691-702
45. Erdman SE, Rao VP, Poutahidis T, Ihrig MM, Ge Z, Feng Y, Tomczak M, Rogers AB, Horwitz BH, Fox JG. CD4+CD25+ Regulatory Lymphocytes Require Interleukin 10 to Interrupt Colon Carcinogenesis in Mice. Cancer Res. 2003;63:6042-50
46. Maloy KJ, Salaun L, Cahill R, Dougan G, Saunders NJ, Powrie F. CD4+CD25+ TR Cells Suppress Innate Immune Pathology Through Cytokine-dependent Mechanisms. J Exp Med. 2003;197(1):9
47. Sredni B, Weil M, Khomenok G, Lebenthal I, Teitz S, Mardor Y, Ram Z, Orenstein A, Kershenovich A, Michowiz S, Cohen YI, Rappaport ZH, Freidkin I, Albeck M, Longo DL, Kalechman Y. Ammonium Trichloro(dioxoethylene-o,o')tellurate (AS101) Sensitizes Tumors to Chemotherapy by Inhibiting the Tumor Interleukin 10 Autocrine Loop. Cancer Research. 2004;64(5):1843-52
Author contact
![]() Corresponding author: WanJun Chen, Tel: (301) 435-7168; Fax: (301) 402-1064; E-mail: wchennidcr.nih.gov
Corresponding author: WanJun Chen, Tel: (301) 435-7168; Fax: (301) 402-1064; E-mail: wchennidcr.nih.gov