Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Clinical evidence of SRC-3...
Mouse model reveals the...
SRC-3 and growth factor signaling
Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(5):664-672. doi:10.7150/ijbs.7.664 This issue Cite
Review
SRC-3 Has a Role in Cancer Other Than as a Nuclear Receptor Coactivator
Gang Ma, Yu Ren, Ke Wang, Jianjun He ![]()
Department of Surgical Oncology, the First Affiliated Hospital, Medical School, Xi'an Jiaotong University, Xi'an, Shaanxi Province, 710061, P. R. China
Received 2011-1-24; Accepted 2011-5-1; Published 2011-5-24
Abstract
Steroid receptor coactivator-3 (SRC-3), also known as AIB1, is a member of the p160 steroid receptor coactivator family. Since SRC-3 was found to be amplified in breast cancer in 1997, the role of SRC-3 in cancer has been broadly investigated. SRC-3 initially was identified as a transcriptional coactivator for nuclear receptors such as the estrogen receptor (ER), involved in the proliferation of hormone-dependent cancers. However, increasing clinical evidence shows that dysregulation of SRC-3 expression in several human hormone-independent cancers is correlated with pathological factors and clinical prognosis. Recently, both in vivo and in vitro studies demonstrate that SRC-3 may influence a number of cancer cellular processes in several ways independent of nuclear receptor signaling. In addition, an SRC-3 transgenic mice model shows that SRC-3 induces tumors in several mouse tissues. These results indicate that the role of SRC-3 in cancer is not just as a nuclear receptor coactivator. The focus of this review is to examine possible SRC-3 roles in cancer, other than as a nuclear receptor coactivator.
Keywords: Steroid receptor coactivator-3, mice model, tumorigenesis, cell cycle, apoptosis, invasion and metastasis
Introduction
Steroid receptor coactivator-3 (SRC-3) is a member of the p160 steroid receptor coactivator family. It was discovered initially to be amplified in breast cancer in 1997 and so was named amplified in breast cancer 1 (AIB1) [1]. SRC-3 is also known as nuclear receptor coactivator-3 (NCoA-3), receptor associated coactivator-3 (RAC3), activator of thyroid hormone and retinoid receptor (ACTR), thyroid hormone receptor activating molecule-1 (TRAM1), and p300/CBP interacting protein (p/CIP). The SRC-3 gene is located on chromosome 20q12-12. The SRC-3 protein is approximately 160 kDa and contains three basic structural domains, consistent with the other two SRC family members (SRC-1 and SRC-2). The N-terminal basic helix-loop-helix-Per/ARNT/Sim domain (bHLH-PAS) mediates the interaction between SRC-3 and other DNA-binding proteins. The receptor-interaction domain (RID) contains three LXXLL motifs, by which SRC-3 binds to the ligand-activated nuclear receptors. The C-terminal domain contains two intrinsic transcriptional activation domains, AD1 and AD2, by which SRC-3 interacts with histone acetyltransferases and histone methyltransferases, respectively. The C-terminal domain of SRC-3 also has histone acetyltransferase activity. The function of SRC-3 as a transcriptional coactivator for nuclear hormone receptors has been described in detail previously [2, 3]. Phosphorylated SRC-3 binds to ligand-activated nuclear receptors via its RID, and then recruits the histone acetyltransferase p300/CBP through AD1. This transcriptional complex subsequently alters chromatin structure and facilitates the transcription of the nuclear receptor target gene. In addition to its role as a nuclear receptor coactivator, SRC-3 is also a transcriptional coactivator for other transcription factors, including E2F1 [4], AP-1 [5], NF-κB [6], STAT6 [7] and PEA3 [8]. Moreover, SRC-3 acts as a translational repressor to regulate proinflammatory cytokine mRNA translation [9].
The correlation between SRC-3 and cancer has been widely discussed since SRC-3 was determined to be amplified in breast cancer in 1997 [1]. Initially, SRC-3 was thought to be involved in the proliferation of hormone-dependent cancers, such as ER-positive breast cancer, primarily by acting as transcriptional coactivator for nuclear receptors such as ER. However, more and more studies have shown that hormone-dependent cancers are not the only type of cancer that SRC-3 affects, and SRC-3 has roles in cancer other than being as a nuclear receptor coactivator. Dysregulation of SRC-3 in several hormone-independent cancers correlates with pathological factors and clinical prognosis. Both in vivo and in vitro studies show that SRC-3 affects many aspects of cancer in ways independent of nuclear receptor signaling. The SRC-3 transgenic mice model shows that SRC-3 is a bona fide oncogene which can induce tumorigenesis in many tissues. The focus of this review is to summarize roles of SRC-3 in cancer other than a nuclear receptor coactivator.
Clinical evidence of SRC-3 dysregulation in cancers
The altered expression of SRC-3 in human cancers is summarized in Table 1. These data demonstrate that dysregulation of SRC-3 occurs in hormone-dependent as well as hormone-independent cancers. Moreover, SRC-3 dysregulation is correlated with pathological factors and prognosis of hormone-dependent as well as hormone-independent cancers. These observations indicate that SRC-3 has a key role in the tumorigenesis and progression of several different cancers, and not just a coactivator of nuclear hormone receptor.
SRC-3 dysregulation in cancers and malignant disease
| Changes | Frequency (Refs) | Pathological and Prognostic association (Refs) |
|---|---|---|
| Hormone-dependent cancers | ||
| Breast cancer | ||
| Gene amplification | 1.6-9.5% [1, 9-12] | Large tumor size [9] |
| mRNA overexpression | 13-64% [1, 13, 14] | High tumor grade [13] Shorter DFS and OS [14] |
| Protein overexpression | 16-53% [11, 15-19] | High tumor grade [16, 17], shorter DFS [18], shorter OS [18, 20], longer DFS [19] |
| Prostate cancer | ||
| Protein overexpression | 13.1-37.9% [21, 22] | High tumor grade, stage and shorter DSS [21], PSA recurrence [22] |
| Ovarian cancer | ||
| Gene amplification | 7.4-25.8% [9, 23] | Shorter DFS [23] |
| Protein overexpression | 64% of the high grade tumors [24] | High tumor grade [24] |
| Endometrium cancer | ||
| mRNA overexpression | 17% [10] | High tumor grade and stage, shorter 24-months survival [25] |
| Protein expression | 93% [26] | High tumor grade [26] |
| Hormone-independent cancers and malignant disease | ||
| Esophageal squamous cell cancer (ESCC) | ||
| Gene amplification | 4.9-13% [27, 28] | ND |
| Protein overexpression | 46-64.3% [28, 29] | High tumor grade [28] Shorter PFS and DSS [29] |
| Colorectal cancer | ||
| Gene amplification | 10-32% [30, 31] | ND |
| Protein overexpression | 35-56.5% [30, 32] | High tumor grade [30] Longer OS [32] |
| Hepatocellular cancer (HCC) | ||
| Gene amplification | 41% of the metastatic HCC; 60% of the recurrent HCC [33] | ND |
| Protein overexpression | 67.6% [34] | ND |
| Pancreatic cancer | ||
| Gene amplification | 37% [35] | ND |
| mRNA overexpression | 73.68% [35] | ND |
| Protein overexpression | 64.47% [35] | ND |
| Gastric cancer | ||
| Gene amplification | 41% [36] (7% high level amplification; 34% low level amplification) | High regional lymph metastasis, high tumor grade and stage, poor prognosis [36] |
| mRNA overexpression | 40% [36] | ND |
| Bladder cancer | ||
| Gene amplification | 7.0% [37] | ND |
| Protein overexpression | 32.5% [37] | Shorter survival [37] |
| Nasopharyngeal cancer | ||
| Gene amplification | 7% [38] | ND |
| Protein overexpression | 51% [38] | High lymph metastasis and tumor state [38] |
| Non-small cell lung cancer (NSCLC) | ||
| Gene amplification | 8.2-25% [39, 40] | ND |
| Protein overexpression | 27-48.3% [39-41] | Shorter survival [39] Shorter DFS and OS [40] |
| Meningioma | ||
| Protein expression | 76% [42] | ND |
DFS, disease free survival; OS, overall survival; DSS, disease specific survival; PFS, progression free survival; PSA, prostatic specific antigen; ND, not determined; HCC, hepatocellular cancer; ESCC, esophageal squamous cell cancer; NSCLC, non-small cell lung cancer.
Mouse model reveals the oncogenic property of SRC-3
SRC-3 knockout mice model
To analyze the relationship between SRC-3 and cancer, researchers generated several mouse models of cancers in which SRC-3 is depleted. The results showed that SRC-3 knockout significantly inhibited carcinogen-induced tumorigenesis by inducing down-regulation of other signaling pathways, as well as nuclear receptor signaling. For example, in two different mouse models, SRC-3 knockout totally inhibited the mouse mammary tumor virus-v-Ha-ras (MMTV-v-Ha-ras) or chemical carcinogen 7, 12-dimethyl-benz[a] anthracene (DMBA)-mediated mammary gland tumorigenesis [44, 45]. The down-regulation of the IGF-I signaling detected in both of these models is considered one reason, as is the down-regulation of ER signaling. Moreover, SRC-3 depletion led to the inhibition of the oncogene MMTV-HER2/neu induced mammary gland tumorigenesis in another mouse model, in which HER-2 phosphorylation was suppressed [46]. More recently, it was reported that SRC-3 knockout also prevented the initiation of thyroid cancer induced by dominant-negative mutant thyroid hormone receptor-β (TR-β), and multiple signaling pathways, but not nuclear receptor signaling, were disrupted in this mouse model [47].Taken together, these results indicate that SRC-3 contributes to the tumorigenesis induced by several different carcinogens.
SRC-3 transgenic mice model
To detect whether SRC-3 overexpression could induce tumorigenesis in the mammary gland, Torres-Arzayus and colleagues [48] established the first SRC-3 transgenic mouse model in 2004. In this model, tumors were induced in the mammary gland (48%). Surprisingly, tumors were also found in the pituitary (42%), uterus (18%) and lung (18%). Moreover, 16.7% of the mammary gland tumors were ERα-negative. Further studies revealed that activation of IGF-I signaling was enhanced in this SRC-3-tg mouse model, indicating that SRC-3 induced tumorigenesis by up-regulating IGF-I signaling. More recently, the same group developed another estrogen receptor signaling-eliminated SRC-3 transgenic mouse model by two methods, ovariectomy (ovx) and ERα null mutant [49]. In both groups, ectopic SRC-3 overexpression still induced tumorigenesis in lung, pituitary, skin, and bone tissues. Meanwhile, in the ovx group, although the ovariectomy attenuated estrogen receptor signaling and inhibited mammary gland development, some mice still developed mammary hyperplasia and ductal carcinoma. Taken together, the data from these two SRC-3 transgenic mouse models strongly indicate that SRC-3 triggers tumorigenesis through either other nuclear receptor signals or in a nuclear receptor signaling-independent manner, although the details of the mechanism are not fully understood.
SRC-3 is involved in cancers in a nuclear receptor signaling-independent manner
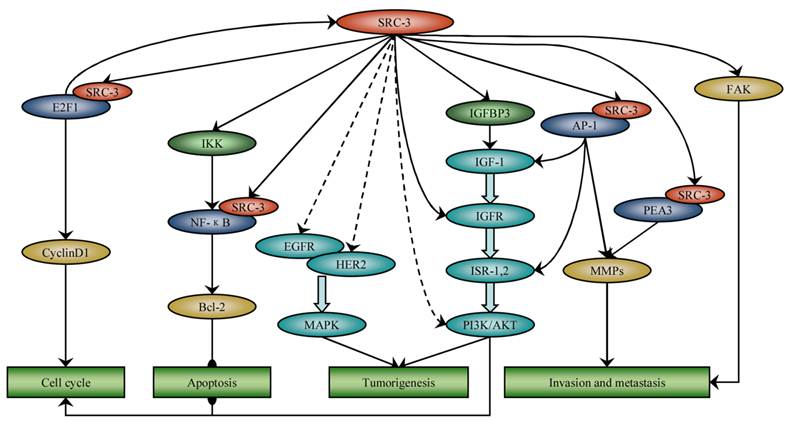
Several investigations indicated that SRC-3 is implicated in cancer through nuclear receptor signaling-independent as well as nuclear receptor signaling-dependent mechanisms. Here, we intend to focus on the role of SRC-3 in cancer other than as a nuclear receptor coactivator (Figure 1).
SRC-3 is involved in many cancer processes independent of nuclear receptor signaling. SRC-3 promotes cell cycle progression by coactivating E2F1 and activating Akt. SRC-3 up-regulates IGF-I and EGF signaling by phosphorylating Akt, EGFR and HER2, respectively, and induces tumorigenesis. SRC-3 inhibits apoptosis through activation of NF-ΚB and Akt signaling. SRC-3 promotes invasion and metastasis by coactivating AP-1 and PEA3 and activating FAK.
SRC-3 is involved in cell cycle control
Cancer is considered to be a disease of the cell cycle, and compromised cell cycle control is detected frequently in several cancers [50]. There is evidence that SRC-3 may promote the cancer cell cycle. For example, SRC-3 overexpression maintained the S phase cell number in fulvestrant-treated breast cancer cell line T47D [4]. In contrast, the SRC-3 stable knockdown by siRNA decreased S phase cell number and increased G1 phase cell number both in human embryonic lung fibroblast and HCC cell lines [35, 51]. Moreover, SRC-3 depletion induced a significant increase in G1/G0 phase cells and a decrease in G2/M phase cells in TR-β-induced thyroid cancer mouse model [47]. Another proof was that SRC-3 nuclear translocation was coincident with the G1/S phase transition of cancer cell [51, 52].
Other research examined the mechanism by which SRC-3 modulates cell cycle control in cancer cells. One group found that SRC-3 acted as a transcriptional coactivator for the G1/S phase transition-associated transcription factor E2F1, and enhance the transcription of the E2F1 target genes, which are mostly G1/S cycle transition-related proteins [4, 51]. Therefore, SRC-3 dysregulation eventually led to cell cycle activation through E2F1 signaling. This conclusion is supported by the fact that SRC-3 overexpression induced proliferation and transformation of the ER-negative human mammary epithelial cell line MCF10A in an E2F1-dependent manner [51]. Interestingly, other studies show that SRC-3 may promote its own transcription with E2F1 [53, 54]. This positive feedback regulatory loop may enhance the influence SRC-3 exerted on cell cycle control. In addition, SRC-3 also influenced cell cycle control through activation of the Akt signaling pathway. For example, SRC-3 inhibited cell cycle inhibitor p21Cip1/Waf1 expression by activating Akt signaling in HCC cells [35]. Taken together, these data demonstrate that SRC-3 is involved in cell cycle control-related signaling. The dysregulation of SRC-3 may disturb normal cell cycle progression and promote cancer initiation and proliferation.
SRC-3 and growth factor signaling
SRC-3 regulates the insulin-like growth factor I (IGF-I) signaling
IGF-I signaling is involved in many important cell processes related to cancer [55]. Many studies showed that SRC-3 is associated with the regulation of IGF-I signaling in cancer. In vivo, SRC-3 knockout down-regulated IGF-I mRNA and protein expression, and thus inhibited tumorigenesis [44, 56, 57]. In contrast, ectopic SRC-3 overexpression up-regulated IGF-I mRNA and protein expression and induced tumorigenesis in a mouse model [48]. Moreover, SRC-3 expression also influenced the expression of many IGF-I signaling components, such as IGF-I receptor β (IGF-IRβ) [45], IRS-1 and IRS-2 [44, 45]. In accordance with in vivo studies, the in vitro studies also demonstrated that SRC-3 could regulate the expression of IGF-I signaling components [58, 59]. In addition, SRC-3 modulated the activation of IGF-I signaling components in vitro and in vivo. For example, SRC-3 overexpression enhanced Akt phosphorylation in two prostate cancer cell lines, PC3 and LNCaP [58]. The cancer cells derived from the SRC-3 transgenic mice showed excessive activation of components of the IGF-I/PI3K/Akt/mTOR pathway, in particular Akt [48]. Both in vivo and in vitro studies indicate that SRC-3 is tightly correlated with IGF-I signaling in cancers.
Quite a few studies have been performed on the mechanism of SRC-3 regulating the IGF-I signaling in cancers. SRC-3 can bind to the transcription factor AP-1 and enhance AP-1-mediated transcription of IGF-I and IRS-1 [5]. IGF-binding protein 3 (IGFBP3) inhibited the degradation of serum IGF-I by forming a complex with IGF-I [60]. A recent study showed that SRC-3 could affect the IGF protein level through regulation of IGFBP3 expression [61]. In this study, mutation of the phosphorylation sites of SRC-3 increased IGF-I expression by up-regulating IGFBP3, and resulted in tumorigenesis in the mouse liver. These results can account partly for the association between SRC-3 and IGF-I signaling. However, the mechanism by which SRC-3 modulates the expression and activation of IGF-I signaling components, in particular the activation of Akt, needs further exploration. Collectively, the above results indicate that dysregulation of SRC-3 influences the expression and activation of IGF-I signaling components.
SRC-3 regulates EGF signaling
EGF signaling is important for the initiation and progression of cancer [62]. Clinical studies showed that SRC-3 overexpression correlated with expression of the EGFR family member HER2 in breast cancer specimens, and the expression of both of these proteins correlated with shorter survival and primary tamoxifen resistance in breast cancer patients [13, 20]. A recent in vitro study showed that endogenous SRC-3 knockdown by siRNA in lung, pancreatic and breast cancer cell lines reduced EGF-mediated phosphorylation of EGFR and HER-2, and thereby inhibited the activation of EGF signaling [63]. As a result, cancer cell proliferation was inhibited. An in vivo study showed that SRC-3 depletion completely inhibited mammary tumorigenesis induced by the oncogene MMTV-HER2/neu [46]. Further studies revealed that SRC-3 depletion strongly prevented HER-2 phosphorylation and thus inhibited the HER2-induced activation of EGF/MAPK signaling. Although neither of these studies thoroughly elucidated the mechanism by which SRC-3 regulate EGFR and HER2 phosphorylation, they still confirm that SRC-3 regulates EGF signaling.
SRC-3 and apoptosis
Apoptosis is a normal physiological process of the cell; altered regulation of apoptosis is common in cancer [64]. Many studies showed that SRC-3 is implicated in the cancer cell apoptosis. In vitro, overexpression of SRC-3 reduced H2O2-mediated apoptosis in the HEK293 cell line [65]. In contrast, SRC-3 down-regulation by siRNA restored the sensitivity of chronic myeloid leukemia cell line K562 to tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis [66], decreased apoptosis inhibitor Bcl-2 expression in the prostate cancer cell line PC3 [23], and increased the level of cleaved caspase-7 in the NSCLC cell line H1299 [23, 41]. Moreover, in vivo studies also showed that SRC-3 possessed anti-apoptotic properties [47, 48].
NF-κB signaling inhibits apoptosis by regulating the transcription of many anti-apoptotic proteins, such as Bcl-2 and the FLICE inhibitory protein (FLIP) [67, 68]. SRC-3 activated NF-κB signaling in coordination with IκB kinase (IKK) and acted as a transcriptional coactivator for NF-κB in cancer cells [6, 69]. Therefore, researchers examined whether SRC-3 affected apoptosis through NF-κB signaling. SRC-3 knockdown by siRNA indeed reduced activation of NF-κB signaling and resulted in apoptosis in human chronic myeloid leukemia K562 cells [66], while another study showed that SRC-3 overexpression activated NF-κB-mediated anti-apoptosis gene transcription in HEK293 cells [65]. More recently, another study showed that SRC-3 down-regulation induced by deguelin in Jurkat cells inhibited the expression of NF-κB target genes known to be anti-apoptotic [70]. All of these results demonstrate that SRC-3 may decrease apoptosis by activating NF-κB signaling in cancer cells. In addition, SRC-3 also disturbed apoptosis through Akt signaling, which is involved in multiple apoptosis signaling pathways [71-73]. An in vitro study showed that SRC-3 knockout reduced Akt activation, and thus attenuated Akt-mediated JNK suppression, resulting in apoptosis of DT40 chicken B-lymphocytes [74]. Another study revealed that inhibiting SRC-3 expression by gambogic acid in the human leukemia cell line K562 also inactivated Akt signaling, thereby decreasing Bcl-2 expression [75]. In contrast, SRC-3 overexpression in HEK293 cells prevented apoptosis in part by activating Akt signaling [65]. These data demonstrate that SRC-3 suppression of apoptosis in several cancer cells is through activation of NF-κB and Akt signaling.
SRC-3 promotes invasion and metastasis
Invasion and metastasis play critical roles in cancer progression [76]. Several studies indicated that SRC-3 promoted cancer invasion and metastasis. Clinically, SRC-3 overexpression was associated with local invasion [25, 26, 34, 77] and lymph node metastasis [30, 37, 39, 77]. In vitro, SRC-3 modulated cell invasion and migration in a nuclear receptor signaling-independent manner [25, 77, 78]. In an SRC-3-tg mouse model, most cancers induced by ectopic SRC-3 overexpression were invasive and metastatic [48, 49]. In contrast, the SRC-3 knockout mouse model showed that loss of SRC-3 inhibited cancer invasion and metastasis [8, 44, 47, 79].
There have been many studies on the mechanism by which SRC-3 regulates cancer invasion and metastasis. Some studies focused on the correlation between SRC-3 and matrix metalloproteinases (MMPs). One study found that SRC-3 stimulated MMP7 and MMP10 expression in ER-negative MDA-MB-231 breast cancer cells by acting as coactivator for transcription factor AP-1 [78]. Another study found that SRC-3 promoted MMP2 and MMP13 expression in LNCaP prostate cancer cells by coactivating PEA3, an Ets transcription factor family member, as well as AP-1 [77]. In accordance with the in vitro studies, an in vivo study also demonstrated that SRC-3 enhanced MMP2 and MMP9 expression by functioning as a transcriptional coactivator for PEA3 [8]. In addition to regulating the expression of MMPs, SRC-3 also was shown to activate focal adhesion kinase (FAK) signaling, and thus enhance cell motility, which is critical to the cancer invasion and metastasis. A study showed that SRC-3 promoted FAK phosphorylation and thus induced tumor cell migration [77]. More recently, another study showed that SRC-3Δ4, a nuclear localization signal (NLS)-deleted isoform of SRC-3, could function as an adaptor protein for signal transmission from EGFR to FAK [80]. Taken together, the above studies demonstrated that SRC-3 promotes cancer invasion and metastasis by mechanisms other than nuclear receptor signaling.
Conclusion
Accumulating evidence indicates that the role of SRC-3 in cancer is more than as a nuclear receptor coactivator. Clinical investigations showed that SRC-3 is not only associated with hormone-dependent cancers. In the SRC-3-tg mouse model, SRC-3 ectopic overexpression induced tumorigenesis in several organs, even when a prototypical nuclear hormone receptor signaling is eliminated. In contrast, SRC-3 knockout in mice inhibited tumorigenesis induced by multiple different carcinogens. Both in vitro and in vivo studies demonstrated that SRC-3 is involved in many cancer processes through several mechanisms, in particular the nuclear receptor-independent signaling. SRC-3 controlled cell cycle progression by coactivating E2F1, up-regulated IGF-I and EGF signaling, inhibited apoptosis through activation of NF-κB and Akt signaling and promoted invasion and metastasis by coactivating AP-1 and PEA3 and activating FAK.
A recent study showed that SRC-3 deficiency reduced apoptosis by up-regulating NF-κB signaling in T and B lymphocytes, and eventually lead to lymphoma [81]. This indicated that the mechanisms independent of nuclear receptor signaling by which SRC-3 is involved in cancers is complex and requires further investigation in the future. All of the above data demonstrate that SRC-3 has roles in cancer initiation and progression other than being as a nuclear receptor coactivator. Therefore, SRC-3 may be an attractive target of cancer therapy.
Acknowledgements
This work was supported by the Shaanxi Province Natural Science Foundation grant 2007C247 and Shaanxi Province Science and Technology Research and Development grant 2009K12-01.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Anzick SL, Kononen J, Walker RL. et al. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965-8
2. Liao L, Kuang SQ, Yuan Y, Gonzalez SM, O'Malley BW, Xu J. Molecular structure and biological function of the cancer-amplified nuclear receptor coactivator SRC-3/AIB1. J Steroid Biochem Mol Biol. 2002;83:3-14
3. Amazit L, Pasini L, Szafran AT. et al. Regulation of SRC-3 intercompartmental dynamics by estrogen receptor and phosphorylation. Mol Cell Biol. 2007;27:6913-32
4. Louie MC, Zou JX, Rabinovich A, Chen HW. ACTR/AIB1 functions as an E2F1 coactivator to promote breast cancer cell proliferation and antiestrogen resistance. Mol Cell Biol. 2004;24:5157-71
5. Yan J, Yu CT, Ozen M, Ittmann M, Tsai SY, Tsai MJ. Steroid receptor coactivator-3 and activator protein-1 coordinately regulate the transcription of components of the insulin-like growth factor/AKT signaling pathway. Cancer Res. 2006;66:11039-46
6. Werbajh S, Nojek I, Lanz R, Costas MA. RAC-3 is a NF-kappa B coactivator. FEBS Lett. 2000;485:195-9
7. Arimura A, vn PM, Schroder AJ, Rothman PB. The transcriptional co-activator p/CIP (NCoA-3) is up-regulated by STAT6 and serves as a positive regulator of transcriptional activation by STAT6. J Biol Chem. 2004;279:31105-12
8. Qin L, Liao L, Redmond A. et al. The AIB1 oncogene promotes breast cancer metastasis by activation of PEA3-mediated matrix metalloproteinase 2 (MMP2) and MMP9 expression. Mol Cell Biol. 2008;28:5937-50
9. Yu C, York B, Wang S, Feng Q, Xu J, O'Malley BW. An essential function of the SRC-3 coactivator in suppression of cytokine mRNA translation and inflammatory response. Mol Cell. 2007;25:765-78
10. Bautista S, Valles H, Walker RL. et al. In breast cancer, amplification of the steroid receptor coactivator gene AIB1 is correlated with estrogen and progesterone receptor positivity. Clin Cancer Res. 1998;4:2925-9
11. Glaeser M, Floetotto T, Hanstein B, Beckmann MW, Niederacher D. Gene amplification and expression of the steroid receptor coactivator SRC3 (AIB1) in sporadic breast and endometrial carcinomas. Horm Metab Res. 2001;33:121-6
12. Iwase H, Omoto Y, Toyama T. et al. Clinical significance of AIB1 expression in human breast cancer. Breast Cancer Res Treat. 2003;80:339-45
13. Kirkegaard T, McGlynn LM, Campbell FM. et al. Amplified in breast cancer 1 in human epidermal growth factor receptor - positive tumors of tamoxifen-treated breast cancer patients. Clin Cancer Res. 2007;13:1405-11
14. Bouras T, Southey MC, Venter DJ. Overexpression of the steroid receptor coactivator AIB1 in breast cancer correlates with the absence of estrogen and progesterone receptors and positivity for p53 and HER2/neu. Cancer Res. 2001;61:903-7
15. Zhao C, Yasui K, Lee CJ. et al. Elevated expression levels of NCOA3, TOP1, and TFAP2C in breast tumors as predictors of poor prognosis. Cancer. 2003;98:18-23
16. List HJ, Reiter R, Singh B, Wellstein A, Riegel AT. Expression of the nuclear coactivator AIB1 in normal and malignant breast tissue. Breast Cancer Res Treat. 2001;68:21-8
17. Hudelist G, Czerwenka K, Kubista E, Marton E, Pischinger K, Singer CF. Expression of sex steroid receptors and their co-factors in normal and malignant breast tissue: AIB1 is a carcinoma-specific co-activator. Breast Cancer Res Treat. 2003;78:193-204
18. Thorat MA, Turbin D, Morimiya A. et al. Amplified in breast cancer 1 expression in breast cancer. Histopathology. 2008;53:634-41
19. Alkner S, Bendahl PO, Grabau D. et al. AIB1 is a predictive factor for tamoxifen response in premenopausal women. Ann Oncol. 2010;21:238-44
20. Osborne CK, Bardou V, Hopp TA. et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst. 2003;95:353-61
21. Harigopal M, Heymann J, Ghosh S, Anagnostou V, Camp RL, Rimm DL. Estrogen receptor co-activator (AIB1) protein expression by automated quantitative analysis (AQUA) in a breast cancer tissue microarray and association with patient outcome. Breast Cancer Res Treat. 2009;115:77-85
22. Gnanapragasam VJ, Leung HY, Pulimood AS, Neal DE, Robson CN. Expression of RAC 3, a steroid hormone receptor co-activator in prostate cancer. Br J Cancer. 2001;85:1928-36
23. Zhou HJ, Yan J, Luo W. et al. SRC-3 is required for prostate cancer cell proliferation and survival. Cancer Res. 2005;65:7976-83
24. Tanner MM, Grenman S, Koul A. et al. Frequent amplification of chromosomal region 20q12-q13 in ovarian cancer. Clin Cancer Res. 2000;6:1833-9
25. Yoshida H, Liu J, Samuel S, Cheng W, Rosen D, Naora H. Steroid receptor coactivator-3, a homolog of Taiman that controls cell migration in the Drosophila ovary, regulates migration of human ovarian cancer cells. Mol Cell Endocrinol. 2005;245:77-85
26. Sakaguchi H, Fujimoto J, Sun WS, Tamaya T. Clinical implications of steroid receptor coactivator (SRC)-3 in uterine endometrial cancers. J Steroid Biochem Mol Biol. 2007;104:237-40
27. Balmer NN, Richer JK, Spoelstra NS, Torkko KC, Lyle PL, Singh M. Steroid receptor coactivator AIB1 in endometrial carcinoma, hyperplasia and normal endometrium: Correlation with clinicopathologic parameters and biomarkers. Mod Pathol. 2006;19:1593-605
28. Fujita Y, Sakakura C, Shimomura K. et al. Chromosome arm 20q gains and other genomic alterations in esophageal squamous cell carcinoma, as analyzed by comparative genomic hybridization and fluorescence in situ hybridization. Hepatogastroenterology. 2003;50:1857-63
29. Xu FP, Xie D, Wen JM. et al. SRC-3/AIB1 protein and gene amplification levels in human esophageal squamous cell carcinomas. Cancer Lett. 2007;245:69-74
30. He LR, Liu MZ, Li BK. et al. Overexpression of AIB1 predicts resistance to chemoradiotherapy and poor prognosis in patients with primary esophageal squamous cell carcinoma. Cancer Sci. 2009;100:1591-6
31. Xie D, Sham JS, Zeng WF. et al. Correlation of AIB1 overexpression with advanced clinical stage of human colorectal carcinoma. Hum Pathol. 2005;36:777-83
32. Lassmann S, Weis R, Makowiec F. et al. Array CGH identifies distinct DNA copy number profiles of oncogenes and tumor suppressor genes in chromosomal- and microsatellite-unstable sporadic colorectal carcinomas. J Mol Med. 2007;85:293-304
33. Grivas PD, Tzelepi V, Sotiropoulou-Bonikou G, Kefalopoulou Z, Papavassiliou AG, Kalofonos H. Estrogen receptor alpha/beta, AIB1, and TIF2 in colorectal carcinogenesis: do coregulators have prognostic significance. Int J Colorectal Dis. 2009;24:613-22
34. Wang Y, Wu MC, Sham JS, Zhang W, Wu WQ, Guan XY. Prognostic significance of c-myc and AIB1 amplification in hepatocellular carcinoma. A broad survey using high-throughput tissue microarray. Cancer. 2002;95:2346-52
35. Xu Y, Chen Q, Li W. et al. Overexpression of transcriptional coactivator AIB1 promotes hepatocellular carcinoma progression by enhancing cell proliferation and invasiveness. Oncogene. 2010;29:3386-97
36. Henke RT, Haddad BR, Kim SE. et al. Overexpression of the nuclear receptor coactivator AIB1 (SRC-3) during progression of pancreatic adenocarcinoma. Clin Cancer Res. 2004;10:6134-42
37. Sakakura C, Hagiwara A, Yasuoka R. et al. Amplification and over-expression of the AIB1 nuclear receptor co-activator gene in primary gastric cancers. Int J Cancer. 2000;89:217-23
38. Luo JH, Xie D, Liu MZ. et al. Protein expression and amplification of AIB1 in human urothelial carcinoma of the bladder and overexpression of AIB1 is a new independent prognostic marker of patient survival. Int J Cancer. 2008;122:2554-61
39. Liu MZ, Xie D, Mai SJ. et al. Overexpression of AIB1 in nasopharyngeal carcinomas correlates closely with advanced tumor stage. Am J Clin Pathol. 2008;129:728-34
40. He LR, Zhao HY, Li BK. et al. Overexpression of AIB1 negatively affects survival of surgically resected non-small-cell lung cancer patients. Ann Oncol. 2010;21:1675-81
41. Cai D, Shames DS, Raso MG. et al. Steroid receptor coactivator-3 expression in lung cancer and its role in the regulation of cancer cell survival and proliferation. Cancer Res. 2010;70:6477-85
42. Wang H, Zhang D, Wu W. et al. Overexpression and gender-specific differences of SRC-3 (SRC-3/AIB1) immunoreactivity in human non-small cell lung cancer: an in vivo study. J Histochem Cytochem. 2010;58:1121-7
43. Carroll RS, Brown M, Zhang J, DiRenzo J, De Mora J F, Black PM. Expression of a subset of steroid receptor cofactors is associated with progesterone receptor expression in meningiomas. Clin Cancer Res. 2000;6:3570-5
44. Kuang SQ, Liao L, Zhang H, Lee AV, O'Malley BW, Xu J. AIB1/SRC-3 deficiency affects insulin-like growth factor I signaling pathway and suppresses v-Ha-ras-induced breast cancer initiation and progression in mice. Cancer Res. 2004;64:1875-85
45. Kuang SQ, Liao L, Wang S, Medina D, O'Malley BW, Xu J. Mice lacking the amplified in breast cancer 1/steroid receptor coactivator-3 are resistant to chemical carcinogen-induced mammary tumorigenesis. Cancer Res. 2005;65:7993-8002
46. Fereshteh MP, Tilli MT, Kim SE. et al. The nuclear receptor coactivator amplified in breast cancer-1 is required for Neu (ErbB2/HER2) activation, signaling, and mammary tumorigenesis in mice. Cancer Res. 2008;68:3697-706
47. Ying H, Willingham MC, Cheng SY. The steroid receptor coactivator-3 is a tumor promoter in a mouse model of thyroid cancer. Oncogene. 2008;27:823-30
48. Torres-Arzayus MI, de Mora J F, Yuan J. et al. High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell. 2004;6:263-74
49. Torres-Arzayus MI, Zhao J, Bronson R, Brown M. Estrogen-dependent and estrogen-independent mechanisms contribute to AIB1-mediated tumor formation. Cancer Res. 2010;70:4102-11
50. Park MT, Lee SJ. Cell cycle and cancer. J Biochem Mol Biol. 2003;36:60-5
51. Louie MC, Revenko AS, Zou JX, Yao J, Chen HW. Direct control of cell cycle gene expression by proto-oncogene product ACTR, and its autoregulation underlies its transforming activity. Mol Cell Biol. 2006;26:3810-23
52. Avivar A, Garcia-Macias MC, Ascaso E, Herrera G, O'Connor JE, de Mora JF. Moderate overexpression of AIB1 triggers pre-neoplastic changes in mammary epithelium. FEBS Lett. 2006;580:5222-6
53. Mussi P, Yu C, O'Malley BW, Xu J. Stimulation of steroid receptor coactivator-3 (SRC-3) gene overexpression by a positive regulatory loop of E2F1 and SRC-3. Mol Endocrinol. 2006;20:3105-19
54. Hsia EY, Kalashnikova EV, Revenko AS, Zou JX, Borowsky AD, Chen HW. Deregulated E2F and the AAA+ coregulator ANCCA drive proto-oncogene ACTR/AIB1 overexpression in breast cancer. Mol Cancer Res. 2010;8:183-93
55. Larsson O, Girnita A, Girnita L. Role of insulin-like growth factor 1 receptor signalling in cancer. Br J Cancer. 2005;92:2097-101
56. Wang Z, Rose DW, Hermanson O. et al. Regulation of somatic growth by the p160 coactivator p/CIP. Proc Natl Acad Sci U S A. 2000;97:13549-54
57. Xu J, Liao L, Ning G, Yoshida-Komiya H, Deng C, O'Malley BW. The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc Natl Acad Sci U S A. 2000;97:6379-84
58. Zhou G, Hashimoto Y, Kwak I, Tsai SY, Tsai MJ. Role of the steroid receptor coactivator SRC-3 in cell growth. Mol Cell Biol. 2003;23:7742-55
59. Oh A, List HJ, Reiter R. et al. The nuclear receptor coactivator AIB1 mediates insulin-like growth factor I-induced phenotypic changes in human breast cancer cells. Cancer Res. 2004;64:8299-308
60. Rajah R, Bhala A, Nunn SE, Peehl DM, Cohen P. 7S nerve growth factor is an insulin-like growth factor-binding protein protease. Endocrinology. 1996;137:2676-82
61. York B, Yu C, Sagen JV. et al. Reprogramming the posttranslational code of SRC-3 confers a switch in mammalian systems biology. Proc Natl Acad Sci U S A. 2010;107:11122-7
62. Normanno N, De Luca A, Bianco C. et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2-16
63. Lahusen T, Fereshteh M, Oh A, Wellstein A, Riegel AT. Epidermal growth factor receptor tyrosine phosphorylation and signaling controlled by a nuclear receptor coactivator, amplified in breast cancer 1. Cancer Res. 2007;67:7256-65
64. Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis. 2000;21:485-95
65. Colo GP, Rubio MF, Nojek IM. et al. The p160 nuclear receptor co-activator RAC3 exerts an anti-apoptotic role through a cytoplasmatic action. Oncogene. 2008;27:2430-44
66. Colo GP, Rosato RR, Grant S, Costas MA. RAC3 down-regulation sensitizes human chronic myeloid leukemia cells to TRAIL-induced apoptosis. FEBS Lett. 2007;581:5075-81
67. Sheikh MS, Huang Y. Death receptor activation complexes: it takes two to activate TNF receptor 1. Cell Cycle. 2003;2:550-2
68. Pajak B, Orzechowski A. [FLIP--an enemy which might lose the battle against the specific inhibitors of translation]. Postepy Hig Med Dosw (Online). 2005;59:140-9
69. Wu RC, Qin J, Hashimoto Y. et al. Regulation of SRC-3 (pCIP/ACTR/AIB-1/RAC-3/TRAM-1) Coactivator activity by I kappa B kinase. Mol Cell Biol. 2002;22:3549-61
70. Li R, Chen Y, Shu WX, Chen Z, Ke WJ. Involvement of SRC-3 in deguelin-induced apoptosis in Jurkat cells. Int J Hematol. 2009;89:628-35
71. Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22:8983-98
72. Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9:59-71
73. Vandermoere F, El YI, Adriaenssens E, Lemoine J, Hondermarck H. The antiapoptotic effect of fibroblast growth factor-2 is mediated through nuclear factor-kappaB activation induced via interaction between Akt and IkappaB kinase-beta in breast cancer cells. Oncogene. 2005;24:5482-91
74. Horiguchi K, Arai S, Nishihara T, Nishikawa J. AIB1 promotes DNA replication by JNK repression and AKT activation during cellular stress. J Biochem. 2006;140:409-19
75. Li R, Chen Y, Zeng LL. et al. Gambogic acid induces G0/G1 arrest and apoptosis involving inhibition of SRC-3 and inactivation of Akt pathway in K562 leukemia cells. Toxicology. 2009;262:98-105
76. Duffy MJ, McGowan PM, Gallagher WM. Cancer invasion and metastasis: changing views. J Pathol. 2008;214:283-93
77. Yan J, Erdem H, Li R. et al. Steroid receptor coactivator-3/AIB1 promotes cell migration and invasiveness through focal adhesion turnover and matrix metalloproteinase expression. Cancer Res. 2008;68:5460-8
78. Li LB, Louie MC, Chen HW, Zou JX. Proto-oncogene ACTR/AIB1 promotes cancer cell invasion by up-regulating specific matrix metalloproteinase expression. Cancer Lett. 2008;261:64-73
79. Chung AC, Zhou S, Liao L, Tien JC, Greenberg NM, Xu J. Genetic ablation of the amplified-in-breast cancer 1 inhibits spontaneous prostate cancer progression in mice. Cancer Res. 2007;67:5965-75
80. Long W, Yi P, Amazit L. et al. SRC-3Delta4 mediates the interaction of EGFR with FAK to promote cell migration. Mol Cell. 2010;37:321-32
81. Coste A, Antal MC, Chan S. et al. Absence of the steroid receptor coactivator-3 induces B-cell lymphoma. EMBO J. 2006;25:2453-64
Author contact
![]() Corresponding author: Jianjun He, Department of Surgical Oncology, the First Affiliated Hospital, Medical School, Xi'an Jiaotong University, Xi'an, Shaanxi Province, 710061, P. R China. Email: chinahjjcom. Tel and Fax: (86-29)8532-4609
Corresponding author: Jianjun He, Department of Surgical Oncology, the First Affiliated Hospital, Medical School, Xi'an Jiaotong University, Xi'an, Shaanxi Province, 710061, P. R China. Email: chinahjjcom. Tel and Fax: (86-29)8532-4609