Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(6):691-699. doi:10.7150/ijbs.7.691 This issue Cite
Research Paper
The DNA Binding and Bending Activities of Truncated Tail-less HMGB1 protein are Differentially Affected by Lys-2 and Lys-81 Residues and Their Acetylation
Ivan Elenkov1, Petar Pelovsky1, Iva Ugrinova1, Masayuki Takahashi2, Evdokia Pasheva1 ![]()
1. Institute of Moleculat Biology, Bulgarian Academy of Sciences, 1113 Sofia, Bulgaria
2. UMR 6204 CNRS, Université de Nantes, Nantes, France
Received 2011-2-28; Accepted 2011-5-24; Published 2011-6-1
Abstract
The role of lysines 2 and 81 as target sites for acetylation in full-length HMGB1 and truncated tail-less protein, respectively, has been studied by mutation analysis for the abilities of these proteins to bind and bend DNA. The DNA bending ability of truncated tail-less HMGB1 containing Lys-2 mutated to alanine does not differ from that of the wild-type protein, while the same mutation of Lys-81 reduced the bending capacity of the mutant protein. These data demonstrate that Lys-81 is critical for the DNA bending ability of truncated HMGB1. Such a conclusion is further confirmed by the experiments carried out with CBP-acetylated proteins: acetylation of Lys-2 in mutant protein K81/A81 alleviated DNA bending and induced DNA end-joining. On the contrary, the acetylation of Lys-81 in the mutant K2/A2 enhanced the bending potential of HMGB1∆C. Regarding the ability of HMGB1 to specifically bind bent DNA, the individual mutations of either K2 or K81 as well as the double mutation of both residues to alanine were found to completely abolish binding of truncated tail-less HMGB1 to cisplatin-modified DNA. We conclude that unlike the case with the bending ability of truncated HMGB1, where Lys-81 has a primary function, Lys-2 and Lys-81 are both critical for the protein's binding to cisplatin-modified DNA. The mutation K2/A2 in full-length HMGB1 and acidic tail removal induce the same conformational changes. Any further substitutions at the acetylable lysines in the truncated form of HMGB1 do not have an additional effect.
Keywords: HMGB1 protein, tail-less HMGB1 protein, Lys-2 and Lys-81 residues, acetylation, binding to platinated DNA, DNA bending.
Introduction
The high mobility group box protein 1 (HMGB1) is an abundant non-histone protein of chromatin with rather unusual biology. It is composed of two tandem DNA binding HMG-box domains (N-terminal A and central B), and an acidic C-terminal tail, mediating protein-protein interactions [1-3]. This structure confers HMGB1 the peculiar feature to recognize and specifically bind DNA structures, containing sharp bends or kinks, such as four way junctions [4], DNA damaged by the anticancer drug cisplatin [5, 6] and by UV-light [7], or to induce bending in linear duplex DNA [8, 9]. The structure-specific DNA binding is attributed to the A box [9, 10] while the DNA bending capacity is primarily related to the B domain [11]. The abilities of HMGB1 to bend DNA and to bind bent DNA define this protein as an architectural factor, promoting the assembly of nucleoprotein complexes [2]. This crucial role explains the implication of HMGB1 in mediating fundamental cellular events such as transcription, recombination, replication and repair [3]. One way for regulation of such a role is the posttranslational modification(s), mainly acetylation and phosphorylation. Regarding acetylation, in spite of the very early identification of Lys-2 as a main acetylation site in the protein from calf thymus [12], attempts to evaluate the consequences of this modification for the properties of HMGB1 were initiated much later. In vivo modified protein, isolated from butyrate-treated cells and shown to be monoacetylated at Lys-2 [13], demonstrated an enhanced binding affinity to distorted DNA structures [13], inability to bend DNA and, instead, an increased capacity to stimulate joining of DNA fragments via their ends [14]. Furthermore, acidic tail removal by tryptic cleavage was found to create another site for acetylation, Lys-81 [15]. In vitro acetylation of this target site in truncated tail-less HMGB1 restored the protein's bending ability and strongly reduced the DNA end-joining activity [14]. Acetylation of Lys-81, therefore, completely alleviated the effects obtained upon acetylation of Lys-2.
Given the data on the role of acetylation of HMGB1 protein for its architectural properties, our next aim was to assess the individual contribution of Lys-2 and Lys-81 in this regard. To this end, recombinant full-length and truncated tail-less HMGB1 were obtained as well as mutant proteins in which the acetylatable lysine residues were replaced by alanine: Lys-2 in full length protein and Lys-2 or/and Lys-81 in the truncated tail-less HMGB1. The proteins generated were analysed to explore their contribution to DNA binding specificity and bending ability. The data obtained suggest a critical role for K81 in the linker region between A and B box for the architectural capacity of the tail-less HMGB1. It should be stressed here, that the truncated tail-less protein should not be regarded solely as an artificial, in vitro generated protein. A trypsin-like chromatin-bound protease has been identified that specifically cleaves the acidic tail of HMGB1 [16]. Moreover, proteins with truncated C-terminal domain called HMG3 were purified from calf thymus [17] and various tumor cell lines [18].
Materials and methods
Polymerase chain reaction (PCR)
DNA constructs for full-length recombinant HMGB1, its truncated tail-less molecule (HMGB1∆C), their single mutants HMGB1(K2/A2), HMGB1(К81/А81), HMGB1∆C(К2А/A2), HMGB1∆C(К81/А81) and the double mutant HMGB1∆C(К2/А2, K81/A81) were prepared by PCR amplification of cDNA encoding full-length rat HMGB1 (lib.N 961, RZPD) as described [19]. The primers used introduce EcoR1 and XhoI cloning sites for the full length HMGB1 forward 5'-TGCACTGGAATTCATGGGCAAAGGAGATCC-3' and reverse 5'-CAGTGCACTCGAGTTATTCATCATCATCATCTTC-3' and for the truncated form HMGB1∆C forward 5'-TGCACTGGAATTCATGGGCAAAGGAGATCC-3' and reverse 5'-CTTCTTTTTCTTGCTTTTTTCAGCCTTG-3' respectively. The substitution of K2 to A2 in full length HMGB1(K2/A2) and tail-less HMGB1∆C(К2/A2) proteins was generated by primers bearing the appropriate mutation: forward 5'-TGCACTGGAATTCATGGGCGCAGGAGATCC-3' for both proteins and reverse 5'-CAGTGCACTCGAGTTATTCATCATCATCATCTTC-3' for the full length and reverse 5'-CTTCTTTTTCTTGCTTTTTTCAGCCTTG-3' for the truncated protein respectively. For the substitution of K81 to A81 in full length HMGB1(K81/A81) and tail-less HMGB1∆C(К281/A81) proteins several successive PCRs were carried out. First the DNA coding amino acid sequences from 1 to 86 for both protein preparations was amplified with forward primer bearing EcoR1 restriction site (5'-TGCACTGGAATTCATGGGCAAAGGAGATCC-3') and a reverse one bearing a mutation at Lys 81 (5'-CTTTTTTGTTTCCCCTGCGGGAGGGATATAGGT -3'). In a second PCR a DNA fragment coding amino acids from 74 to 186 was synthesized by a forward primer introducing a mutation at Lys 81 (5'-ACCTATATCCCTCCCGCAGGGGAAACAAAAAAG-3') and a reverse one with a Xho1 restriction sequence (5'-CAGTGCACTCGAGTTATTCATCATCATCATCTTC-3') for the full length HMGB1 and (5'-CAGTGCACTCGAGCTTCTTTTTCTTGCTTTTTTCAGCC-3') for the truncated form respectively. The overlapping DNA fragments were used as a template for a third PCR resulting in the entire DNA length for HMGB1(K81/A81) and HMGB1∆C(К281/A81) by incubating it with primers forward 5'-TGCACTGGAATTCATGGGCAAAGGAGATCC-3' and reverse 5'-CAGTGCACTCGAGTTATTCATCATCATCATCTTC-3' and forward 5'-TGCACTGGAATTCATGGGCAAAGGAGATCC-3' and reverse 5'-CTTCTTTTTCTTGCTTTTTTCAGCCTTG-3' respectively. The double mutant HMGB1ΔCK2/A2K81/A81 was created as above except that for the amplification of the first fragment (1-86 a.a.) a forward primer bearing a mutation at Lys2 and EcoR1 restriction site was used (5'-TGCACTGGAATTCATGGGCGCAGGAGATCC-3'). The PCR was carried out in a 50 µl final volume using QB-96 thermocycler (LKB). The reaction mixture contained: 0.125 mM MgCl2, 0.25 mM dNTP, 2 µM of the appropriate primers, 50 ng matrix DNA, 2 units Taq polymerase (Pharmacia) and 5 µl 10 x PCR buffer. The purity of all DNA preparations was checked by agarose gel electrophoresis and the accuracy of the obtained constructs was proved by sequencing analysis on ABI PRISM 3100 Genetic Analyzer.
Expression of recombinant proteins
The PCR products were treated with the restriction enzymes EcoR1 and XhoI, cloned in an expression vector pET28a+ and expressed in modified Escherichia coli BL21 Poly Lys S. His-tagged protein samples were purified on a HIS-Select HF Nickel Affinity gel (Sigma). Before His-tag removal the proteins were loaded on the Nickel Affinity gel equilibrated with 20 mM Tris-HCl, pH 7.9 containing 60 mM imidazole, 50 mM NaCl and the slurry was washed with thrombin cleavage buffer (20 mM Tris-HCl, pH 8.4, 150 mM NaCl, 2.5 mM CaCl2) supplemented with 0.5 units of thrombin (human plasma, Calbiochem) per milligram recombinant protein and incubated for 4 h. at room temperature. After brief centrifugation the supernatant containing the His-tag depleted protein was collected and used for further experiments. The purity of all protein preparations was checked by polyacrylamide gel electrophoresis containing sodium dodecyl sulfate (SDS- PAGE).
Acetylation assay
Expression and purification of recombinant histone acetyltransferase CBP together with the protocol of the acetylation procedure in the presence of acetyl CoA are described in [15]. For each protein preparation the histone acetyltransferase (HAT) assay was first carried out on filters using radioactive [14C] acetyl CoA to confirm the proteins' modification. Briefly, the filter binding assay was performed in 20µl of reaction mixture using 100 pmol of [14C] acetyl-CoA (3.5 mCi/mmol, Amersham Life Science Inc.). All protein substrates including histones H3/H4 were added to a concentration of 0.033µg/ml. After incubation at 30 °C for 40 min, the reaction mixture was spotted onto a Whatman P-81 phosphocellulose filter, and following four washes with sodium carbonate buffer, pH 9.2, the filters were dried and counted in a liquid scintillation counter. The level of acetylation of the tail-less HMGB1 protein and its mutant forms corresponded to the previously reported [19], compared to the modification level of recombinant H3/H4 and presented on Fig.2. For the ligase-mediated circularization assay and electrophoretic mobility shift assay all protein samples were acetylated in the presence of cold acetyl CoA.
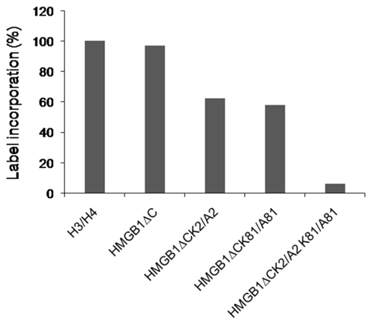
The acetylation level of the truncated HMGB1 protein and its mutant forms. The protein samples are subjected to acetylation by CBP using [14C] acetyl CoA.The reaction mixtures were spotted onto a Whatman P-81 phosphocellulose filters,dried and counted in a liquid scintillation counter.
Ligase-mediated circularization assay
The DNA probe used was a 111-bp fragment obtained by BamH1 and PvuII digestion of pUC19 and labeled with [α-32P]dCTP. The assay generally followed a procedure described elsewhere [8]. After termination of ligation, the samples were fractionated by 5% PAGE as described [15]. After the electrophoresis, the gels were dried and autoradiographed. Generated circles were identified in the gel by their resistance to exonuclease III digestion.
Electrophoretic mobility shift assay (EMSA)
The analysis was carried out as described previously [7] using a 32P-labeled 40-bp oligonucleotide, site specifically platinated at a single GpG inserted in a restriction site for BamH1 endonuclease [13].
Circular Dichroism measurements (CD)
CD spectra were measured with step mode (bandwidth 2 nm, data interval 0.1 nm, response time 0.125 sec) in a J-810 CD spectrometer (Jasco, Japan). The temperature of measurements was set at 150C using Peltier effect temperature controller. The spectra were averaged over 3 scans to increase the signal to noise ratio. Thermal unfolding of proteins was monitored by change in CD signal at 222 nm (bandwidth 5nm, data interval 1/sec, response time 0.5 sec) upon temperature elevation from 15 to 700C with a rate 100C/min. A 1x0.2 cm mini quartz cell with four windows (Hellma, Germany) was used. The cell permits to measure the CD signal either with 1 cm or 0.2 cm path length. The spectra were measured with 0.2 cm path length to get signal also at far-UV region and the thermal folding with 1 cm path length to get larger signal.
Results and Discussion
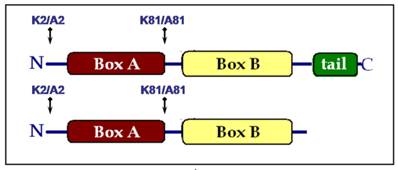
Lysine residues 2 and 81 were mutated to alanine either individually (K2/A2 or K81/A81) or together (K2/A2,K81/A81) in the truncated HMGB1 protein lacking the acidic C-domain (HMGB1∆C). Regarding the full-length protein, a K2/A2 mutant was obtained as Lys-2 was the only acetylable site proved so far [13]. We generated also a K81/A81 substitution in the molecule of HMGB1, despite the fact that K81 was not modified by the acetyltransferase CBP, in order to estimate the contribution of Lys-81 to the architectural properties of the protein. As a result, seven recombinant HMGB1 proteins were created and the introduced mutations are schematically presented in Fig.1: wild-type full length HMGB1 and its single mutants HMGB1K2/A2 and HMGB1K81/A81; wild-type truncated HMGB1 (HMGB1∆C), its single mutants HMGB1∆C(K2/A2) and HMGB1∆C(K81/A81), as well as the double mutant HMGB1∆C(K2/A2, K81/A81).
Schematic presentation of the mutations generated in the full length and tail-less HMGB1 protein.
The role of Lys-2 and Lys-81 and their acetylation for the DNA bending activity of truncated tail-less HMGB1
Previous study on acetylation of HMGB1 from calf thymus identified lysine 2 as a main target site [12] but more recent in vitro experiments demonstrated that the removal of the acidic C-terminal tail exposed lysine 81 to this modification [15]. Our attention was focussed on K2 and K81 because they were the sites for acetylation in the truncated HMGB1. The acetylation level of the truncated mutant samples were tested as in [19] and the results presented in Fig.2. The label incorporation in HMGB1∆C was comparable to that of the classical CBP substrates H3/H4. The single mutant forms were less acetylated (~ 60%) which can be explained by the fact that one of the target lysine resdue (K2 or K81) was omitted. Longer incubation time did not result in higher percentage of incorporated radioactivity. The double mutant exhibited a background modification level.
The consequences of the mutations K2/A2 or K81/A81 as well as the double mutation K2/A2,K81/A81 and the contribution of the individual acetylation sites for the DNA bending ability of truncated HMGB1 lacking the acidic tail were analysed at two protein concentrations by the ring closure assay in the presence of DNA ligase (Fig.3).
DNA bending ability of various recombinant tail-less HMGB1 proteins (∆C), non-modified or in vitro CBP acetylated, assayed by ligase-mediated circularization of linear 111 bp DNA. Panel A. Generation of circular monomers (CM) and linear dimmers (LD) in the presence of various truncated tail-less HMGB1 proteins (HMGB1∆C). To simplify the presentation, ∆C stands for HMGB1∆C. ∆C, purified protein (lane 3); ∆C, di-acetylated at K2 and K81 (lanes 4, 5); lane 6, control treatment with ExoIII to identify circular DNA; ∆CK2/A2 mutant, either non-modified (lane 7) or in vitro acetylated (lanes 8, 9); ∆CK81/A81 mutant, either non-modified (lane 10) or in vitro acetylated (lanes 11,12); lane 13, control treatment with ExoIII to identify linear DNA; ∆CK2/A2,K81/A81 double mutant, either non-modified (lane 14) or acetylated (lanes 15, 16). Panel B. Generation of CM by full-length wild-type HMGB1 (lane 2), K2/A2 mutant (lane 3), K81/A81 mutant (lane 4), control treatment with ExoIII (lane 5), free DNA (lane 1).
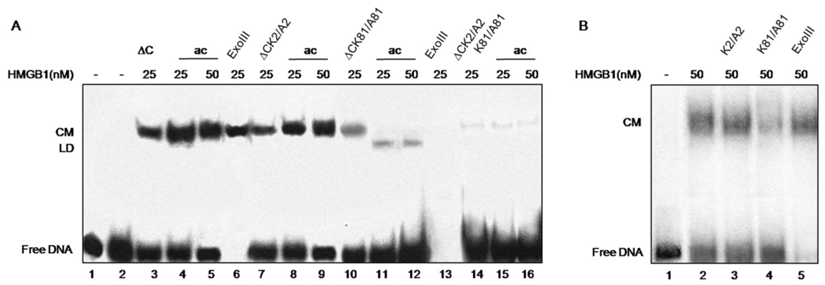
The tail-less protein showed an efficient bending ability (Fig.3, lane 3) in agreement with previous data that the acidic tail is not necessary for the bending effect [20]. Further in vitro acetylation of truncated HMGB1 at both K2 and K81 with CBP was found to enhance the protein's bending activity (Fig.3, compare lanes 4, 5 with lane 3). This finding confirms our earlier data that the acetylated tail-less protein is more effective in inducing DNA bending than its unmodified counterpart [15]. Next, we analysed the effects of single mutations (K2/A2 or K81/A81) and double mutation (K2/A2, K81/A81) for the DNA bending ability of truncated HMGB1. The data obtained are rather informative regarding the role for Lys-2 and Lys-81 in DNA bending. The bending activity of mutant protein containing K2 mutated to alanine (HMGB1∆CK2/A2) is similar to that of wild type protein (Fig. 3, panel A, compare lanes 7 and 3), thus demonstrating that K2 is not essential for the bending capacity of truncated HMGB1. This conclusion is confirmed by the data obtained with truncated HMGB1∆C in which K81 is mutated to alanine (HMGB1∆CK81/A81): the mutant is much less efficient in inducing DNA bending than the wild-type truncated HMGB1 (HMGB1∆C) or the truncated protein containing K2 mutated to alanine (Fig. 3, compare lane 10 to lane 3 and lane 7, respectively). The protein containing double mutation (HMGB1∆CK2/A2, K81/A81) strongly reduced DNA bending activity (Fig. 3, lane 14). Some circular monomers were still visible which was not surprising as the functional B domain, responsible for the property of the protein to induce sharp angles in DNA, remains intact. Together, this analysis and particularly the individual mutations of Lys-2 and Lys-81 to alanine clearly demonstrate that Lys-81 is critical for the DNA bending ability of truncated HMGB1, the contribution of Lys-2 being much less. Such a conclusion is further confirmed by the experiments carried out with CBP-acetylated proteins. Acetylation of K2 in mutant protein HMGB1∆CK81/A81 alleviated DNA bending and induced DNA end-joining (Fig. 3, lanes 11, 12; see also ref. [14]). On the contrary, the acetylation of Lys-81 in the mutant HMGB1∆CK2/A2 enhanced the bending potential of HMGB1 (Fig. 3, compare lane 8 and 9 with lane 7). As expected, CBP acetylation of double mutant was without any effect on its properties (Fig.3, lanes 15, 16).
The fact that the acetylable lysine residue at position 2 is not essential for the bending ability of the full length HMGB1 was shown also by a ring closure assay carried out with the recombinant HMGB1 and its mutated form HMGB1K2/A2 (Fig. 3, panel B, compare lanes 2 and 3). In the HMGB1 molecule the C terminus overlaps the two HMG boxes and shields the Lys-81 from acetylation so it is not considered acetylable target site but still remains critical for the property of the protein to induce kinks in DNA (Fig. 3, panel B, compare lanes 4 and 2).
Following the demonstration that HMGB1 can efficiently bend DNA (8, 10, 11), this property of the protein was associated primarily with the B domain and its N-terminal and C-terminal flanking sequences (the full B domain, see ref. [11]). The investigations were mainly focused on the DNA binding motifs of the protein, its HMG boxes, and less attention was paid to the behaviour of the linker basic sequences 81KGETKKKFK89. The length of the inter-box linker in UBF (Upstream Binding Factor) was found critical for the relative box positioning and thus for ensuring an in-phase bend [21]. This might be the case with HMGB1 protein as its linker's length in different HMGB1 homologues is almost preserved (between −2 and +3 amino acid residues) and the basic, lysine-rich sequence is conserved. The basic flanking sequences were shown to stimulate the DNA binding of the HMG box by providing additional DNA contacts, and thus required for optimal bending [22]. A more detailed analysis on the linker/DNA interactions was reported by Stott et al. [23]. A protein construct that included the structured regions of the HMG box of SRY (sex-determining region on the Y chromosome) and the B domain of HMGB1 plus the basic linker region was generated. The interaction of the SRY.B di-domain protein with DNA containing a SRY binding sequence ensured the formation of a well defined protein/DNA complex. The NMR analysis proved that the linker was bound to the minor groove and especially that Lys-81 was in close contact with the DNA, accomplished through deoxyribose and base interactions. This finding may support one possible explanation for the essential role of Lys-81 regarding the efficient DNA bending abilities of HMGB1. Any substitution or acetylation of Lys-81 will change its contacts with DNA thus affecting the property of the protein to induce DNA kink. For the moment we cannot definitely say on which step the acetylation of Lys-81 exerts its effect, whether the modification of K81 enhances HMGB1 binding to linear DNA or influences the HMGB1 bending property once the protein molecule is associated to DNA. We carried out gel-shift experiments with all protein preparations in their non-acetylated and acetylated forms but at the concentration used for the ring closure assay no complex with linear DNA was ever detected (data not shown).
Binding of full-length and truncated tail-less HMGB1 to cisplatin-damaged DNA is abolished upon mutation of Lys-2 and/or Lys-81 to alanine
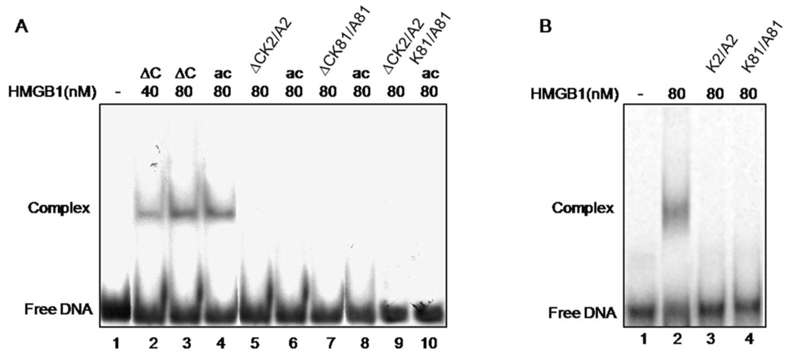
Cisplatin-treated DNA is a widely used substrate for illustration the ability of HMGB1 protein to preferentially bind distorted DNA structures [1]. Here, this approach was used to evaluate the role of two lysine residues, K2 and K81 and their acetylation, for this particular property of HMGB1 and its truncated form. The individual mutation of either K2 or K81 or double mutation of both residues to alanine were found to completely abolish binding of truncated tail-less HMGB1 to cisplatin-modified DNA as judged by EMSA (Fig.4, lanes 5, 7, 9). As far as these lysines are target sites for acetylation, a modification reported to strongly enhance the affinity of HMGB1 to cisplatinated DNA [13] we next attempted acetylation of the mutants with the transacetylase CBP, shown to acetylate the tail-less HMGB1 but not the full length molecule [15]. As far as the single mutation of K2 or K81 to alanine should not affect the possibility for acetylation of the intact, non-mutated residue, it was intriguing to know whether such a possibility has been actually realized. This was tested by gel retardation assay. Interestingly, following in vitro acetylation with CBP of each single mutant, unlike the data obtained with wild type tail-less protein (Fig.4, lanes 2-4), band shifts have not been observed (Fig.4, lanes 6, 8, 10). Moreover, it turned out that mutant truncated HMGB1 was unable to bind damaged DNA even though the non-mutated lysine of the protein had been acetylated (see Fig.2). All these data demonstrate that the acetylatable lysine residues 2 and 81 in the tail-less HMGB1 are critical for the protein's binding to cisplatin-modified DNA. It is worth mentioning, that the same results have been registered with the single mutants K2/A2 and K81/A81 of full-length HMGB1 (Fig. 4, panel B, compare lane 2 with 3 and 4).
Electrophoretic mobility shift assay of the binding of various HMGB1 proteins to cisplatin-damaged DNA. Panel A. Interaction of wild-type tailless (∆C) protein (lanes 2 and 3), in vitro acetylated ∆C at K2 (lane 4), ∆CK2/A2 mutant (lane 5), acetylated ∆CK2/A2 mutant (lane 6), ∆C K81/A81 mutant (lane 7), acetylated ∆CK81/A81 mutant (lane 8), ∆ K2/A2,K81/A81 double mutant (lane 9) and acetylated ∆CK2/A2K81/A81 mutant (lane 10). Lane 1 shows the position of free DNA. Panel B. Interaction of full-length HMGB1 (lane 2) and HMGB1K2/A2 and HMGB1K81/A81 mutants (lanes 3 and 4 respectively) with cis-platinated DNA. Lane 1 shows the position of free DNA.
Mutations of Lys-2 have recently been used to analyse the role of this residue for the binding of HMGB1 to distorted DNA structures [24]. Single mutation of either K2 or K11 was reported to reduce the binding affinity relative to the wild type protein, reaching full inhibition with double mutant protein. One explanation for the observed discrepancies might be the use of different substrates in the binding assays: cisplatin-damaged DNA (our experiment), and four-way DNA junctions [24]. Considering the different effects of Lys-2/Lys-11 double mutation on the binding of the AB di-domain to four-way junction and to minicircles (a little effect, and ~15-fold reduction of binding, respectively, see ref. 24) of one hand and, of the other, the observation that the same mutation abolished binding of A domain to both four-way junctions and minicircles, such an explanation seems quite likely.
CD and thermal stability analysis
The effects of mutations and C-terminal deletion on the protein structure were examined by CD measurements, which are informative of the protein's secondary structure. All analysed proteins exhibited similar CD spectra (Fig. 5, panel A), indicating that neither the base substitution nor the acidic tail deletion affected the protein secondary structure. Two negative peaks, one at 222 nm and another one at 205 nm were observed, corresponding well with alpha helical folding. The absence of significant change in CD spectra indicates that the acidic tale has not particular secondary structure. The protein structure was further investigated by measuring the protein thermal stability, reflecting domain/domain contacts. Weakening of these contacts usually decreases the protein stability. Wild-type protein was unfolded around 450 with one main transition (Fig. 5, panel B), suggesting that the protein is folded in one domain or in domains nicely connected. The mutation of K2 to alanine decreased the thermal stability by about 60C, but there was always one main transition. The substitution decreased the global stability of protein but not the local stability of helical folding at the N-terminal region where the substitution was made.
The effect of C-tail removal and the substitution of acetylable lysines on the CD spectra and thermal stability of HMGB1protein. Panel A. CD spectra of wild type recombinant HMGB1 (black line), HMGB1K2/A2 (red up-triangles), recombinant HMGB1∆C (magenta down-triangles), HMGB1∆CK2/A2,K81/A81 (cyan crosses). Panel B. Thermal stability of wild type recombinant HMGB1 (black squares), HMGB1K2/A2 (red up-triangles), recombinant HMGB1∆C (magenta down-triangles), HMGB1∆CK2/A2,K81/A81 (cyan crosses).
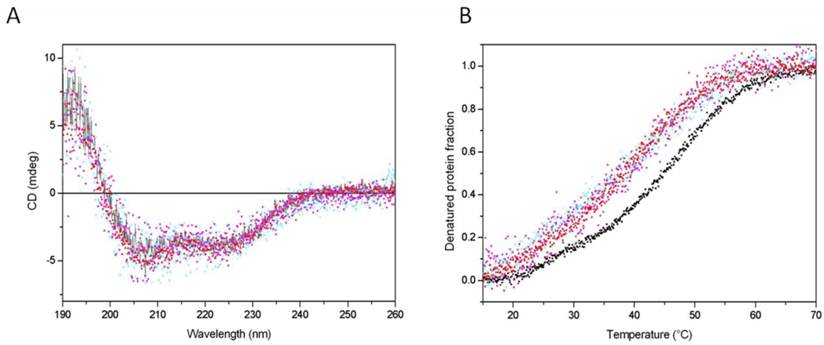
The deletion of the acidic tail also decreased the stability in a very similar manner as did the substitution at the N-terminal part. Interestingly, the double mutation did not further destabilize the protein folding, suggesting that both modifications, K2 substitution and C-tail removal, destabilize the protein via the same mechanism. One probable explanation is that the two parts are involved in a contact between the C- and the N-terminal regions. The results, therefore, are in agreement with the idea that the acidic C-terminal tail makes extensive intramolecular contacts with the DNA binding surfaces of both HMG boxes [25-27] as well as with the recently reported evidence that the acidic tail is in physical contacts with Lys-2 [28]. It is reasonable to expect that acetylation of Lys-2 or its substitution with alanine may disrupt this contact.
Conclusions
Our results demonstrate that Lys-81 is critical for the ability of HMGB1 protein to induce sharp angles in DNA. The CBP acetylation of Lys-81 enhances the bending potential of the truncated protein lacking the acidic C-tail. The acetylable lysine residue at position 2 is not important for the bending properties of HMGB1 and its modification by CBP in the truncated form stimulates DNA end-joining. Both lysine residues, Lys-2 and Lys-81, turned out to be essential for the formation of a stable complex between HMGB1 and DNA damaged by the anti-tumor drug cis-platinum. Their individual substitution by alanine entirely abolished the affinity of the protein to cis-platinated DNA. Neither the acetylation at Lys-2 in HMGB1∆K81/A81 nor at Lys-81 in HMGB1∆CK81/A81 was able to overcome the inhibitory effect.
There is increasing evidence for the modulating effect of the post-synthetic acetylation of HMGB1 protein especially concerning its properties as an “architectural” factor. The individual impact of each of the acetylable lysines, modified or not, for the ability of the protein to bend and to bind with high affinity to distorted DNA structures will provide some clues for better understanding the role of HMGB1 in structural and functional aspect.
Acknowledgements
The work was partially supported by Bulgarian-French program Rila3/14 financed by EGIDE and Bulgarian Ministry of Education.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Bustin M. Regulation of DNA-dependent activities of the functional motifs of the high- mobility- group chromosomal proteins. Mol. Cell Biol. 1999;19:5237-5246
2. Thomas JO, Travers A. HMG1 and 2 and related “architectural" DNA-binding proteins. Trends Biochem. Sci. 2001;26:167-174
3. Travers A, Thomas JO. Chromosomal HMG-box proteins. In: (ed.) Zlatanova J, Leuba SH. Chromatin structure and dynamics: state-of-the-art, New Comprehensive Biochemistry. Amsterdam: Elsevier Science. 2004:103-134
4. Bianchi ME, Beltrame M, Paonessa G. Specific recognition of cruciform DNA by nuclear protein HMG1. Science. 1989;243:1056-1059
5. Pil PM, Lippard SJ. Specific binding of chromosomal protein HMG1 to DNA damaged by the anticancer drug cisplatin. Science. 1992;256:234-237
6. Hughes E, Engelsberg BN, Billings PC. Purification of nuclear proteins that bind to cisplatin damaged DNA: identity with high mobility group proteins 1 and 2. J. Biol. Chem. 1992;267:13520-13527
7. Pasheva E, Pashev IG, Favre A. Preferential binding of high mobility group 1 protein to UV damaged DNA. J. Biol. Chem. 1998;273:24730-24736
8. Pil PM, Chow CS, Lippard SJ. High-mobility-group 1 protein mediates DNA bending as determined by ring closures. Proc. Natl. Acad. Sci. USA. 1993;90:9465-9469
9. Paull T, Haykinson MJ, Johnson RC. The non-specific DNA-binding and -bending proteins HMG1 and HMG2 promote the assembly of complex nucleoprotein structures. Genes Dev. 1993;7:1521-1534
10. Webb M, Thomas JO. Structure-specific binding of the two tandem HMG boxes of HMGB1 to four-way junction DNA is mediated by the A-domain. J. Mol. Biol. 1999;294:373-387
11. Teo S, Grasser KD, Thomas JO. Differences in the DNA binding properties of the HMG-box domains of HMGB1 and the sex-determining factor SRY. Eur. J. Biochem. 1995;230:943-950
12. Sterner R, Vidali G, Allfrey V. Studies of acetylation and deacetylation in high mobility group proteins: identification of the sites of acetylation in HMG-1. J. Biol. Chem. 1979;254:11577-11583
13. Ugrinova I, Pasheva E, Armengaud J. et al. In vivo acetylation of HMGB1 protein enhances its binding affinity to distorted DNA structures. Biochemistry. 2001;40:14655- 14660
14. Ugrinova I, Mitkova E, Moskalenko C. et al. The DNA bending versus DNA end-joining activity of HMGB1 protein is modulated in vitro by acetylation. Biochemistry. 2007;46:2111-2117
15. Pasheva E, Sarov M, Bidjekov K. et al. In vitro acetylation of HMGB-1 and -2 proteins by CBP: the role of the acidic tail. Biochemistry. 2004;43:2935-2940
16. Dyson M, Walker JM. Chromatin associated protease from calf thymus, Int. J. Pept. Protein Res. 1984;24:201-207
17. Elton T, Reeves R. Micro heterogeneity of the mammalian high mobility group (HMG) proteins 1 and 2 investigated by reverse-phase high performance liquid chromatography. Anal. Biochem. 1985;144:403-416
18. Guggenheim ER, Xu D, Zhang C. et al. Photo-affinity isolation and identification of proteins in cancer cell extracts that bind to platinum-modified DNA. Chembiochem. 2009;10:141-157
19. Pelovski P, Pashev I, Pasheva E. Interplay between in vitro acetylation and phosphorylation of tailless HMGB1 protein, Biochem. Biophys. Res. Communs. 2009;380:138-142
20. Stros M. DNA bending by the chromosomal protein HMG1 and its high mobility group box domains. J. Biol. Chem. 1998;273:10355-10361
21. Stefanovsky VY, Guillaume P, Bazett-Jones DP. et al. DNA looping in the RNA polymerase I enhancesome is the result of non-cooperative in-phase bending by two UBF molecules. Nucl. Acids Res. 2001;29:3241-3247
22. Lnenicek-Allen M, Read C M, Crane-Robinson C. The DNA bend angle and binding affinity of an HMG box increased by the presence of short terminal arms. Nucl. Acids Res. 1994;24:1047-1051
23. Stott K, Tang GS, Lee KB. et al. Structure of a complex of tandem HMG boxes and DNA. J Mol Biol. 2006;360:90-104
24. Assenberg R, Webb M, Connolly E. et al. A critical role in structure-specific DNA binding for the acetylatable lysine residues in HMGB1. Biochem J. 2008;411:553-61
25. Knapp S, Muller S, Digilio G. et al. The long acidic tail of high mobility group box 1 (HMGB1) protein forms an extended and flexible structure that interacts with specific residues within and between the HMG boxes. Biochemistry. 2004;43:11992-11997
26. Watson M, Scott K, Thomas JO. Mapping intramolecular interactions between domains in HMGB1 using a tail-truncated approach. J. Mol. Biol. 2007;374:1286-1297
27. Wang Q, Zeng M, Wang W. et al. The HMGB1 acidic tail regulates HMGB1 DNA binding specificity by a unique mechanism. Biochem. Biophys. Res. Communs. 2007;360:14-19
28. Ueda T, Chou H, Kawase T. et al. Acidic C-tail of HMGB1 is required for its target binding to nucleosome linker DNA and transcription stimulation. Biochemistry. 2004;43:9901-9908
Author contact
![]() Corresponding author: Tel. + 3592 979 2668; e-mail: evabas.bg
Corresponding author: Tel. + 3592 979 2668; e-mail: evabas.bg