Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(6):792-804. doi:10.7150/ijbs.7.792 This issue Cite
Research Paper
The Architecture and Complete Sequence of Mitochondrial Genome of an Assassin Bug Agriosphodrus Dohrni (Hemiptera: Reduviidae)
Hu Li1, Jianyu Gao1, Haiyu Liu1, Hui Liu1, Aiping Liang2, Xuguo Zhou3, ![]() , Wanzhi Cai1,
, Wanzhi Cai1, ![]()
1. Department of Entomology, China Agricultural University, Beijing 100193, China
2. Institute of Zoology, Chinese Academy of Sciences, Beijing 100101, China
3. Department of Entomology, University of Kentucky, Lexington, KY 40546-0091, USA
Received 2010-12-9; Accepted 2011-6-18; Published 2011-7-1
Abstract
The 16, 470 bp nucleotide sequence of the mitochondrial genome (mitogenome) of an assassin bug from the reduviid subfamily Harpactorinae, Agriosphodrus dohrni, has been revealed. The entire genome encodes for two ribosomal RNA genes (rrnL and rrnS), 22 transfer RNA (tRNA) genes, 13 protein-coding genes, and a control region. The nucleotide composition is biased toward adenine and thymine (A+T = 72.2%). Comparative analysis with two other reduviid species Triatoma dimidiata and Valentia hoffmanni, exhibited highly conserved genome architectures including genome contents, gene order, nucleotide composition, codon usage, amino acid composition, as well as genome asymmetry. All protein-coding genes use standard mitochondrial initiation codons (methionine and isoleucine), except that nad1 starts with GTG. All tRNAs have the classic clover-leaf structure, except that the dihydrouridine (DHU) arm of tRNASer(AGN) forms a simple loop. Secondary structure comparisons of the two mitochondrial ribosomal subunits among sequenced assassin bugs show that the sequence and structure of rrnL is more conservative than that of rrnS. The presence of structural elements in the control region is also discussed, with emphasis on their implications in the regulation of replication and/or transcription of the reduviid mitogenome. The phylogenetic analyses indicated that within Reduviidae, Harpactorinae is a sister group to the Salyavatinae + Triatominae clade.
Keywords: Mitogenome, Agriosphodrus dohrni, Reduviidae, genome architecture, phylogenetic relationship
Introduction
Mitogenomes have become a major resource for comparative genomics and play an important role in metabolism, apoptosis, disease and aging [1]. In insects, mtDNA is typically a small double-stranded circular molecule of 14-20 kb in length. It encodes 13 protein-coding genes (PCGs), two rRNA genes, and 22 tRNA genes [1, 2]. Additionally, insect mitogenome contains a major non-coding region known as the A+T-rich region that plays a role in initiation of transcription and replication [2]. The length of this region is highly variable among different insects due to its high rate of nucleotide substitution, insertions/deletions, and the presence of a variable number of tandem repeats [3, 4]. For the past two decades, mtDNA has been widely regarded as the molecular marker of choice for the phylogenetic analysis in metazoans because of its abundance in animal tissues, the small genome size, faster rate of evolution, low or absence of sequence recombination, and evolutionary conserved gene products [5-7], although there are some criticisms on using mtDNA for phylogenetics, especially at the deeper level [8, 9].
To date, mitogenomes of 236 insect species across 22 orders have been sequenced and deposited in the GenBank and the insect mitogenomes have been used extensively as molecular markers for population genetics, phylogeographic analyses, and the reconstruction of phylogenetic relationships at different taxonomic levels. Reduviidae, the largest family of terrestrial predatory Hemiptera, consists of approximately 7000 species [10]. Both mitochondrial and nuclear ribosomal genes have been used as molecular markers to resolve the phylogenetic relationship in Reduviidae [10, 11]. However, the number of subfamilies and their phylogenetic relationship are still at the point of juncture because the resolution is rather limited using specific gene markers [10]. The utility of mitogenome data may provide new insights into reduviid higher-level systematics. Up to date, mitogenomes of two assassin bugs, Triatoma dimidiata from subfamily Triatominae [12] and Valentia hoffmanni from subfamily Salyavatinae [13], have been sequenced. T. dimidiata is a medically important insect pest species. It feeds on vertebrate blood and has been known as the vector of Chagas' disease in humans and other mammals.
In this report, we present the complete mitogenome of Agriosphodrus dohrni, a representative of Harpactorinae (the largest subfamily of Reduviidae) and a potential biocontrol agent in orchards and forests [14]. Additionally, we compared the sequence and genome architecture of A. dohrni with blood-feeding T. dimidiata and V. hoffmanni. Finally, phylogenetic analysis of the three known reduviid species (including A. dohrni from this study) with representative heteropterans at the mitogenome level is provided to reexamine the feasibility of mitogenome data to resolve infraordinal relationships of Heteroptera.
Materials and Methods
Agriosphodrus dohrni and genomic DNA extraction
Adult A. dohrni samples were collected from Hanzhong, Shaanxi Province, China in May 2009. All collections were initially preserved in 95% ethanol in the field, and transferred to -20℃ for the long-term storage upon the arrival at the China Agricultural University (CAU). The genomic DNA was extracted from muscle tissues of the thorax using a cetyl-trimethylammonium bromide (CTAB)-based method [15]. Voucher specimen (No. VHem-00201) was deposited at the Entomological Museum of CAU.
PCR amplification, cloning and sequencing
The mitogenome of A. dohrni was generated by amplification of overlapping PCR fragments (Supplementary Material: Table S1). Initially, 13 fragments were amplified using the universal primer sets from [6] (Fig. 1). Nine perfectly matched primers were designed based on the read of these short fragments for the secondary PCRs (Supplementary Material: Table S1).
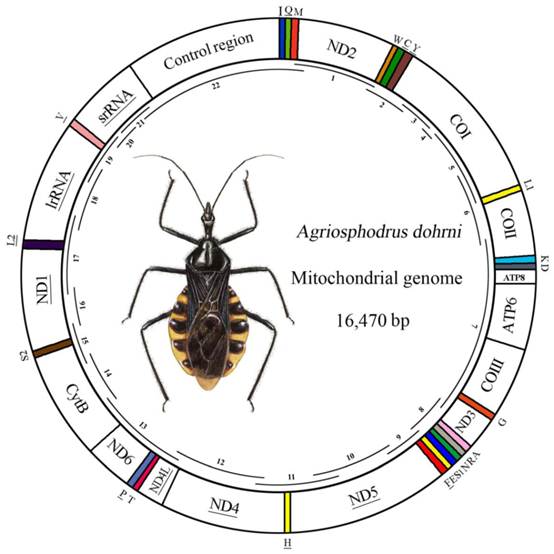
Map of the A. dohrni mitogenome. The tRNAs are denoted by the color blocks and are labeled according to the IUPACIUB single-letter amino acid codes. Gene name without underline indicates the direction of transcription from left to right, and with underline indicates right to left. Overlapping lines within the circle denote PCR fragments amplified used for cloning and sequencing.
Short PCRs (< 1.5 kb) were carried out using Qiagen Taq DNA polymerase (Qiagen, Beijing, China) with the following cycling conditions: 5 min at 94℃, followed by 35 cycles of 50 s at 94℃, 50 s at 48-55℃, 1-2 min at 72℃ depending on the size of amplifications, and the subsequent final elongation step at 72℃ for 10 min. Long PCRs (> 1.5 kb) were performed using NEB Long Taq DNA polymerase (New England BioLabs, Ipswich, MA) under the following cycling conditions: 30s at 95℃, followed by 45 cycles of 10 s at 95℃, 50s at 48-55℃, 3-6 min at 68℃ depending on the size of amplicons, and the final elongation step at 68℃ for 10 min . The quality of PCR products were evaluated by spectrophotometry and agarose gel electrophoresis.
The PCR fragments were ligated into the pGEM-T Easy Vector (Promega) and resulting plasmid DNAs were isolated using the TIANprp Midi Plasmid Kit (Qiagen, Beijing, China). All fragments were sequenced in both directions using the BigDye Terminator Sequencing Kit (Applied Bio Systems) and the ABI 3730XL Genetic Analyzer (PE Applied Biosystems, San Francisco, CA, USA) with two vector-specific primers and internal primers for primer walking.
Secondary structure prediction and sequence analysis
DNA sequences were proof-read and aligned into contigs in BioEdit version 7.0.5.3 [16]. Protein-coding regions and rRNA genes were identified by sequence homology with published insect mitochondrial sequences from public domains (e.g., GenBank). The tRNA genes were identified by tRNAscan-SE Search Server v.1.21 [17] with default setting. Some tRNA genes that could not be determined by tRNAscan-SE were identified by comparing to the tRNA coding regions in other hemipterans. Secondary structures of the small and large subunits of rRNAs were inferred using models predicted for Drosophila melanogaster and D. virilis [18], Apis mellifera [19], Manduca sexta [20] and Ruspolia dubia [21]. Stem-loops were named according to the convention of Gillespie et al. (2006) [19], as well as Cameron et al. (2008) [20]. Protein-coding genes were aligned with Clustal X [22]. A+T content and codon usage were calculated using MEGA version 4.0 [23]. The putative control region was determined using the mfold web server at http://mfold.rna.albany.edu/ [24] to locate regions with potential inverted repeats or palindromes. Strand asymmetry was calculated using the formulas: AT skew= [A−T]/ [A+T] and GC skew= [G−C]/ [G+C] [25].
Phylogenetic analysis
Phylogenetic analysis was carried out based on the 33 complete or nearly complete mitogenomes of true bugs from GenBank (Supplementary Material: Table S2).Two species from Auchenorrhyncha were selected as outgroups. A DNA alignment was inferred from the amino acid alignment of the thirteen protein-coding genes using MEGA version 4.0 [23], which can translate between DNA and amino acid sequences within alignments. Alignments of individual genes were then concatenated excluding the stop codon. Model selection was done with MrModeltest 2.3 [26] and ModelTest 3.7 [27] for Bayesian inference and ML analysis, respectively. According to the Akaike information criterion, the GTR+I+G model was optimal for analysis with nucleotide alignments. MrBayes Version 3.1.1 [28] and a PHYML online web server [29] were employed to analyze this data set under the GTR+I+G model. In Bayesian inference, two simultaneous runs of 1,000,000 generations were conducted for the matrix. Each set was sampled every 200 generations with a burnin of 25%. Trees inferred prior to stationarity were discarded as burn-in, and the remaining trees were used to construct a 50% majority-rule consensus tree. In ML analysis, the parameters were estimated during analysis and the node support values were assessed by bootstrap resampling (BP) [30] calculated using 100 replicates.
Results
The complete mitogenome of A. dohrni was sequenced and determined to be 16, 470 bp in size and it contains 13 protein-coding genes, 22 tRNA genes, two rRNA genes, and a control region (Fig.1). The sequence was deposited in GenBank under the accession number HM071001.
Protein coding genes
All but one protein-coding genes of A. dohrni initiate with ATN as the start codon (eight with ATG, two with ATT, one with ATC and one with ATA) (Table 1). The only exception is the ND1 gene, which likely uses GTG as a start codon. Conventional stop codon (TAA) has been assigned to majority of the protein-coding genes of A. dohrni. COI, COIII and ND5, however, terminate with a single T residue which adjacent directly to the tRNA and non-coding region. Similar arrangement in which the termination codon is believed to be generated by the polyadenylation process has been observed for other insect species [31, 32].
Organization of the A. dohrni mitogenome
| Gene | Direction | Location (bp) | Size (bp) | Anticodon | Start Codon | Stop Codon | Intergenic Nucleotidea |
|---|---|---|---|---|---|---|---|
| tRNAIle | F | 1-64 | 64 | 30-32 GAT | |||
| tRNAGln | R | 62-130 | 69 | 98-100 TTG | -3 | ||
| tRNAMet | F | 130-198 | 69 | 160-162 CAT | -1 | ||
| ND2 | F | 199-1203 | 1005 | ATT | TAA | 0 | |
| tRNATrp | F | 1202-1267 | 66 | 1232-1234 TCA | -2 | ||
| tRNACys | R | 1260-1327 | 68 | 1291-1293 GCA | -8 | ||
| tRNATyr | R | 1369-1434 | 66 | 1400-1402 GTA | 41 | ||
| COI | F | 1446-2979 | 1534 | ATG | T- | 11 | |
| tRNALeu(UUR) | F | 2980-3044 | 65 | 3009-3011 TAA | 0 | ||
| COII | F | 3045-3728 | 684 | ATC | TAA | 0 | |
| tRNALys | F | 3730-3799 | 70 | 3760-3762 CTT | 1 | ||
| tRNAAsp | F | 3802-3867 | 66 | 3833-3835 GTC | 2 | ||
| ATP8 | F | 3869-4027 | 159 | ATT | TAA | 1 | |
| ATP6 | F | 4021-4704 | 684 | ATG | TAA | -7 | |
| COIII | F | 4691-5471 | 781 | ATG | T- | -14 | |
| tRNAGly | F | 5472-5534 | 63 | 5502-5504 TCC | 0 | ||
| ND3 | F | 5535-5888 | 354 | ATA | TAA | 0 | |
| tRNAAla | R | 5889-5950 | 62 | 5918-5920 TGC | 0 | ||
| tRNAArg | F | 5954-6019 | 66 | 5984-5986 TCG | 3 | ||
| tRNAAsn | F | 6026-6093 | 68 | 6058-6060 GTT | 6 | ||
| tRNASer(AGN) | F | 6093-6161 | 69 | 6120-6122 GCT | -1 | ||
| tRNAGlu | F | 6163-6227 | 64 | 6194-6196 TTC | 1 | ||
| tRNAPhe | R | 6230-6294 | 65 | 6260-6262 GAA | 2 | ||
| ND5 | R | 6296-8006 | 1711 | ATG | T- | 1 | |
| tRNAHis | R | 8008-8070 | 63 | 8038-8040 GTG | 1 | ||
| ND4 | R | 8071-9402 | 1332 | ATG | TAA | 0 | |
| ND4L | R | 9396-9680 | 285 | ATG | TAA | -7 | |
| tRNAThr | F | 9687-9750 | 64 | 9718-9720 TGT | 6 | ||
| tRNAPro | R | 9752-9817 | 66 | 9785-9787 TGG | 1 | ||
| ND6 | F | 9831-10328 | 498 | ATG | TAA | 13 | |
| CytB | F | 10328-11464 | 1137 | ATG | TAA | -1 | |
| tRNASer(UCN) | F | 11474-11542 | 63 | 11504-11506TGA | 9 | ||
| ND1 | R | 11706-12629 | 924 | GTG | TAA | 163 | |
| tRNALeu(CUN) | R | 12630-12694 | 65 | 12663-12665TAG | 0 | ||
| lrRNA | R | 12695-13962 | 1268 | 0 | |||
| tRNAVal | R | 13963-14031 | 67 | 14000-14002 TAC | 0 | ||
| srRNA | R | 14032-14827 | 796 | 0 | |||
| Control region | 14828-16470 | 1643 | 0 |
Transfer RNAs
The whole set of 22 tRNAs typical of arthropod mitogenomes were found in A. dohrni, and schematic drawings of their respective secondary structures were shown in Supplementary Material: Figure S1. A. dohrni tRNA genes fold into a classic clover-leaf structure, with the exception of tRNASer(AGN) , in which its dihydrouridine (DHU) arm simply forms a loop. Based on the secondary structure, a total of 25 unmatched base pairs are found in the A. dohrni tRNAs. Twenty-one of them are G-U pairs, which from a weak bond, locate in the AA stem (11-bp), the T stem (2-bp), and the DHU stem (8-bp). The remaining four pairs include U-U mismatches at the AA stem of tRNAAla and tRNAPro, respectively, U-C mismatche at the AA stem of tRNAAla, and A-A mismatch at the AC stem of tRNALys (Supplementary Material: Figure S1).
Ribosomal RNAs
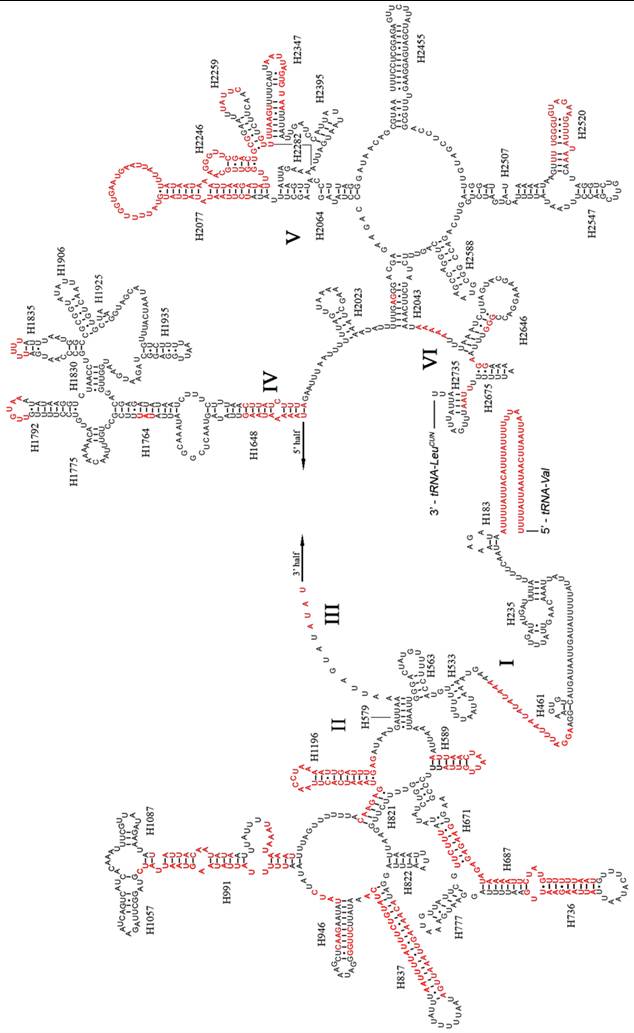
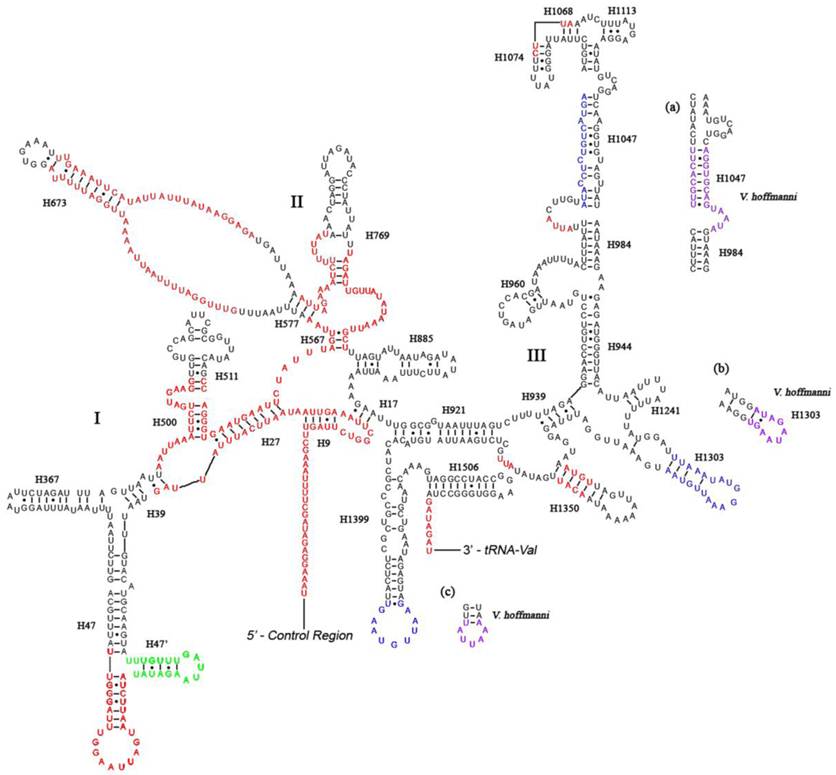
Like other insect mitogenomes, the large and small rRNA subunits (rrnL and rrnS) in A. dohrni are located at tRNALeu(CUN) - tRNAVal and tRNAVal - control region, respectively (Fig. 1 and Table 1). The length of rrnL and rrnS was determined to be 1, 268 bp and 796 bp, respectively. Both rrnL and rrnS are in congruent with the secondary structure models proposed for other insects [18-21, 40, 41]. The secondary structure of A. dohrni rrnL consists of 6 structural domains (domain III is absent in arthropods) (Fig. 2). The structures of H991 region within the domain II were determined according to the models for M. sexta [20] and R. dubia [21], although they are highly variable and difficult to predict. Domain III of A. dohrni is apparently shorter than other insects. The secondary structure of rrnS contains three domains (Fig. 3). Helix-47 region is highly variable among different insects, and in A. dohrni can be folded into two helices, H47 and H47' (Fig. 3).
Predicted secondary structure of the rrnL gene in the A. dohrni mitogenome. Regions in red indicate the high variability in the three assassin bugs. Roman numerals denote the conserved domain structure. Dashed (-) indicate Watson-Crick base pairing and dot (•) indicate G-U base pairing.
Predicted secondary structure of the rrnS gene in the A. dohrni mitogenome. Regions in red indicate high variability in the three assassin bugs. Region in green displays a new predicated helix (H47') compared with A. mellifera [19]. Regions in blue show sequence variability (base insertions and deletions) in the three assassin bugs. Regions in purple and parts (a-c) show the new proposed structures in V. hoffmanni. Roman numerals denote the conserved domain structure. Dashed (-) indicate Watson-Crick base pairing and dot (•) indicate G-U base pairing. Structural annotations follow Fig. 2.
AT contents and codon usage
The nucleotide composition of A. dohrni mitogenome is biased toward adenine and thymine (A+T = 72.2%), ranging from 71.7% in protein-coding genes, 73.1% in rRNA genes, 73.4% in tRNA genes, to 71.9% in the control region (Table 3). The genome-wide bias toward AT was well documented in the codon usage table. At the third codon position, A or T are overwhelmingly overrepresented than G or C. The overall pattern was very similar among the three assassin bugs, with similar frequency of occurrences of various codons within a single codon family. There is a strong bias toward AT-rich codons with the five most prevalent codons in A. dohrni, as in order, TTA (Leu), TTT (Phe), ATT (Ile), ATA (Met) and AAT (Asn).
Nucleotide composition of the A. dohrni (A.), T. dimidiata (T.) and V. hoffmanni (V.) mitogenomes
| Feature | %A+T | AT Skew | GC Skew | ||||||
|---|---|---|---|---|---|---|---|---|---|
| A. | T. | V. | A. | T. | V. | A. | T. | V. | |
| Whole genome | 72.2 | 69.5 | 73.8 | 0.08 | 0.16 | 0.12 | -0.12 | -0.27 | -0.21 |
| protein-coding genes | 71.7 | 68.8 | 73.4 | -0.14 | -0.17 | -0.16 | -0.005 | -0.02 | 0.004 |
| First codon position | 73.5 | 69.2 | 74.6 | -0.09 | -0.13 | -0.10 | 0.01 | -0.03 | 0.01 |
| Second codon position | 70.0 | 68.0 | 72.3 | -0.18 | -0.24 | -0.20 | 0.01 | 0.03 | 0.02 |
| Third codon position | 71.5 | 69.3 | 73.4 | -0.16 | -0.17 | -0.16 | -0.04 | -0.08 | -0.02 |
| rRNA genes | 74.8 | 73.1 | 75.7 | -0.09 | -0.18 | -0.11 | 0.27 | 0.31 | 0.30 |
| tRNA genes | 73.4 | 74.3 | 76.0 | 0.02 | 0.03 | 0.01 | 0.11 | 0.15 | 0.14 |
| Control region | 71.9 | 66.0 | 69.9 | 0.05 | 0.20 | 0.03 | -0.17 | -0.29 | -0.25 |
Non-coding region
The non-coding region of insect mitogenome consists of a control region and short intergenic spacers. In A. dohrni, a long control region (1, 643bp) and 14 intergenic spacers have been identified (Table 1). Majority of the intergenic spacers are only 1- to 13-bp long, however, the longest intergenic spacer located between tRNASer(UCN) and ND1 is 163 bp in length. It has two 58-bp tandem repeats and one 47-bp copy of the anterior portion of the repeat unit. This 163-bp intergenic spacer has no similarity to any existing sequences in the GenBank, and we suspected a similar function to the 314-bp intergenic spacer from T. dimidiata that is believed to be the other origin of replication [12, 42].
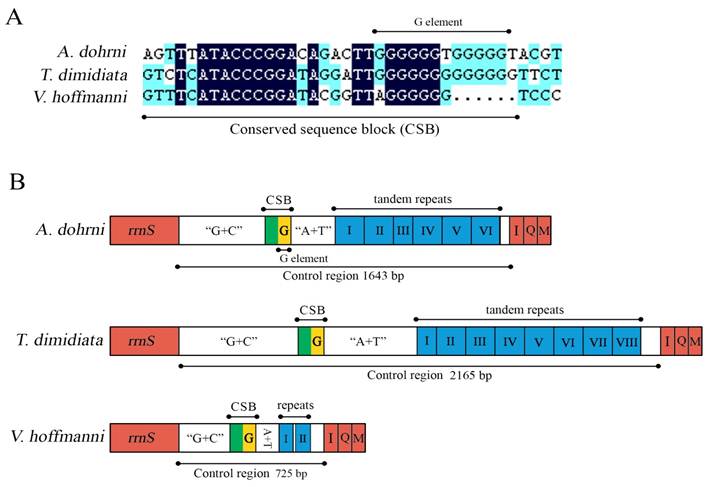
The control region of A. dohrni mitogenome is located at the conserved position between rrnS and tRNAIle- tRNAGln- tRNAMet gene cluster (Fig. 1). The A+T content of this region is one of the lowest in the A. dohrni mitogenome and can be divided into four parts: 1) a 410-bp region that is bordered by rrnS and a conserved region, of which the G+C content (43.4%) is higher than the average of the entire genome; 2) a short conserved sequence block (CSB) 3) a 188-bp region heavily biased toward A+T (79.2%); 4) a region composed of six long tandem repeats; and 5) the remainder of the control region (Fig. 4B).
Control regions of the A. dohrni, V. hoffmanni and T. dimidiata mitogenomes. (A) The conserved sequence block (CSB) and the G element of mitochondrial control regions of the three assassin bugs. (B) A schematic drawing of the structural organization of mitochondrial control regions of the three assassin bugs. The control regions flanking genes rrnS, trnI (I), trnQ (Q), and trnM (M) are highlighted in red boxes. The blue boxes with Roman numerals indicate the tandem repeat region. “G+C” indicates high G+C content region. “A+T” indicates high A+T content region. The yellow box indicates G element.
Phylogenetic relationships
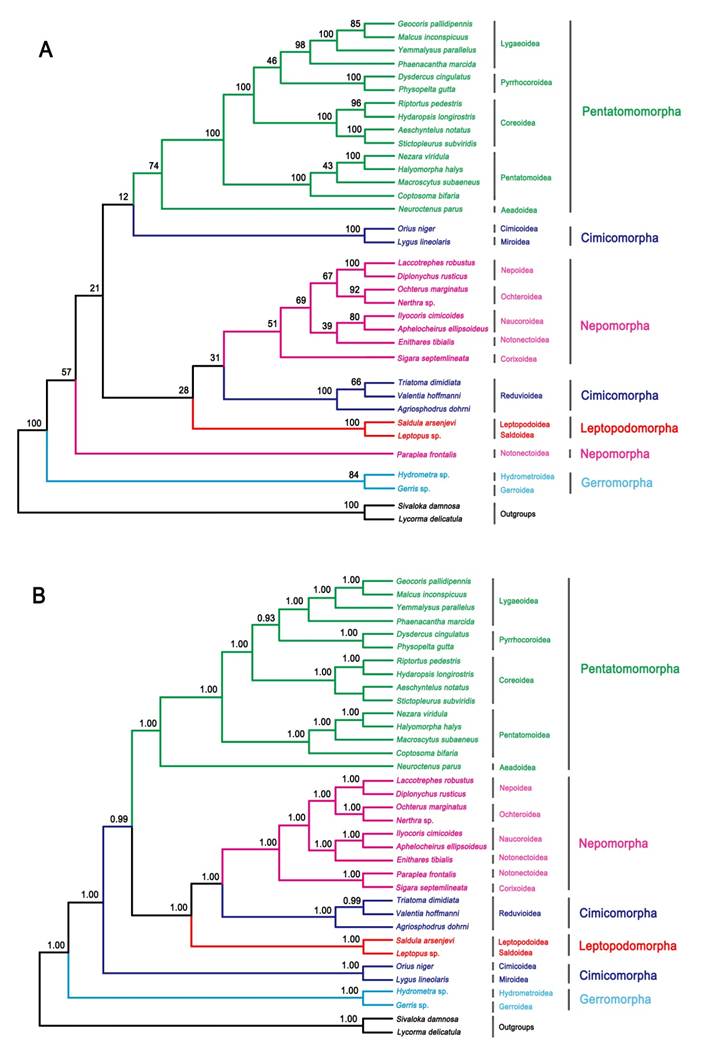
ML and BI analysis performed on the nucleotide dataset generate similar tree topologies (Fig. 5). The five Pentatomomorpha superfamilies (15 taxa) are monophyletic with the following relationships: Aradoidea + (Pentatomoidea + (Pyrrhocoroidea + (Lygaeoidea + Coreoidea))). The sister groups' relationship of five Nepomorpha superfamilies are confirmed, but the position of Pleoidea is unstable. Two Leptopodomorpha superfamilies are monophyletic with the sister relationship to Nepomorpha. Cimicomorpha is paraphyletic consisting of two groups (Reduvioidea and (Cimicoidea and Miroidea)). Two Gerromorpha superfamilies are monophyletic in the basal position of these five infraorders. The infraordinal relationships tend to be poorly resolved with low support in ML analysis. The assassin bug subfamily Harpactorinae presents a sister position to the (Salyavatinae + Triatominae) highly supported by ML and Bayesian inferences.
Phylogenetic tree inferred from the mitogenomes of 33 heteropterans. (A) ML analysis inferred from all codon positions of 13 PCGs. Bootstrap support values are indicated at each node. (B) Bayesian analysis inferred from all codon positions of 13 PCGs. Bayesian posterior probabilities are indicated at each node.
Discussion
The organization and characteristics of A. dohrni mitogenome
The architecture of A. dohrni mitogenome including genome content, gene order, and genome asymmetry is consistent with two other reduviid species T. dimidiata and V. hoffmanni. The mitogenomes of the three assassin bugs share the same genome content (37 genes and 1 control region) and gene order, and have exactly the same genome asymmetry with a gene strand asymmetry (GSA) rate of 0.24 [GSA = (Number of genes on the major-strand - Number of genes on the minor-strand)/Number of total genes] [7]. The size differences among reduviid mitogenomes (A. dohrni, 16, 470 bp; T. dimidiata, 17, 019 bp; and V. hoffmanni, 15, 625 bp) is mainly due to the variable number of repeats in the control regions. It is also worth noting that the evolutionary conserved genome architecture shared among assassin bugs is also considered ancestral to both insects and crustaceans [7, 33].
Among three assassin bugs, six of the protein-coding genes (COI, ATP6, COIII, ND4, ND6 and CytB) have the same Met start codons (ATG or ATA) (Table 2). The other five genes use either Met or Ile as the start codon (ATT, ATC, ATG and ATA). In T. dimidiata, the start codon for ND1 is ATA, whereas, A. dohrni and V. hoffmanni have GTG as their start codon. On the contrary, for ND5, T. dimidiata starts with GTG, whereas, A. dohrni and V. hoffmanni use ATG and ATT as their respective start codons (Table 2).
Start and stop codons of the A. dohrni, T. dimidiata and V. hoffmanni mitogenomes
| Gene | Start Codon | Stop Codon | ||||
|---|---|---|---|---|---|---|
| A. dohrni | T. dimidiata | V. hoffmanni | A. dohrni | T. dimidiata | V. hoffmanni | |
| ND2 | ATT | ATC | ATG | TAA | TAG | TAA |
| COI | ATG | ATG | ATG | T- | T- | T- |
| COII | ATC | ATA | ATA | TAA | T- | T- |
| ATP8 | ATT | ATA | ATC | TAA | TAA | TAA |
| ATP6 | ATG | ATG | ATG | TAA | TAA | TAG |
| COIII | ATG | ATG | ATG | T- | TA- | T- |
| ND3 | ATA | ATA | ATT | TAA | TA- | T- |
| ND5 | ATG | GTG | ATT | T- | TA- | T- |
| ND4 | ATG | ATG | ATG | TAA | TAA | TAA |
| ND4L | ATG | ATG | ATT | TAA | TAA | TAA |
| ND6 | ATG | ATA | ATA | TAA | TAA | TAA |
| CytB | ATG | ATG | ATG | TAA | T- | TAG |
| ND1 | GTG | ATA | GTG | TAA | TAA | TAA |
Dihydrouridine (DHU) arm of A. dohrni tRNASer(AGN) simply forms a loop as seen in the two reduviid species T. dimidiata and V. hoffmanni as well as other insects [2, 13, 34-39]. The anticodon (AC) stem of tRNASer(AGN) among three reduviid species is very conservative, with a long base pairing (9-bp instead of the common 5-bp) and a bulged nucleotide in the middle. The length of A. dohrni tRNAs ranges from 62-69 bp and is similar to the size of T. dimidiata (63-70 bp) and V. hoffmanni (59-70 bp). tRNAs from the three reduviid species possess invariable lengths for the aminoacyl (AA) stem (7-bp), the AC loop (7 nucleotides), and the AC stem (5-bp). Most of the size variations stem from the DHU and T arms, within which the loop size (3-9bp) is more variable than the stem size (3-5bp, except for tRNASer(AGN)). The A. dohrni anticodons are identical to those observed in T. dimidiata and V. hoffmanni.
The information regarding inferred rRNA secondary structures in assassin bugs and true bugs has been limited. Therefore, it is unclear how much variation exists in the rRNA structure and whether differences in length are due to the loss of helices or the reduction in helix sizes. Although the sizes of rrnL and rrnS in A. dohrni (1, 268 bp and 796 bp, respectively) are similar to those of T. dimidiata (1, 270 bp and 781 bp, respectively) and V. hoffmanni (1,256 bp and 777 bp, respectively), the sequence variation is too high in some regions for meaningful structural comparisons (Fig. 2). Overall, the 5' end before the helix H183, and domain-II and -V are the most variable regions of the rrnL, and their length and secondary structure of the loop are more conserved than the stem (Fig. 2). For rrnS, both 5' and 3' ends, and domain-I and -II are more variable. Three regions (Fig. 3a-c) of the V. hoffmanni rrnS which have lengthy base deletions were highlighted in blue and the new proposed structures were highlighted in purple (Fig. 3). Region-a, which was proposed to form a helix (H1047), with a 15-bp deletion, can be folded into a new helix (Fig. 3a). In region-b, helix H1303 had a 12-bp deletion and led to the loss of its stem-loop structure (Fig. 3b). In region-c, the 9-bp loop of helix H1399 reduced to 5-bp and formed a shorter loop (Fig. 3c). To summarize, the size and structure of rrnL is more conserved than rrnS in three sequenced reduviids.
The AT bias is consistent in these three assassin bugs and the nucleotide skew statistics [25] for the whole mitogenome of these three species show that 1) the entire genome and control region are moderately A/C skewed; 2) protein-coding genes and codons lack significant G or C skew but moderate T skew; 3) rRNA genes are significant T/G skewed; and 4) tRNA genes are A/G skewed. Overall, the nucleotide composition of all three reduviid species representing three different subfamilies, respectively, is consistent and likely conserved in the family Reduviidae. It is worth noting that the control regions are not the most AT-rich region in the reduviid mitogenomes, and this generalized labeling of “AT-rich region” for the control region should be reconsidered [13].
The sequence alignment of all three assassin bug control regions reveals a CSB (Fig. 4A), including a G element which has been reported in triatomine bugs Rhodnius prolixus and T. dimidiata (referred as Gs) [12], and some dipterans (referred as G islands) [43]. After the CSB region, there is an A+T rich region which potentially can form the stem-loop structure (Fig. 4B). A possible involvement of this unique motif in insect replication and transcription initiation [43-45] is one of the interests for the future research.
The presence of various numbers of tandem repeats is one of the characteristics of the insect control region [46]. In the case of T. dimidiata, the 2, 165 bp control region has eight tandem repeats including one 82-bp, five 140 bp, and two 173 bp repeats (Fig. 4B). Six tandem repeats identified in A. dohrni include two 193 bp, one 41 bp (a partial copy of the anterior repeat unit), and three 194-bp repeats. However, in V. hoffmanni, the 725 bp control region only has two 38-bp repeats, separated by a non-repetitive sequence (Fig. 4B). Repeated sequences are common in the control region for most insects, and length variations due to the various numbers of repeats are not without precedents [12]. Consequently, analysis of the repeat units among individuals from different geographical locations may shed light on the geographical structuring and phylogenetic relationships of species.
Phylogenetic relationships among Heteroptera inferred from mitogenome sequences
Based on both morphological and molecular characterizations, previous studies support the contention that Reduviidae is monophyletic, whereas Reduviinae is polyphyletic in true bugs [10, 57, 58]. However the number of subfamilies and the phylogenetic relationships within Reduviidae has remained a point of discussion. As the mitogenome sequencing becoming a common practice, the utility of the mitochondrial genome data for the resolution of subfamily-level relationships within Reduviidae is promising.
The seven-infraorder classification of the Heteroptera has been accepted by most researchers [49, 50], however, the phylogenetic relationships among infraorders are still controversial [47-51]. The mitogenomes of 32 species in 29 families within five infraorders of Heteroptera have already been sequenced [12, 13, 52, 53] and those data provide a new source for understanding deep-level true bug phylogeny. Based on the analysis of nine nepomorphan mitogenomes, Hua [13] suggested elevating Pleoidea to the infraorder Plemorpha. Within Cimicomorpha, Reduvioidea is paraphyletic with respect to Cimicoidea and Miroidea in this study, and it is incongruent with previous results [54-56]. These discrepancies suggest that the selection of representative taxa at family-level may influence the phylogenetic relationships within the infraorder resolved from the mitogenome data.
In the present study, the sister-relationship within Pentatomomorpha, Nepomorpha, Leptopodomorpha and Gerromorpha are highly supported by BI and ML analysis. In addition, the Gerromorpha clade is stable in the basal position. This result may provide evidence for the feasibility of mitogenome data to resolve infraordinal relationships of Heteroptera, however, the prerequisite is to ensure the integrality and representative of the infraorder-level taxa. Future analyses should focus on including Enicocephalomorpha and Dipsocoromorpha mitogenome data and additional representatives for some poorly sampled clades.
Supplementary Material
Table S1: Primer sequences used in this study; Table S2: General informatics of the taxa used in this study; Figure S1: Inferred secondary structure of 22 tRNAs of the A. dohrni mitogenome.
Acknowledgements
This research is supported by grants from the National Natural Science Foundation of China (Nos. 30825006, 30970394, 31061160186), Beijing Natural Science Foundation (No. 6112013), the Doctoral Program of Higher Education of China (No. 200800190015), the Key Laboratory of the Zoological Systematics and Evolution of the Chinese Academy of Sciences (No. O529YX5105) and the Innovation Program for Ph. D. Students of China Agricultural University (No. 15059211). We are grateful to three anonymous reviewers' comments and suggestions. Specially thank goes to Dr. Song Nan for his helps in many ways.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767-1780
2. Wolstenholme DR. Genetic novelties in mitochondrial genomes of multicellular animals. Curr Opin Genet Dev. 1992;2:918-925
3. Fauron CMR, Wolstenholme DR. Extensive diversity among Drosophila species with respect to nucleotide sequences within the adenine+thymine-rich region of mitochondrial DNA molecules. Nucleic Acids Res. 1980;8:2439-2452
4. Inohira K, Hara T, Matsuura ET. Nucleotide sequence divergence in the A+T-rich region of mitochondrial DNA in Drosophila simulans and Drosophila mauritiana. Mol Biol Evol. 1997;14:814-822
5. Lin CP, Danforth BN. How do insect nuclear and mitochondrial gene substitution patterns differ? Insights from Bayesian analyses of combined datasets. Mol Phylogenet Evol. 2004;30:686-702
6. Simon C, Buckley TR, Frati F, Stewart JB, Beckenbach AT. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu Rev Ecol Evol Syst. 2006;37:545-579
7. Gissi C, Iannelli F, Pesole G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity. 2008;101:301-320
8. Rubinoff D, Holland BS. Between two extremes: mitochondrial DNA is neither the Panacea nor the Nemesis of phylogenetic and taxonomic inference. Syst Biol. 2005;54:952-961
9. Cameron SL, Lambkin CL, Barker SC, Whiting MF. A mitochondrial genome phylogeny of Diptera: whole genome sequence data accurately resolve relationships over broad timescales with high precision. Syst Entomol. 2007;32:40-59
10. Weirauch C, Munro JB. Molecular phylogeny of the assassin bugs (Hemiptera: Reduviidae), based on mitochondrial and nuclear ribosomal genes. Mol Phylogenet Evol. 2009;53:287-299
11. Weirauch C, Forero D, Jacobs DH. On the evolution of raptorial legs-an insect example (Hemiptera: Reduviidae: Phymatinae). Cladistics. 2010;26:1-12
12. Dotson EM, Beard CB. Sequence and organization of the mitochondrial genome of the Chagas disease vector, Triatoma dimidiata. Insect Mol Biol. 2001;10:205-215
13. Hua JM, Li M, Dong PZ, Cui Y, Xie Q, Bu WJ. Phylogenetic analysis of the true water bugs (Insecta: Hemiptera: Heteroptera: Nepomorpha): evidence from mitochondrial genomes. BMC Evol Bio. 2009;9:134
14. Luo XY, Zhou DK, Li H, Cheng W, Cai W. Taxonomic and bionomic notes on Agriosphodrus dohrni (Signoret) (Hemiptera: Reduviidae: Harpactorinae). Zootaxa. 2010;2358:57-67
15. Aljanabi SM, Martinez I. Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Res. 1997;25:4692-4693
16. Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95-98
17. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955-964
18. Cannone JJ. et al. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3:15
19. Gillespie JJ, Johnston JS, Cannone JJ, Gutell RR. Characteristics of the nuclear (18S, 5.8S, 28S and 5S) and mitochondrial (12S and 16S) rRNA genes of Apis mellifera (Insecta: Hymenoptera): Structure, organization and retrotransposable elements. Insect Mol Biol. 2006;15:657-686
20. Cameron SL, Whiting MF. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene. 2008;408:112-123
21. Zhou ZJ, Huang Y, Shi FM. The mitochondrial genome of Ruspolia dubia (Orthoptera: Conocephalidae) contains a short A+T-rich region of 70 bp in length. Genome. 2007;50:855-866
22. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876-4882
23. Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596-1599
24. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406-3415
25. Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. 1995;41:353-358
26. Nylander JAA. MrModeltest v2; Program distributed by the author. Evolutionary Biology Centre, Uppsala University. 2004
27. Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817-818
28. Huelsenbeck JP, Ronquist F. MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754-755
29. Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696-704
30. Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783-791
31. Friedrich M, Muqim N. Sequence and phylogenetic analysis of the complete mitochondrial genome of the flour beetle Tribolium castanaeum. Mol Phylogenet Evol. 2003;26:502-512
32. Lee ES, Shin KS, Kim MS, Park H, Cho S, Kim CB. The mitochondrial genome of the smaller tea tortrix Adoxophyes honmai (Lepidoptera: Tortricidae). Gene. 2006;373:52-57
33. Hwang UW, Friedrich M, Tautz D, Park CJ, Kim W. Mitochondrial protein phylogeny joins myriapods with chelicerates. Nature. 2001;413:154-157
34. Beard CB, Hamm DM, Collins FH. The mitochondrial genome of the mosquito Anopheles gambiae: DNA sequence, genome organization, and comparisons with mitochondrial sequences of other insects. Insect Mol Biol. 1993;22:103-124
35. Kim I, Cha SY, Yoon MH, Hwang JS, Lee SM, Sohn HD, Jin BR. The complete nucleotide sequence and gene organization of the mitochondrial genome of the oriental mole cricket, Gryllotalpa orientalis (Orthoptera: Gryllotalpidae). Gene. 2005;353:155-168
36. Arnoldi FG, Ogoh K, Ohmiya Y, Viviani VR. Mitochondrial genome sequence of the Brazilian luminescent click beetle Pyrophorus divergens (Coleoptera: Elateridae): mitochondrial genes utility to investigate the evolutionary history of Coleoptera and its bioluminescence. Gene. 2007;405:1-9
37. Li X, Ogoh K, Ohba N, Liang X, Ohmiya Y. Mitochondrial genomes of two luminous beetles, Rhagophthalmus lufengensis and R. ohbai (Arthropoda, Insecta, Coleoptera). Gene. 2007;392:196-205
38. Cha SY, Yoon HJ, Lee EM, Yoon MH, Hwang JS, Jin BR, Han YS, Kim I. The complete nucleotide sequence and gene organization of the mitochondrial genome of the bumblebee, Bombus ignitus (Hymenoptera: Apidae). Gene. 2007;392:206-220
39. Song N, Liang AP. Complete mitochondrial genome of the small brown planthopper, Laodelphax striatellus (Delphacidae: Hemiptera), with a novel gene order. Zool Sci. 2009;26:851-860
40. McMahon DP, Hayward A, Kathirithamby J. The mitochondrial genome of the 'twisted-wing parasite' Mengenilla australiensis (Insecta, Strepsiptera): a comparative study. BMC Genomics. 2009;10:603
41. Wei SJ, Shi M, He JH, Sharkey M, Chen XX. The complete mitochondrial genome of Diadegma semiclausum (Hymenoptera: Ichneumonidae) indicates extensive independent evolutionary events. Genome. 2009;52:308-319
42. Crozier RH, Crozier YC. The mitochondrial genome of the honeybee Apis melliferu: complete sequence and genome organization. Genetics. 1993;133:97-117
43. Oliveira MT, Barau JG, Junqueira ACM, Feijao PC. et al. Structure and evolution of the mitochondrial genomes of Haematobia irritans and Stomoxys calcitrans: The Muscidae (Diptera: Calyptratae) perspective. Mol Phylogenet Evol. 2008;48:850-857
44. Saito S, Tamura K, Aotsuka T. Replication origin of mitochondrial DNA in insects. Genetics. 2005;171:1695-1705
45. Asin-Cayuela J, Dustafsson CM. Mitochondrial transcription and its regulation in mammalian cells. Trends Biochem Sci. 2007;32:111-117
46. Zhang DX, Hewitt FM. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem Syst Ecol. 1997;25:199-210
47. Schuh RT. [Review of] Evolutionary trends of Heteroptera. Part II. Mouthpart-structures and feeding strategies, by R. H. Cobben. Syst Zool. 1979;28:653-656
48. Schuh RT. The influence of cladistics on the classification of the Heteroptera. Annu Rev Entomol. 1986;31:67-93
49. Wheeler WC, Schuh RT, Bang R. Cladistic relationships among higher groups of Heteroptera: congruence between morphological and molecular data sets. Entomol Scand. 1993;24:121-137
50. Schuh RT, Slater JA. True bugs of the world (Hemiptera: Heteroptera) classification and natural history. Ithaca, New York: Cornell University Press. 1995
51. Xie Q, Tian Y, Zheng LY, Bu WJ. 18S rRNA hyper-elongation and the phylogeny of Euhemiptera (Insecta: Hemiptera). Mol Phylogenet Evol. 2008;47:463-471
52. Lee W, Kang J, Jung C, Hoelmer K, Lee SH, Lee S. Complete mitochondrial genome of brown marmorated stink bug Halyomorpha halys (Hemiptera: Pentatomidae), and phylogenetic relationships of hemipteran suborders. Mol Cells. 2009;28:155-165
53. Hua JM, Li M, Dong PZ, Cui Y, Xie Q, Bu WJ. Comparative and phylogenomic studies on the mitochondrial genomes of Pentatomomorpha (Insecta: Hemiptera: Heteroptera). BMC Genomics. 2008;9:610
54. Schuh RT, Štys P. Phylogenetic analysis of cimicomorphan family relationships (Heteroptera). J New York Entomol Soc. 1991;99:298-350
55. Schuh RT, Weirauch C, Wheeler WC. Phylogenetic relationships within the Cimicomorpha (Hemiptera: Heteroptera): a total-evidence analysis. Syst Entomol. 2009;34:15-48
56. Tian Y, Zhu WB, Li M, Xie Q, Bu WJ. Influence of data conflict and molecular phylogeny of major clades in cimicomorphan true bugs (Insecta: Hemiptera: Heteroptera). Mol Phylogenet Evol. 2008;47:581-597
57. Weirauch C. Cladistic analysis of Reduviidae (Heteroptera: Cimicomorpha) based on morphological characters. Syst Entomol. 2008;33:229-274
58. Weirauch C, Schuh RT. Systematics and evolution of Heteroptera: 25 years of progress. Annu Rev Entomol. 2011;56:487-510
Author contact
![]() Corresponding author: Dr. Wanzhi Cai, Department of Entomology, China Agricultural University, Yuanmingyuan West Road, Beijing 100193, China. Phone: 86-10-62732885 Fax: 86-10-62732885 Email: caiwzedu.cn. Xuguo Zhou: xuguozhouedu.
Corresponding author: Dr. Wanzhi Cai, Department of Entomology, China Agricultural University, Yuanmingyuan West Road, Beijing 100193, China. Phone: 86-10-62732885 Fax: 86-10-62732885 Email: caiwzedu.cn. Xuguo Zhou: xuguozhouedu.