Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(8):1114-1121. doi:10.7150/ijbs.7.1114 This issue Cite
Research Paper
FRS2α is Essential for the Fibroblast Growth Factor to Regulate the mTOR Pathway and Autophagy in Mouse Embryonic Fibroblasts
Xiang Lin1,2,#, Yongyou Zhang1,*, Leyuan Liu1, Wallace L. McKeehan1, Yuemao Shen2, Siyang Song2, ![]() , Fen Wang1,
, Fen Wang1, ![]()
1. Center for Cancer and Stem Cell Biology, Institute of Biosciences and Technology, Texas A&M Health Science Center, Houston, TX;
2. The Key Laboratory of Education Ministry for Cell Biology and Tumor Cell Engineering; School of Life Sciences, Xiamen University, Xiamen, Fujian 361005, P. R. China.
# Current address: Institute of Life Sciences, Fuzhou University, Fuzhou, Fujian, 350108, P. R. China;
* Current address: Howard Hughes Medical Institute Laboratory, Wolstein Research Building, 2103 Cornell Road, Cleveland, OH 44106-7285.
Received 2011-4-15; Accepted 2011-5-22; Published 2011-9-15
Abstract
Although the fibroblast growth factor (FGF) signaling axis plays important roles in cell survival, proliferation, and differentiation, the molecular mechanism underlying how the FGF elicits these diverse regulatory signals is not well understood. By using the Frs2α null mouse embryonic fibroblast (MEF) in conjunction with inhibitors to multiple signaling pathways, here we report that the FGF signaling axis activates mTOR via the FGF receptor substrate 2α (FRS2α)-mediated PI3K/Akt pathway, and suppresses autophagy activity in MEFs. In addition, the PI3K/Akt pathway regulated mTOR is crucial for the FGF signaling axis to suppress autophagy in MEFs. Since autophagy has been proposed to play important roles in cell survival, proliferation, and differentiation, the findings suggest a novel mechanism for the FGF signaling axis to transmit regulatory signals to downstream effectors.
Keywords: FGF, autophagy, mouse embryonic fibroblast, receptor tyrosine kinase, cell signaling.
Introduction
The FGF family consists of 18 receptor-binding members that regulate a broad spectrum of cellular activities (1). The FGF elicits its regulatory signals via activating the FGFR tyrosine kinases encoded by four highly homologous genes. FRS2α, also called SNT1 for Suc13-associating neurotropic factor target 1, is a broadly expressed membrane-anchored adaptor protein that is required for the FGF to activate the MAP and PI3 kinase pathways, the two major pathways in the FGF signaling cascade (2-4). The FRS2 family has two highly homologous members, FRS2α and FRS2β. Frs2α is broadly expressed in adult and fetal tissues, whereas Frs2β is more restrictively expressed (5-7). Frs2α null mouse embryos die between embryonic (E) 7.0-7.5 days (5). Although FRS2β can compensate for the loss of FRS2α with respect to MAPK activation in mouse embryonic fibroblast (MEF) cells (8), recent reports demonstrate that FRS2α and FRS2β do not always mediate the same signals (9-12).
FRS2α and FRS2β share similar structure domains, which include the N-terminal myristylation site that anchors FRS2 to the cell membrane, the phosphotyrosine-binding (PTB) domain required for binding to the FGFR, and the C-terminal sequence that contains multiple tyrosine and serine/threonine phosphorylation sites. The PTB-binding domain of the FGFR does not include a phosphorylated tyrosine residue, which is different from the PTB-binding sites of other growth factor receptors (13). The PTB domain of FRS2α also binds Cks1, a molecule that triggers degradation of cell cycle regulatory protein p27kip1 during the G1/S transition in the cell cycle (14). Although not essential (15), a VT (valine-threonine) motif encoded by alternatively spliced sequences in the intracellular juxtamembrane domain of FGFR1 and FGFR2 is important for association with the PTB domain (3,13,16-18). Interestingly, the binding of FGFR1 to FRS2α, but not FRS2β, is enhanced by receptor autophosphorylation (15). FRS2α has six tyrosine phosphorylation sites. Among them, Tyr196, Tyr306, Tyr349, and Tyr392 are Grb2-binding sites that have been shown to be important for transmitting the signals to the PI3K/Akt pathway. Tyr436 and Tyr471 are Shp2-binding sites that have been shown important for transmitting the signals to the MAP kinase pathway (19-21). Mice expressing a FRS2α mutant that lacks the Shp2-binding sites exhibit severe developmental defects; those that lack the Grb2-binding sites have less severe defects (19,22). FGF stimulation also causes phosphorylations on multiple serine/threonine residues of FRS2α, which provides a negative feedback for the FGF signaling activity (23). FRS2α phosphorylation appears to be FGFR isoform-specific, which may result in differential recruitments of downstream signaling molecules and, thus, contribute to signaling specificity of the FGFR (24,25). We reported previously that the four Grb2-binding, but not the two Shp2-binding sites, are essential for FGFR1 to activate the FGF-inducible response element (FiRE) of the mouse syndecan 1 gene (15), which is an enhancer required for the FGF to promote syndecan 1 expression and has been widely used as a reporter for the FGF signaling pathway (15,24,26).
Autophagy is a lysosomal-mediated “self-digest” process for degrading and recycling various cellular constituents, such as long-lived proteins and entire organelles. Autophagy initiates with the formation of autophagosomes in which the isolation membrane engulfs cellular constituents (27). Autophagosomes then fuse with lysosomes to form autolysosomes where the contents are degraded by acidic lysosomal hydrolases. As a self-digestion system, autophagy may influence cell survival, proliferation, and differentiation by accelerating turnover of old protein or organelles (28). However, whether autophagy can be regulated by the FGF signaling axis is currently unknown.
Here, we show that FRS2α is required for the FGF signaling axis to activate the mTOR pathway and to suppress the autophagy activity in MEFs. Ablation of Frs2α, as well as inhibition of FGFR or PI3K, but not ERK, kinase activity suppressed FGF to activate the mTOR pathway and to inhibit autophagy. Thus, the results, for the first time, demonstrate that the FRS2α-mediated mTOR pathway is required for the FGF signaling axis to regulate autophagy, and suggest a new mechanism by which the FGF elicits its regulatory signals.
Materials and Methods
Animals. All animals were housed in the Program for Animal Resources of the Institute of Biosciences and Technology, and handled in accordance with the principles and procedures of the Guide for the Care and Use of Laboratory Animals. All experimental procedures were approved by the Institutional Animal Care and Use Committee. The mice carrying Frs2αflox were bred and genotyped as described (29). The embryos were harvested at embryonic day 14.5 for establishing MEF cultures as described (15). Briefly, E14.5 embryos carrying homozygous Frs2αflox were minced in 3 ml ice-cold 0.25% trypsin-EDTA solution (Sigma-Aldrich, St. Louis, MO), incubated at 4 ºC overnight, and then 37 ºC for 30 minutes in the same solution. The isolated cells were propagated at a ratio of 1:3 in 6-well plates and maintained in 5% fetal bovine serum/DMEM until being used. The MEFs were then immortalized by transfection with pRSV-TAg plasmid that carried the SV40 T antigens. Ad5-CMV-Cre-GFP and Ad5-CMV-GFP viruses from the Center for Cell and Gene Therapy (Houston, TX) were used to delete the floxed fragment.
Transfections. Overnight cultured MEFs (1X105 cells in 6-well plates) were transfected with 5 µg of the indicated FRS2α mutants in pcDNAzeo plasmids (Invitrogen Corporation, La Jolla, CA) and 5 µl Lipofectamine (Invitrogen Corporation). Inhibitor for ERK1/2 kinase (SL327) was from BioSource Int., Inc. (Camarillo, CA), and PI3K (LY294002) was from Cell Signaling (Beverly, MA). Bafilomycin A1 was from LC Laboratories (Woburn MA). The cells were incubated at 37 ºC for 24 hours, and the culture media were then changed to a serum-free DMEM containing 10 µg/ml heparin for serum starvation. After incubation at 37 ºC for 12 hours, the cells were stimulated with FGF2 at the indicated concentrations for the indicated times before being lysed with 200 µl RIPA buffer containing the protease-phosphatase inhibitor cocktail (Sigma-Aldrich, Inc. St Louis, MO) for Western blot analyses.
Western blot. The cell lysates equivalent to 50 µg total proteins were separated on SDS-PAGEs and blotted onto PVDF membranes for Western analyses with the indicated antibodies. The sources and dilution of the antibodies are: phosphorylated FRS2α Tyr196 (1:1000), phosphorylated ERK1/2 (1:1000), phosphorylated Akt Thr308 (1:1000), phosphorylated Akt Ser473 (1:1000), and phosphorylated S6K1 Thr389 (1:1000) from Cell Signaling Technology Inc. (Danvers, MA); human LC3 (1:500) from Novus Biologicals, LLC (Littleton, CO); and β-actin (1: 5000) from Santa Cruz Biotechnology, Inc., (Santa Cruz, CA). The specifically bound primary antibodies were detected with the horseradish peroxidase conjugated secondary antibodies and visualized with the ECL-plus Chemoluminescent reagents. The immunoblot results were developed on X-ray film.
Results and Discussion
FGF2 activates the mTOR pathway in a time-dependent manner
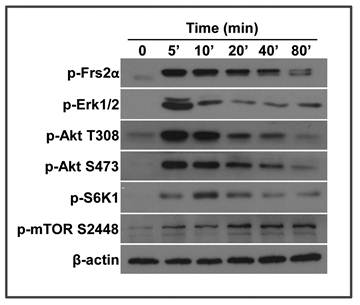
We previously reported that the MEF cells derived from Frs2α floxed embryos are able to respond to FGF signals (15). Since the FRS2α-mediated PI3K/Akt pathway is an upstream regulator of the mTOR pathway, we investigated whether FGF activated mTOR. MEF cells were treated with FGF2 at the final concentration of 5 ng/ml, and lysed at various time points. Activation of the FGF signaling axis and mTOR pathway was analyzed by Western blot. Treating with FGF2 induced FRS2α phosphorylation within 5 minutes. The phosphorylation was then gradually reduced (Fig. 1). Consistently, Akt and Erk1/2, the major downstream molecules of the FGF signaling axis, were also strongly phosphorylated within 5 minutes, which were then gradually reduced. S6K1, the major substrate of the mTOR C1 complex, was also strongly phosphorylated, although the phosphorylation was slightly delayed. Consistently, phosphorylation of mTOR S2448, an S6K1 phosphorylation site (30), was increased at a lagging mode. Together, the results demonstrate that the FGF signaling axis induces activation of the mTOR pathway.
FGF2 activates the mTOR pathway in a time-dependent manner. Wildtype mouse embryonic fibroblasts (MEFs) treated with 5 ng/ml FGF2 were lysed and immunoblotted with the indicated antibodies. β-actin was used as a loading control.
FRS2α is required for FGF2 to activate the mTOR pathway
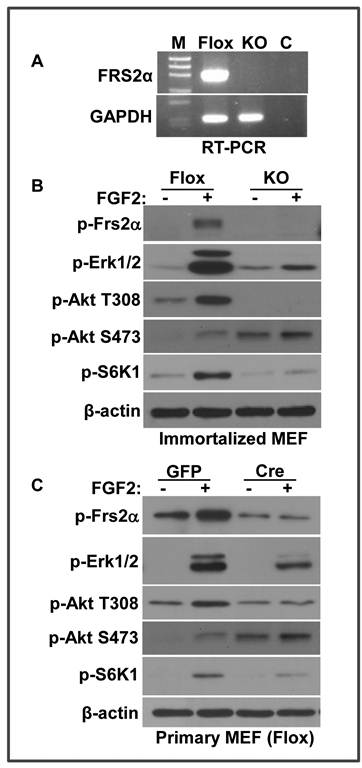
FRS2α is an adaptor protein in the FGF signaling pathway, which recruits multiple downstream pathways, including the ERK1/2 and PI3K/Akt pathways, to the FGFR kinase. To investigate whether FRS2α was required for FGF2 to activate the mTOR pathway, MEFs bearing homozygous Frs2α floxed alleles were treated with adenovirus-Cre/GFP to delete the floxed fragment. The infected cells were isolated with a fluorescence-activated cell sorter. PCR analyses showed that no floxed allele was detected (Fig. 2A). Unlike MEFs bearing floxed Frs2α alleles that exhibited strong activation of ERK1/2, Akt, and S6K1 by FGF2, MEFs bearing Frs2α null alleles failed to respond to FGF2 with respect to ERK1/2, Akt, and S6K1 phosphorylation (Fig. 2B). The weak activation of ERK activation in Frs2α null MEFs were likely due to FRS2α-independent pathways. The results indicated that FRS2α was required for the FGF signaling axis to activate the mTOR pathway. Interestingly, the background phosphorylation of Akt S473, an mTOR-rictor phosphorylation site (31), was significantly increased in Frs2α null MEF. This is not surprising since FRS2α has been shown to play important roles in cell signaling feedback regulation (32). To confirm that the results were not associated with the cloning procedure, primary MEFs were isolated from Frs2α floxed embryos and infected either with adenovirus-GFP or adenovirus-Cre/GFP to repeat the experiments. The results demonstrated that ablation of the Frs2α alleles significantly impaired activation of Akt, ERK, and mTOR by FGF2 (Fig. 2C), which confirmed that FRS2α was required for FGF2 to activate the mTOR pathway. In addition to the FRS2α-independent pathway, incomplete disruption of Frs2α in primary MEFs might also contribute to compromised phosphorylation of ERK1/2 induced by FGF2.
FRS2α is required for FGF2 to activate the mTOR pathway. A. Total RNA was extracted from Frs2α null or wildtype MEF for RT-PCR analysis of Frs2α expression. GAPDH was used as a loading control B. Immortalized MEFs bearing Frs2α floxed (Flox) or null (KO) alleles were treated with FGF2 (5 ng/ml) for 10 minutes, and the phosphorylated proteins were immunoblotted with the indicated antibodies. C. MEFs bearing Frs2α floxed alleles infected with adenovirus bearing GFP (GFP) or Cre/GFP cDNAs were incubated with FGF2 as in A. The phosphorylated proteins were immunoblotted with the indicated antibodies.
FGF2 activates the mTOR pathway via the PI3K/Akt pathway
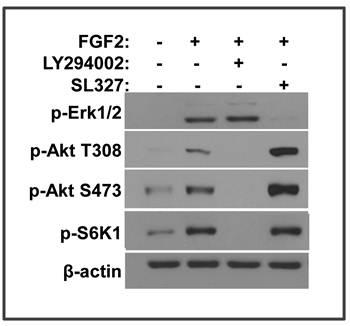
FRS2α has 6 tyrosine phosphorylation sites of which 4 are Grab2 binding that are important for PI3K/Akt activation and 2 are Shp2 binding that are important for MAP kinase activation (19). Both PI3K/Akt and MAP kinase pathways can be linked to the mTOR pathway (33). To determine which pathway is required for FGF2 to activate mTOR in MEFs, inhibitor specific for either PI3K or MEK1/2 was added to block the PI3K/Akt or MAP kinase pathway, respectively. Inhibition of PI3K, but not MEK1/2, abolished the phosphorylation of S6K1, indicating that only the PI3K/Akt pathway was required for FGF2 to activate the mTOR pathway (Fig. 3). Interestingly, inhibition of the MAP kinase pathway significantly increased the phosphorylation of Akt T308 and S473, which were catalyzed by the PI3K/PDPK1 (phosphoinositide dependent kinase 1) and mTOR C2, respectively. Thus, the results suggested that inhibition of the MAP kinase pathway sustained or promoted activation of the Akt-mTOR signaling axis. The results are in line with the reports that sustained activation of the MAP kinase provides a feedback control mechanism of the FGF signaling axis (23).
FGF2 activates the mTOR pathway via the PI3K/Akt pathway. A. Immortalized MEFs bearing Frs2α floxed alleles were treated with 5 ng/ml FGF2 in the presence or absence of the PI3K (10 nM, LY294002) or MEK (10 nM, SL327) inhibitors as indicated. The phosphorylated proteins were immunoblotted with the indicated antibodies.
FRS2α is required for FGF2 to inhibit autophagy through the mTOR pathway
The mTOR pathway is a major signaling pathway that inhibits autophagy (34). To investigate whether FGF signaling regulated autophagy, we then assessed the influence of FGF2 on LC3 II abundance. LC3, also called microtubule associated light chain 3, is a broadly used autophagy indicator. The full-length ProLC3 is processed to its cytosolic form, LC3 I, which is activated by conversion to its lipidated form, LC3 II. The lipidated LC3 II is translocated to preautophagosomes and autophagosomes, which are then degraded after fusing with lysosomes. Thus, the increased abundance of LC3 II reflects enhanced autophagic activity or reduced autophagosome turnover (35,36). To determine whether the FGF signaling axis regulated autophagy, the MEFs with either Frs2α floxed or null alleles were treated with FGF2, and the abundance of LC3 was assessed by Western blot. Treating the Frs2α floxed MEFs with FGF2 enhanced S6K1 phosphorylation and reduced the abundance of the LC3 II isoform, suggesting that FGF2 regulated autophagy, likely through the mTOR pathway. Consistent with the result that FGF2 treatment did not induce S6K1 phosphorylation in Frs2α null MEFs (Fig. 2B), FGF2 treatment also did not reduce LC3 II abundance in Frs2α null MEFs (Fig. 4A), indicating that FGF2 regulated the autophagy activity through FRS2α-mediated pathway(s).
FRS2α is required for FGF2 to inhibit autophagy via the mTOR pathway. A. Overnight cultures of immortalized MEFs bearing Frs2α floxed (Flox) or null (KO) alleles were cultured in a serum-free condition in the presence or absence of 5 ng/ml FGF2 for 2 hours, and the abundance of phosphorylated S6K1 and LC3 II isoforms were immunoblotted as indicated. B&C. Immortalized MEFs bearing Frs2α floxed alleles were treated with FGF2 in the presence or absence of bafilomycin A1 (B) or rapamycin (C), and the abundance of phosphorylated S6K1 and LC3 II isoforms were immunoblotted as indicated.
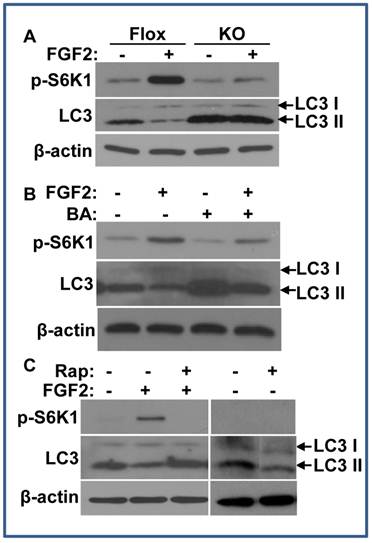
As the abundance of the LC3 II isoform was dynamically controlled, the variation of LC3 II abundance alone may reflect either an inhibition of LC3 I to LC3 II conversion or an activation of LC3 II turnover. ATG1 and ATG13 are substrates of the mTOR C1 kinase that is activated by the PI3K/Akt pathway. Phosphorylation of ATG1 and ATG13 by mTOR C1 changes the conformation of the ATG1 complex to an “open” conformation, resulting in suppression of autophagy (34). As S6K1 is a substrate of the mTOR C1 kinase, which is often used as readout for mTOR C1 activity, the data of enhanced S6K1 phosphorylation in FGF2 treated MEFs inclines toward the possibility that autophagic activity is repressed by increased mTOR C1 activity. To clarify how FGF2 regulated autophagy, the MEFs were treated with bafilomycin A1 to suppress the fusion between autophagosomes and lysosomes (37). Although blocking the degradation of LC3 II by suppression of fusions between autophagosomes and lysosomes increased the abundance of LC3 II either in the presence or absence of FGF2, treating the cells with FGF2 still significantly reduced the abundance of the LC3 II isoform, indicating less LC3 II formation (Fig. 4B). Therefore, the results indicate FGF2 suppresses LC3 I to LC3 II conversion, and thus, the autophagy initiation in cells. To determine whether the mTOR pathway was required for FGF2 to suppress autophagy, rapamycin, an mTOR C1 inhibitor, was added to the MEF cultures. Treating with rapamycin suppressed the phosphorylation of S6K1 and increased the abundance of the LC3 II in FGF2 treated MEFs (Fig. 4C), although it did not affect Akt and Erk1/2 phosphorylation (data not shown). Separate experiments revealed that treating with rapamycin alone did not increase LC3 II abundance in MEFs (Fig. 4C).
The results suggest that activation of mTOR C1 is required for FGF2 to suppress the autophagy activity. Further experiments with other autophagy markers, including expression of P62, ATG5, ATG12, etc., will be carried out to unravel detailed molecular mechanisms underlying this regulation.
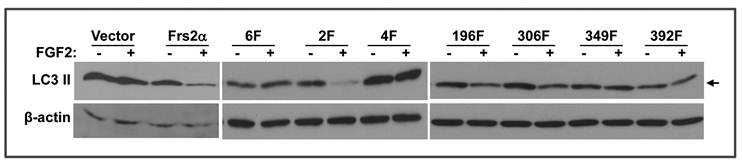
Two Grb2 binding sites of FRS2α, Y349 and Y392, are required for FGF signals to suppress autophagy
Reinstatement of full length FRS2α expression in Frs2α null MEFs by transfection restored regulation of FGF2 on the autophagy activity (Fig. 5). To determine whether the Grb2- or Shp2-binding sites were required for regulating autophagy, mutants lacking the four Grb2- or two Shp2-binding sites were expressed in Frs2α null MEFs as described (15). To better demonstrate the difference in LC3 II abundance, only lightly exposed films were shown. Therefore, LC3 I bands in this figure are too weak to be seen. Consistent with the chemical inhibition experiments demonstrating that the PI3K/Akt, but not ERK, pathway was required for FGF2 to suppress autophagy, expression of the FRS2α mutant lacking the Grb2-binding sites (Y196, Y306, Y349, and Y392) failed to restore the activity, whereas expression of FRS2α lacking the Shp2 binding sites (Y436 and Y471) restored the response to FGF2 with respect to autophagy inhibition. The results again demonstrate that the FGF signaling pathway regulates autophagy through the Grb2-mediated PI3K/Akt pathway. There are four Grb2-binding sites on FRS2α. To further investigate which Grb2-binding phosphorylation sites were required for FRS2α to mediate the autophagy regulation signals, mutant FRS2α carrying an individual Grb2-binding site mutation were expressed in Frs2α null MEFs. Expression of mutants with a substitution of either Y196 or Y306 with phenylalanine rescued the FGF activity to inhibit autophagy activity, whereas expression of the mutant with a substitution of either Y349 or Y392 with phenylalanine failed to rescue such defects (Fig. 5). The results indicate that Y349 and Y392, but not Y196 and Y306, are required for the FGF to suppress autophagy, and imply that each Grb2-binding site may mediate a subset of FGF signals. Activation of PI3-kinase is mediated by assembly of Gab1 to the Grb2/FRS2α complex, which enables tyrosine phosphorylation of Gab1 by FGFR to generate binding sites for p85, the regulatory subunit of PI3-kinase, thus resulting in recruitment and activation of the PI3-kinase (38). Although all four Grb2-binding sites are involved in activation of the PI3K/Akt pathway in 3T3 cells (2), only two Grb2-binding sites are involved in autophagy regulation in MEFs. The molecular mechanism underlying this discrepancy remains elusive and needs to be studied in the future.
Two Grb2 binding sites, Y349 and Y392, are required for FRS2α to mediate FGF signals for regulating autophagy. MEFs bearing Frs2α null alleles transfected with cDNAs encoding the indicated FRS2α mutants were incubated in the presence or absence of FGF2 as indicated, and the abundance of LC3 II isoforms was analyzed by Western blot. Arrows indicate LC3 II isoform. 6F, FRS2α mutant that has mutations on all six tyrosine phosphorylation sites; 2F, FRS2α mutant that has mutations on the two Shp2-binding tyrosine phosphorylation sites, Y436 and Y471; 4F, FRS2α mutant that has mutations on the four Grb2-binding tyrosine phosphorylation sites, Y196, Y306, Y349, and Y392; 196F, 306F, 349F, and 392F, FRS2α mutants that have a single mutation on the indicated tyrosine phosphorylation sites.
The results suggest that the autophagy-suppressing signals of the FGF signaling axis are different from the FiRE-activating signals that are mediated by the four Grb2-binding sites additively, since substitution of each Grb2-binding site only partially reduces FiRE activation activity (15). The FiRE consists of a 170-bp array of five DNA motifs that bind two FGF-inducible Fos-Jun heterodimers, one inducible AP-2-related protein, one constitutively expressed upstream stimulatory factor, and one constitutive 46-kDa transcription factor. It has been shown that FiRE is selectively activated by the FGF, but not by other tyrosine kinase receptor-activating growth factors (26). Furthermore, the FGF activates FiRE independent of its mitogenic activity in prostate cancer cells (24). Here we showed that the signals of FGF to regulate the mTOR/autophagy pathway was different from those for FiRE activation, and further demonstrate mechanistic diversity of the FGF signaling axis.
Although widely used, LC3 lipidation is not a perfect marker for autophagy (39). Furthermore, it has been reported that the LC3 antibody exhibits different detection sensitivities toward LC3 I and LC3 II (40). Therefore, future efforts to monitor expression of other autophagic markers, such as P62, ATG5, and ATG12, etc., will be needed to confirm the findings and to elucidate the molecular mechanism underlying how FGF signaling regulates autophagic activity. Autophagy is a major cellular pathway to degrade bulky subcellular organelles and macromolecules, and plays important roles in development, metabolism, tumorigenesis, and diseases (41). However, how autophagy contributes to development is not understood, although it has been proposed that autophagy may influence cell differentiation either by impairing new protein or organelle formation or by accelerating turnover of old proteins or organelles. In this study, we provided the first in vitro evidence that autophagy can be negatively regulated by the FGF signaling axis. Recently, we also discovered that the FGF regulates cardiac progenitor cell and cardiomyocyte differentiation via controlling the mTOR pathway-regulated autophagy, and that the FRS2α-mediated pathways are required for the FGF to suppress premature differentiation of heart progenitor cells. Inhibition of autophagic activity suppresses cardiomyocyte differentiation both in the second heart field progenitors and in embryoid body (EB) cultures (42).
In summary, the FGF signaling axis activated the mTOR kinase and repressed autophagy by activating the FRS2α-mediated PI3K/Akt pathway. Suppression of mTOR activation abolished the repression activity of FGF2 on autophagy, indicating that FGF regulated autophagy via the mTOR pathway. This is the first report that the FGF signaling axis plays a crucial role in autophagy regulation, thus, sheding new light on cell signaling mechanisms.
Abbreviations
FGF: fibroblast growth factor; FGFR: FGF receptor; FRS2α: FGFR substrate 2α; MEF: mouse embryonic fibroblast.
Acknowledgements
We thank Mary Cole for proofreading and Kerstin McKeehan for excellent technical support. The work is supported in part by grants CA96824 and P50 CA140388 from the NCI, and RP110555 from Cancer Prevention & Research Institute of Texas.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. McKeehan WL, Wang F, Luo Y. The fibroblast growth factor (FGF) signaling complex; Handbook of Cell Signaling, 2nd ed; Volume I, Chapter 38. New York: Academic/Elsevier Press. 2009:253-259
2. Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, Schlessinger JA. lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell. 1997;89:693-702
3. Xu H, Lee KW, Goldfarb M. Novel recognition motif on fibroblast growth factor receptor mediates direct association and activation of SNT adapter proteins. J Biol Chem. 1998;273:17987-17990
4. Ong SH, Lim YP, Low BC, Guy GR. SHP2 associates directly with tyrosine phosphorylated p90 (SNT) protein in FGF-stimulated cells. Biochemical and biophysical research communications. 1997;238:261-266
5. Hadari YR, Gotoh N, Kouhara H, Lax I, Schlessinger J. Critical role for the docking-protein FRS2 alpha in FGF receptor-mediated signal transduction pathways. Proc Natl Acad Sci U S A. 2001;98:8578-8583
6. McDougall K, Kubu C, Verdi JM, Meakin SO. Developmental expression patterns of the signaling adapters FRS-2 and FRS-3 during early embryogenesis. Mech Dev. 2001;103:145-148
7. Zhou L, McDougall K, Kubu CJ, Verdi JM, Meakin SO. Genomic organization and comparative sequence analysis of the mouse and human FRS2, FRS3 genes. Mol Biol Rep. 2003;30:15-25
8. Gotoh N, Laks S, Nakashima M, Lax I, Schlessinger J. FRS2 family docking proteins with overlapping roles in activation of MAP kinase have distinct spatial-temporal patterns of expression of their transcripts. FEBS letters. 2004;564:14-18
9. Avery AW, Figueroa C, Vojtek AB. UNC-51-like kinase regulation of fibroblast growth factor receptor substrate 2/3. Cellular signalling. 2006;19:177-184
10. Harada A, Katoh H, Negishi M. Direct interaction of Rnd1 with FRS2 beta regulates Rnd1-induced down-regulation of RhoA activity and is involved in fibroblast growth factor-induced neurite outgrowth in PC12 cells. J Biol Chem. 2005;280:18418-18424
11. Huang L, Gotoh N, Zhang S, Shibuya M, Yamamoto T, Tsuchida N. SNT-2 interacts with ERK2 and negatively regulates ERK2 signaling in response to EGF stimulation. Biochemical and biophysical research communications. 2004;324:1011-1017
12. Huang L, Watanabe M, Chikamori M, Kido Y, Yamamoto T, Shibuya M, Gotoh N, Tsuchida N. Unique role of SNT-2/FRS2beta/FRS3 docking/adaptor protein for negative regulation in EGF receptor tyrosine kinase signaling pathways. Oncogene. 2006Oct;25(49):6457-66
13. Dhalluin C, Yan K, Plotnikova O, Lee KW, Zeng L, Kuti M, Mujtaba S, Goldfarb MP, Zhou MM. Structural basis of SNT PTB domain interactions with distinct neurotrophic receptors. Molecular cell. 2000;6:921-929
14. Zhang Y, Lin Y, Bowles C, Wang F. Direct Cell Cycle Regulation by the Fibroblast Growth Factor Receptor (FGFR) Kinase through Phosphorylation-dependent Release of Cks1 from FGFR Substrate 2. J Biol Chem. 2004;279:55348-55354
15. Zhang Y, McKeehan K, Lin Y, Zhang J, Wang F. Fibroblast growth factor receptor 1 (FGFR1) tyrosine phosphorylation regulates binding of FGFR substrate 2alpha (FRS2alpha) but not FRS2 to the receptor. Molecular endocrinology (Baltimore Md). 2008;22:167-175
16. Brackenridge S, Wilkie AO, Screaton GR. Efficient use of a 'dead-end' GA 5' splice site in the human fibroblast growth factor receptor genes. Embo J. 2003;22:1620-1631
17. Paterno GD, Ryan PJ, Kao KR, Gillespie LL. The VT+ and VT- isoforms of the fibroblast growth factor receptor type 1 are differentially expressed in the presumptive mesoderm of Xenopus embryos and differ in their ability to mediate mesoderm formation. J Biol Chem. 2000;275:9581-9586
18. Burgar HR, Burns HD, Elsden JL, Lalioti MD, Heath JK. Association of the signaling adaptor FRS2 with fibroblast growth factor receptor 1 (Fgfr1) is mediated by alternative splicing of the juxtamembrane domain. J Biol Chem. 2002;277:4018-4023
19. Gotoh N, Ito M, Yamamoto S, Yoshino I, Song N, Wang Y, Lax I, Schlessinger J, Shibuya M, Lang RA. Tyrosine phosphorylation sites on FRS2alpha responsible for Shp2 recruitment are critical for induction of lens and retina. Proc Natl Acad Sci U S A. 2004;101:17144-17149
20. Ong SH, Guy GR, Hadari YR, Laks S, Gotoh N, Schlessinger J, Lax I. FRS2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors. Mol Cell Biol. 2000;20:979-989
21. Xu H, Goldfarb M. Multiple effector domains within SNT1 coordinate ERK activation and neuronal differentiation of PC12 cells. J Biol Chem. 2001;276:13049-13056
22. Yamamoto S, Yoshino I, Shimazaki T, Murohashi M, Hevner RF, Lax I, Okano H, Shibuya M, Schlessinger J, Gotoh N. Essential role of Shp2-binding sites on FRS2{alpha} for corticogenesis and for FGF2-dependent proliferation of neural progenitor cells. Proc Natl Acad Sci U S A. 2005Nov;102(44):15983-8
23. Lax I, Wong A, Lamothe B, Lee A, Frost A, Hawes J, Schlessinger J. The Docking Protein FRS2alpha Controls a MAP Kinase-Mediated Negative Feedback Mechanism for Signaling by FGF Receptors. Molecular cell. 2002;10:709-719
24. Wang F, McKeehan K, Yu C, McKeehan WL. Fibroblast growth factor receptor 1 phosphotyrosine 766: molecular target for prevention of progression of prostate tumors to malignancy. Cancer Res. 2002;62:1898-1903
25. Xian W, Schwertfeger KL, Rosen JM. Distinct roles of fibroblast growth factor receptor 1 and 2 in regulating cell survival and epithelial-mesenchymal transition. Molecular endocrinology (Baltimore Md). 2007;21:987-1000
26. Jaakkola P, Vihinen T, Maatta A, Jalkanen M. Activation of an enhancer on the syndecan-1 gene is restricted to fibroblast growth factor family members in mesenchymal cells. Mol Cell Biol. 1997;17:3210-3219
27. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313-326
28. Cecconi F, Levine B. The role of autophagy in mammalian development: cell makeover rather than cell death. Developmental cell. 2008;15:344-357
29. Lin Y, Zhang J, Zhang Y, Wang F. Generation of an Frs2alpha conditional null allele. Genesis. 2007;45:554-559
30. Chiang GG, Abraham RT. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. The Journal of biological chemistry. 2005;280:25485-25490
31. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science (New York). 2005;307:1098-1101
32. Wong A, Lamothe B, Lee A, Schlessinger J, Lax I. FRS2 alpha attenuates FGF receptor signaling by Grb2-mediated recruitment of the ubiquitin ligase Cbl. Proc Natl Acad Sci U S A. 2002;99:6684-6689
33. Chiang GG, Abraham RT. Targeting the mTOR signaling network in cancer. Trends Mol Med. 2007;13:433-442
34. Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS letters. 2010;584:1287-1295
35. Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36:2503-2518
36. Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. The Journal of pathology. 2010;221:117-124
37. Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33-42
38. Ong SH, Hadari YR, Gotoh N, Guy GR, Schlessinger J, Lax I. Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc Natl Acad Sci U S A. 2001;98:6074-6079
39. Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A. et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151-175
40. Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods in molecular biology. 2008;445:77-88
41. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069-1075
Author contact
![]() Corresponding author: Fen Wang: Center for Cancer and Stem Cell Biology, Institute of Biosciences and Technology, Texas A&M Health Science Center, Houston, TX 77030-3303. E-mail: fwangtamhsc.edu; Siyang Song, School of Life Sciences, Xiamen University, Xiamen, Fujian 361005, P. R. China, sysongedu.cn.
Corresponding author: Fen Wang: Center for Cancer and Stem Cell Biology, Institute of Biosciences and Technology, Texas A&M Health Science Center, Houston, TX 77030-3303. E-mail: fwangtamhsc.edu; Siyang Song, School of Life Sciences, Xiamen University, Xiamen, Fujian 361005, P. R. China, sysongedu.cn.