Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(2):236-248. doi:10.7150/ijbs.8.236 This issue Cite
Research Paper
Simvastatin Modulates Remodeling of Kv4.3 Expression in Rat Hypertrophied Cardiomyocytes
Feifei Su1*, Miaoqian Shi1*, Zhiqiang Yan2, Dongbo Ou 1, Juntang Li 3, Zifan Lu4, Qiangsun Zheng1 ![]()
1. Department of Cardiology, Tangdu Hospital, Fourth Military Medical University, Xi'an 710038, China
2. Department of Neurosurgery, Urumqi General Hospital of Lanzhou Military Command, Urumqi 830000, China
3. Department of Immunology, Fourth Military Medical University, Xi'an 710033, China
4. Department of Biochemistry and Molecular Biology, Fourth Military Medical University, Xi'an 710032, China
* These authors contributed equally to this work.
Received 2011-9-15; Accepted 2011-12-31; Published 2012-1-6
Abstract
Objectives: Hypertrophy has been shown to be associated with arrhythmias which can be caused by abnormal remodeling of the Kv4-family of transient potassium channels. Inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase (statins) have recently been shown to exert pleiotropic protective effects in cardiovascular diseases, including anti-arrhythmias. It is hypothesized that remodeling of Kv4.3 occurs in rat hypertrophied cardiomyocytes and is regulated by simvastatin.
Methods: Male Sprague-Dawley rats and neonatal rat ventricular myocytes (NRVMs) underwent abdominal aortic banding (AAB) for 7 weeks and angiotensin II (AngII) treatment, respectively, to induce cardiac hypertrophy. Kv4.3 expression by NRVMs and myocardium (subepicardial and subendocardial) in the left ventricle was measured. The transient outward potassium current (Ito) of NRVMs was recorded using a whole-cell patch-clamp method.
Results: Expression of the Kv4.3 transcript and protein was significantly reduced in myocardium (subepicardial and subendocardial) in the left ventricle and in NRVMs. Simvastatin partially prevented the reduction of Kv4.3 expression in NRVMs and subepicardial myocardium but not in the subendocardial myocardium. Hypertrophied NRVMs exhibited a significant reduction in the Ito current and this effect was partially reversed by simvastatin.
Conclusions: Simvastatin alleviated the reduction of Kv4.3 expression, Ito currents in hypertrophied NRVMs and alleviated the reduced Kv4.3 expression in subepicardial myocardium from the hypertrophied left ventricle. It can be speculated that among the pleiotropic effects of simvastatin, the anti-arrhythmia effect is partly mediated by its effect on Kv4.3.
Keywords: Potassium channels, Kv4.3, arrhythmias, hydroxymethylglutaryl-CoA reductase inhibitors, hypertrophy.
Introduction
Sudden cardiac death is commonly caused by ventricular tachyarrhythmias [1]. Studies have indicated several alterations in cellular electrophysiology in hypertrophied myocardium, including prolongation of the action potential duration [2], which may lead to ventricular arrhythmias. Previous investigations have highlighted the importance of potassium channel remodeling in the prolongation of action potential durations in models of hypertrophy [3, 4]. Furthermore, it is believed that suppression of repolarizing potassium currents leading to action potential prolongation provides an explanation for QT prolongation/dispersion, thereby accounting for a major mechanism of arrhythmogenesis [1]. Potassium channels include diverse classes of ion channels in the heart [5]. The transient outward potassium currents (Ito), which can be classified as voltage-gated currents, contribute to cardiac action potential duration, Ca2+ handling, contractility and hypertrophy. Cardiac disease is invariably associated with Ito and changes in Ito have been linked to a long QT-interval and Brugada syndrome arrhythmias [6-11]. Kv4.3, which is the pore-forming subunit of Ito, is uniformly expressed in the ventricle [12, 13]. Expression of the Kv4.3 gene is decreased by cardiac hypertrophy induced by congestive heart failure in humans [14, 15] and downregulated by cardiac hypertrophy in rats [1].
Simvastatin, one of the hydroxymethylglutaryl coenzyme A reductase inhibitors (statins), has been shown to reduce cardiovascular-related morbidity and mortality in clinical trials independent of its cholesterol-lowering function [16, 17] and has been demonstrated to impact cardiac remodeling, including hypertrophy [18-20]. Interestingly, recent studies have also shown that simvastatin decreases arrhythmogenesis [21-24] and the potential link between Kv4.3 and simvastatin is suggested by these studies of hypertrophy. However, little is known about the effect of simvastatin on cardiac Kv4.3 expression. Therefore, modulation of Kv4.3 expression by simvastatin was investigated in this study using the abdominal aortic banding (AAB) rat model of cardiac hypertrophy. Specifically, the effects of simvastatin on Kv4.3 expression were investigated in the subepicardial and subendocardial myocardium of AAB rats. Furthermore, the effects of simvastatin on Kv4.3 expression and Ito in neonatal rat ventricular myocytes (NRVMs) were investigated. It was observed that simvastatin partially suppressed the reduction in Kv4.3 expression in hypertrophied NRVMs and the subepicardial myocardium, but not in the subendocardial myocardium, with an associated recovery in Ito.
Materials and methods
AAB surgical procedures
Male Sprague-Dawley (SD) rats were purchased from the Animal Center of the Fourth Military Medical University (FMMU). The experimental protocol was approved by the Institutional Care and Use Committee of the FMMU, which conforms to the Guidelines for the Care and Use of Laboratory Animals of the US National Institutes of Health (NIH publication No. 85-23, revised 1996). Simvastatin was purchased from Merck, Sharp & Dohme Inc. Other chemicals were of laboratory grade.
Rats (180-220 g; aged 9-12 weeks) were divided into four groups (n = 10): Sham group, where rats underwent surgery without constriction of the aorta; Sham group treated with simvastatin (40 mg/kg *day) (SS group); abdominal aortic banding (AAB group); and AAB rats treated with simvastatin (AABS group). All rats were anesthetized with an intraperitoneal injection of pentobarbital sodium 45 mg/kg. AAB was performed as previously described [25] with minor modifications. In brief, after anesthetization, rats were restrained and a midline abdominal incision was performed under sterile conditions. The abdominal aorta above the renal arteries was dissected away. A ligature (4-0 silk suture) was positioned around the aorta and a blunted 22 gauge needle, which was then withdrawn, leading to constriction (approximately 50%) of the aorta. After surgery, each rat received penicillin 30 kU intramuscularly as antibiotic therapy. Simvastatin was dissolved in 0.9% NaCl solution. All rats received placebo (0.9% NaCl solution) or simvastatin by intragastric administration every day for 7 weeks after surgery.
2-D echocardiography, hemodynamic and left ventricular-weight index (LVWI) measurements
Echocardiograms were performed kindly by Dr. Yunyan Duan (Department of Ultrasonography, Xijing hospital, Xi'an, China), as previously reported [26, 27]. In brief, rats were anesthetized as previously described. A 2-D axis view of the left ventricle was obtained with a 7.5-MHz transducer and the M-mode tracings were then recorded to measure the indexes of left ventricle: left ventricular posterior wall thickness of diastasis (LVPWTd), interventricular septal thickness of diastasis (IVSTd) and left ventricular fractional shortening (LVFS).
For hemodynamic measurements, the right carotid of anesthetized rats was cannulated with a catheter connected to a microtip pressure transducer (Biological Instruments, ChengDu, China). The transducer was connected to a recording system (Rm6280C, Biological Instruments, ChengDu, China). The catheter was then advanced into the left ventricle for measuring the left ventricular end-systolic pressure (LVESP) and the left ventricular end-diastolic pressure (LVEDP) [28].
Blood samples were collected and rats were sacrificed by cervical vertebrae dislocation. The hearts were removed and the left ventricles were dissected. The left ventricle was weighed and divided by body weight in order to calculate the LVWI.
Triglyceride (TG) and total cholesterol (TC) measurement
Serum was isolated from blood samples by centrifugation for measurement of TG and TC levels (performed by Yan Xu at the Department of Clinical Laboratory, Xijing Hospital, FMMU, Xian, Shaanxi, China).
Cell culture and treatment
Monolayer cultures of NRVMs were prepared as described previously [29]. Ventricular myocardium from neonatal SD rats (aged 1 d, FMMU) were homogenized and dissociated with collagenase II. The cell suspension was plated onto a 10 cm dish for 1 h, allowing enrichment for cardiomyocytes by differential adhesion. The supernatant was then plated on to new dishes with Dulbecco's modified Eagle's medium (DMEM, Gibco-BRL) containing 1% penicillin-streptomycin and 10% fetal bovine serum (FBS) under 5% CO2 at 37°C. The remaining fibroblasts were minimized by the addition of 10 µM 5-bromodeoxyuridine for 24 h. NRVMs were divided into four groups: control (Ctrl group); Ctrl treated with simvastatin (Ctrl+Sim group); cells treated with AngII (AngII group); and cells pretreated with simvastatin before being incubated with AngII (AngII+Sim group). Cells were treated with simvastatin and Ang II according as previously described [30]. Briefly, after 24 h serum starvation, cells were treated with 1 µM simvastatin (sodium salt, MERCK Biosciences) in treatment medium (DMEM containing 0.5% serum). AngII (ALEXIS BIOCHEMICALS) was added to a final concentration of 100 nM 12 h after the addition of simvastatin. Cells were then incubated in the presence of both drugs for a further 24 h [30].
Measurement of NRVMs protein content
NRVMs were trypsinized, counted using a cell counting chamber and then lysed. Cell lysates were prepared to determine protein content by Bradford protein assay. The protein content of cells was determined by dividing the total amount of protein by the number of cells.
Electrophysiology
Whole-cell patch-clamp recordings were performed at room temperature as previously described (Axon Instruments) [31]. The intracellular solution contained potassium glutamate 130 mM, KCl 9 mM, NaCl 10 mM, MgCl2 0.5 mM MgATP 5 mM and HEPES 10 mM (pH 7.2). The extracellular solution contained NaCl 138 mM, KCl 4 mM, CaCl2 2mM, MgCl2 1 mM, Na2HPO4 0.33 mM, HEPES 10 mM and glucose 10 mM (pH7.4). The liquid junction potential between the pipette solution and the bath solution was estimated to be approximately 10 mV, as reported previously [32].
Immunofluorescence staining of NRVMs
Immunoconfocal fluorescence was used to assess Kv4.3 expression in NRVMs. Cultured cells were fixed and permeabilized with 0.5% TritonX-100. After washing with phosphate-buffered saline (PBS), cells were preincubated with 1% bovine serum albumin (BSA) to block non-specific binding. Monoclonal anti-cardiac α-actin antibody (1:500; Sigma) was used to identify cardiac myocytes. Cells were then incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (1:100; BD Pharmingen). For double immunolabeling, secondary labeling was performed with 1:500 anti-potassium-channel Kv4.3 antibody followed by Cy3-conjugated goat anti-rabbit IgG (1:100; BD Pharmingen). Cells were then treated with 4,6-Diamidino-2-phenylindole dihydrochloride hydrate (DAPI) to stain cell nuclei and analyzed by fluorescence microscopy (Department of Anatomy, FMMU).
Western blots
Each sample of left ventricular tissue was divided into subepicardial and subendocardial myocardium from the equivalent region of the animal heart. NRVMs and tissue samples were lysed in lysis buffer containing 50 mM Tris, 150 mM NaCl, 1% Nonidet P-40, 0.25% superoxide dismutase, 1 mM EDTA, 1 mM NaF, 1 mM Na3VO3 and 1 mM phenylmethylsulphonyl fluoride and included a proteinase inhibitor cocktail (Roche). Samples were evaluated by SDS-PAGE. Briefly, the total protein of the samples was determined before being subjected to polyacrylamide gel electrophoresis and being transferred to nitrocellulose (NC) membrane. The NC membrane was immunoblotted with anti-Kv4.3 antibody (Sigma; 1:1,000), and anti-GAPDH antibody (Sigma; 1:10,000). GAPDH protein expression served as a loading control. Bands were evaluated by densitometry using the Odyssey infrared imaging system (LI-COR).
Quantitative real-time RT-PCR
Total RNA from NRVMs, subepicardial and subendocardial regions of the ventricle tissue was extracted with TRIzol Reagent (Invitrogen). RNA was quantified sectrophotometrically at 260 nm. A 20 µl reaction mixture (Invitrogen) was used for reverse transcription. The reverse transcription products were then used as templates for PCR with gene-specific primers according to previously reported methods [32, 33] (Table 1). Real-time PCR was performed using the 7500 Sequence Detector Real-Time PCR System (Applied Biosystems). Cycling conditions were as follows: 10 min at 95°C as an initial step, 15 sec at 95°C and 1 min at 60°C for 40 cycles. Fluorescence signals of genes were recorded during the elongation phase of each PCR cycle. Melting curve analysis was used to confirm the amplification specificity. GAPDH expression served as the internal control. Each gene was quantified in duplicate. Real-time PCR data were analyzed by the comparative CT method as reported [34].
Primers for real-time RT-PCR
| Gene | Sense 5'-3' | Antisense 5'-3' |
|---|---|---|
| Kv4.3 | CCACGAGTTTATTGATGAGCAGAT | TGTTTTGCAGTTTGGTCTCAGTC |
| ANP | AGTGCGGTGTCCAACACAGAT | TTCTCCTCCAGGTGGTCTAGCA |
| GAPDH | TGCACCACCAACTGCTTAG | GATGCAGGGATGATGTTC |
Calcineurin phosphatase activity assay
Calcineurin phosphatase activity was measured by the dephosphorylation rate of a synthetic phosphopeptide substrate using a calcineurin assay kit (Enzo Life Sciences International) according to the instructions provided by the manufacturer. The level of released free phosphate was detected colorimetrically at 620 nm using a plate reader.
Statistical analysis
Data are presented as the mean ± SEM. One-way ANOVA was used for statistical analysis and P<0.05 was considered statistically significant.
Results
Effects of simvastatin on ventricular hypertrophy
In this study, AAB caused cardiac left ventricular hypertrophy in vivo 7 weeks after surgery (Fig. 1 and Table 2). The LVWI was significantly increased (46 %) in the AAB group. Compared with the sham group, the LVPWTd and IVSTd in the AAB group were increased by 42% and 49%, respectively. Compared with the AAB group, application of simvastatin decreased the LVWI, LVPWTd and IVSTd by 20%, 14.7% and 20%, respectively.
Echocardiograms showing the vascular and left ventricular profile. Panel A, representative cardiac dilatation. Panel B, representative abdominal aortic banding vascular ultrasound imaging. Color Doppler flow and narrow frequency spectrum image of AAB indicating angiostegnosis and accelerating blood flow. Panel C, representative echocardiographic M-mode images. All images shown were obtained in the rat.
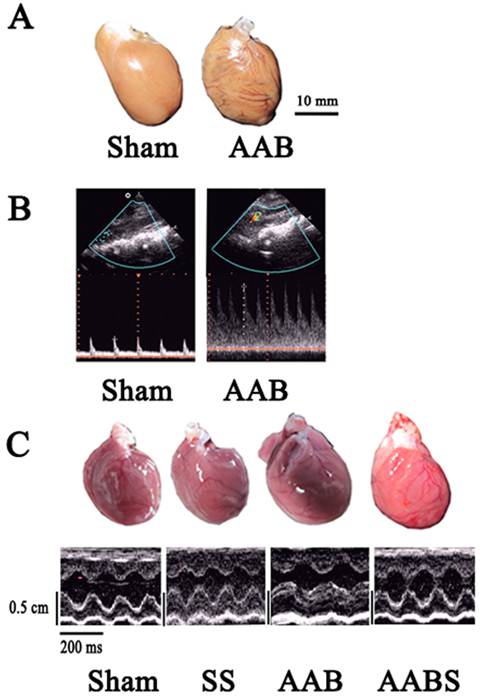
Comparison of left ventricular hypertrophy among four groups.
| LVWI g/kg | LVPWTd mm | IVSTd mm | LVFS % | |
|---|---|---|---|---|
| Sham | 2.235±0.081 | 0.1415±0.0205 | 0.1600±0.0159 | 58.98±11.75 |
| SS | 2.229±0.084 | 0.1352±0.0233 | 0.1638±0.0154 | 67.33±6.19 |
| AAB | 3.269±0.066* | 0.2007±0.0349* | 0.2377±0.0255* | 44.06±10.77* |
| AABS | 2.631±0.168# * | 0.1712±0.0175# * | 0.1907±0.0067# * | 56.61±6.31# * |
Left ventricular profile in rats from different groups. Data are expressed as the mean ± SEM, n = 10. * P<0.05 vs. Sham group; # P<0.05 vs. AAB group.
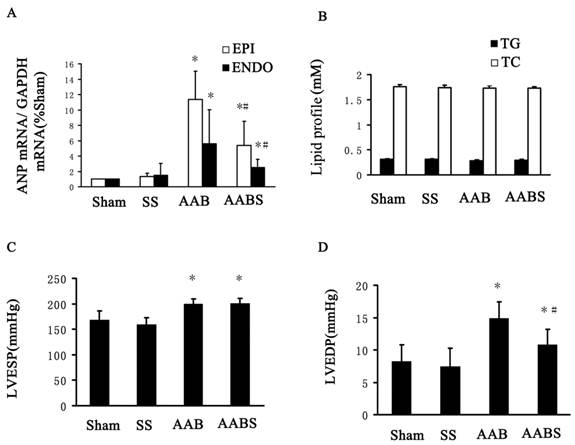
In the AAB and AABS group, atrial natriuretic peptide (ANP) mRNA levels in subendocardial and subepicardial myocardium were increased significantly compared with the sham group. In the AABS group, the ANP mRNA levels were decreased compared with the AAB group. No significant difference in ANP mRNA was observed between the sham and SS groups (Fig. 2A).
Bar graphs showing ANP mRNA expression, TC, TG, LVESP and LVEDP. Panel A, mRNA expression of ANP in the left ventricle. Panel B, lipid profile (including TC and TG) from blood samples. Panel C and D, LVESP and LVEDP. All results shown were obtained in the rat. Data are expressed as the mean ± SEM, n = 10. * P<0.05 vs. Sham group; # P<0.05 vs. AAB group. EPI, subepicardial myocardium; ENDO, subendocardial myocardium.
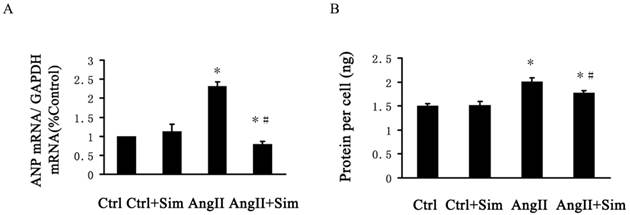
The protein content per cell in AngII-treated NRVMs was increased by 34% compared with the control group. The increased protein content was partially reversed by 12% in the AngII+Sim group compared with the AngII group (Fig. 3B). No significant difference in protein content per cell was observed between the control group and the Ctrl+Sim group. Furthermore, pretreatment with simvastatin inhibited the AngII induced increases in ANP mRNA expression, which is a genetic marker of cardiac hypertrophy, in NRVMs (Fig. 3A).
Bar graphs showing ANP mRNA expression and protein per cell in NRVMs. Data are expressed as the mean±SEM, n = 3. * P<0.05 vs. Ctrl group; # P<0.05 vs. AngII group.
Effects of simvastatin on lipid profiles (TC, TG), LVESP and LVEDP
In this study, no significant differences were observed in blood TG and TC levels among all groups of animals studied (Fig. 2B). LVEDP and LVESP measurements are shown in Fig. 2C and Fig. 2D. In the AAB group, LVESP and LVEDP were increased by 19% and 81% respectively compared with the sham group. Interestingly, LVEDP, but not LVESP was significantly reduced by 27% in the AABS group compared with the AAB group. In accordance with the prior study [35], simvastatin-treated rats exhibited a reduction in LVWI and a significant reduction in LVEDP, which suggests that simvastatin has beneficial hemodynamic effects.
Effects of simvastatin on Kv4.3 expression
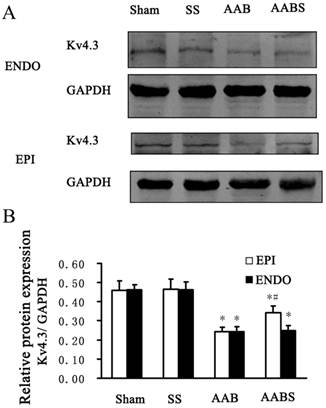
In the AAB group, Kv4.3 protein levels were consistently reduced in subendocardial and subepicardial myocardium (by 47% in both groups) compared with the sham group. Compared with the AAB group, the reduction in subepicardial myocardium Kv4.3 expression appeared to be partially reversed by 40% in the AABS group. However, there was no significant difference in subendocardial myocardium Kv4.3 expression between AAB and AABS groups (Fig. 4).
Representative Western blots (Panel A) and relative bar graphs (Panel B) showing alterations in Kv4.3 expression in the rat left ventricle. The band intensities of Kv4.3 were normalized against that of GAPDH. Data are expressed as the mean ± SEM, n = 10. * P<0.05 vs. Sham; # P<0.05 vs. AAB. EPI, subepicardial myocardium; ENDO, subendocardial myocardium.
In the AAB group, the Kv4.3 mRNA levels in the subepicardial and subendocardial myocardium were significantly reduced by 90% and 92%, respectively, compared with the sham group (Fig. 5A). Compared with the AAB group, a 4-fold increase in Kv4.3 mRNA was observed in subepicardial myocardium in the AABS group. Interestingly, no significant difference was detected in the Kv4.3 levels in subendocardial myocardium in the AABS group compared with the AAB group (Fig. 5A).
Real-time RT-PCR bar graphs showing alterations of relative Kv4.3 mRNA expression in the rat left ventricle and in NRVMs. Panel A shows the relative mRNA expression of Kv4.3 in the rat left ventricle. Data are expressed as the mean±SEM, n = 10. * P<0.05 vs. Sham of respective group; # P<0.05 vs. AAB of respective groups. Panel B represents the relative expression of Kv4.3 mRNA in NRVMs. Data are expressed as the mean ± SEM, n = 3. * P<0.05 vs. Ctrl group; # P<0.05 vs. AngII group. EPI, subepicardial myocardium; ENDO, subendocardial myocardium.
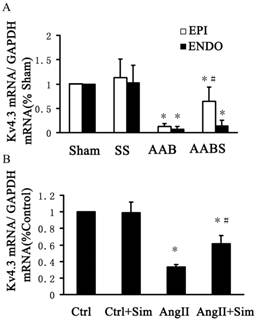
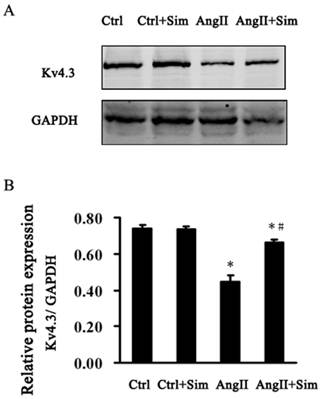
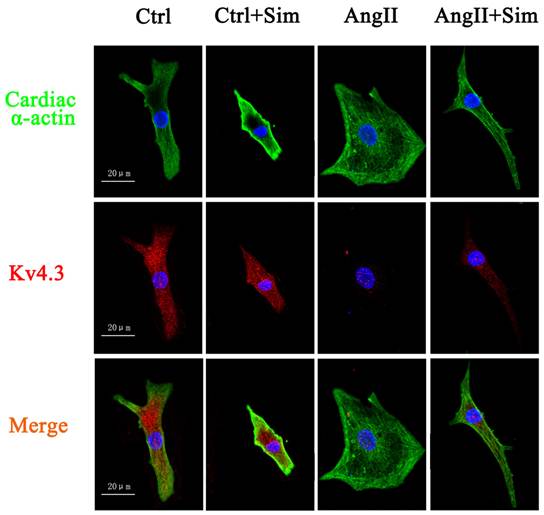
Furthermore, Kv4.3 protein and mRNA expression was detected in NRVMs. The protein and mRNA expression of Kv4.3 in the AngII group was significantly decreased by 39% and 67%, respectively, compared with control group. Pretreatment with simvastatin partially suppressed the reduction in Kv4.3 protein and mRNA expression by 86% and 48% respectively compared with the AngII group (Fig. 6 and Fig. 5, both B panels). Immunofluorescence staining of NRVMs also showed that the reduction in Kv4.3 protein expression was reversed in the AngII+Sim group compared with the AngII group (Fig. 7).
Representative Western blots (Panel A) and relative bar graphs (Panel B) showing alterations of Kv4.3 in NRVMs. Data are expressed as the mean ± SEM, n = 3. * P<0.05 vs. Ctrl group; # P<0.05 vs. AngII group.
Expression of Kv4.3 protein in NRVMs. Immunoreactivity for Kv4.3 (red) and cardiac α-actin (green) was assessed by immunofluorescence 24 h after AngII treatment. Blue staining represents DAPI-stained nuclei. Photographs are representative of nine cultures from more than three separate experiments.
Effects of simvastatin on Ito in NRVMs
Simvastatin has been reported to significantly reduce vulnerability to ventricular fibrillation and shortens the duration of ventricular tachycardia [23, 24]. However, the effects of simvastatin on electrophysiology remodeling in hypertrophied heart are not fully understood. Therefore, the effects of simvastatin on Ito in NRVMs were investigated in this study.
Following the blockade of INa and ICa by TTX and CdCl2, the outward K+ current densities were recorded (Fig. 8A and Fig. 8B). Ito was quantified as the difference in the peak current minus Isus, which was recorded at the end of the 500-ms voltage step [32]. AngII suppressed Ito density at voltages ranging from +20 to +60 mV compared with control (Fig. 8A, 8B, 8C and 8D). At +60 mV, Ito was decreased by 59% from 11.85 ± 0.75 pA/pF in the Ctrl group to 4.91 ± 0.67 pA/pF in the AngII group. Simvastatin partially prevented AngII induced Ito suppression at voltages ranging from +40 to +60 mV. At +60 mV, simvastatin reversed Ito by 48% from 4.91 ± 0.67 pA/pF in the AngII group to 7.26 ± 0.97 pA/pF in the AngII+Sim group. Simvastatin alone had no effect on Ito compared with the control group (Fig. 8E and 8F).
Simvastatin participates in the regulation of Ito in NRVMs. Panels A and B, typical current traces of outward K+ in the control (Panel A) and AngII-treated (Panel B) groups. Panel C, Ito (peak current densities minus Isus) between the control and AngII-treated groups at voltages ranging from -40 to +60 mV. Panel D, average current density at +60 mV between the control and AngII-treated groups. Panel E, Ito of four experimental groups at voltages ranging from-40 to +60mV. Panel F, average current density at +60 mV in different groups. Data are expressed as the mean ± SEM, n = 10. * P<0.05 vs. Ctrl group; # P<0.05 vs. AngII group.
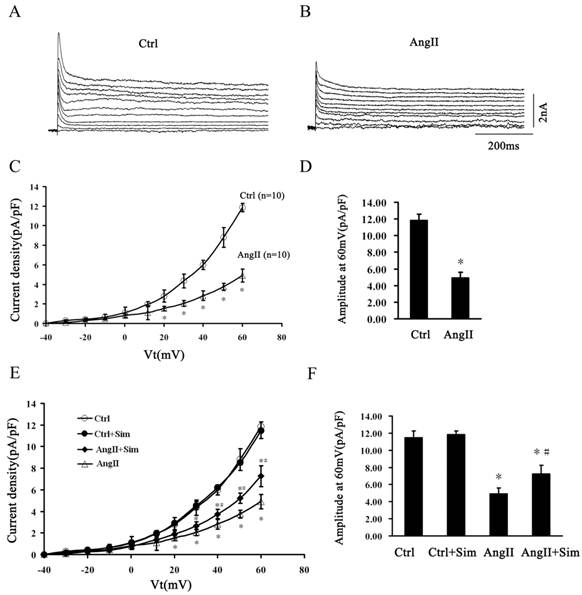
These data suggest that simvastatin alleviated Kv4.3 downregulation and Ito current suppression in hypertrophied NRVMs.
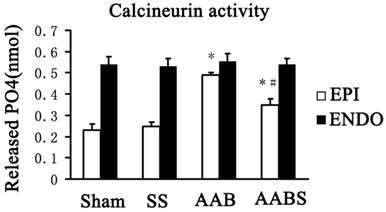
Calcineurin activity
The data obtained in this study revealed that simvastatin reversed Kv4.3 downregulation in the subepicardial myocardium but not the subendocardial myocardium. However, the molecular mechanisms of this effect remain to be elucidated. Previous study has linked calcineurin with changes in a variety of ion channels although the therapeutic potential of targeting calcineurin for correcting cardiac electrical disturbance has not yet been formulated [36]. Therefore, calcineurin activity was assayed in this study. It was observed that simvastatin treatment suppressed the increase in subepicardial myocardium calcineurin activity in the AAB group. No differences in subendocardial myocardium calcineurin activity were detected between the four groups (Fig. 9).
Bar graphs showing calcineurin activity in the rat left ventricle. Data are expressed as the mean ± SEM, n = 3. * P<0.05 vs. Sham of respective groups; # P<0.05 vs. AAB of respective groups. EPI, subepicardial myocardium; ENDO, subendocardial myocardium.
Discussion
In this study, the main findings were as follows: 1) rats exhibited significant left ventricular hypertrophy seven weeks after AAB, which was associated with a clear decrease in Kv4.3 expression in subendocardial and subepicardial myocardium of the left ventricle; 2) simvastatin partially reversed the downregulation in Kv4.3 expression in subepicardial myocardium but not in subendocardial myocardium and was associated with inhibition of cardiac hypertrophy; and 3) simvastatin partially alleviated the AngII induced hypertrophy, Kv4.3 downregulation and Ito current reduction in NRVMs. Therefore, it is hypothesized that simvastatin modulates Ito currents by regulating Kv4.3 expression in hypertrophied cardiomyocytes.
Left ventricular hypertrophy (LVH) in response to elevated systemic pressure forms an adaptive and reversible mechanism that allows the heart to maintain cardiac output in the face of external stress [2, 36, 37]. However, a large number of studies demonstrate that patients suffering from LVH have a greater risk of cardiac failure compared with normotensive patients and left ventricular regression is clearly associated with a reduction in the risk of adverse cardiovascular events [38-42]. LVH is also the most common cardiac manifestation followed by arrhythmias. It has been reported recently that hypertrophy is associated with significant repolarization delays and increased mechanical function in a rat model of pressure overload hypertrophy. Furthermore, the pressure overload hypertrophy secondary to aortic constriction promotes the incidence of arrhythmias [2].
In this study, the AAB model was used to generate LVH [43], which is characterized by increased LVWI, LVPWTd, IVSTd and ANP mRNA expression in the left ventricle. ANP is used as a marker of cardiac hypertrophy due to its expression in cardiac hypertrophy rather than in normal adult myocardium [33]. Furthermore, in the present study, simvastatin suppression of LVH has been shown in AAB rats. This study showed that Kv4.3 expression was reduced in AAB rats and that this effect was partially reversed by simvastatin, predominantly in subepicardial myocytes. Interestingly, this reduction in Kv4.3 expression levels was contradictory to a report that demonstrated that Kv4.3 mRNA but not protein expression was significantly reduced in aldosterone-induced hypertrophy [1]. Thus, although some responses to AAB mimic the response of pathological stimuli, such as hypertrophy and electrical remodeling, differences are observed in the signaling pathways and eventual outcomes of physiological and pathological stimuli [44]. These so-called complexities may reflect that diverse mechanisms underlie the modulation of Kv4.3 in different animal models.
Total Kv4.3 protein levels are not always consistent with Ito currents. Therefore, in order to assess the regulatory effect of simvastatin on the electrophysiological profile of cardiomyocytes, the Ito density in single cardiomyocytes under whole-cell patch-clamp conditions was investigated and immunofluorescence staining was used to assess Kv4.3 expression in the cytolemma (Fig. 7 and 8). In accordance with previous reports [44-46], this study showed that Ito density was reduced in response to hypertrophic stimuli. A reduction in membrane Kv4.3 expression was also observed in hypertrophied NRVMs and simvastatin partially reversed the reduction in Ito density and Kv4.3 expression. Simvastatin alone did not alter Ito density and Kv4.3 expression in AngII-free cells. Kv4.3 is the tetraethylammonium chloride-sensitive cardiac transient outward current Ito subunit. Ito mediates phase I repolarization in cardiac action potential (AP) and influences the overall AP configuration and duration [32]. Moreover, mice that are devoid of ventricular Ito are highly susceptible to arrhythmias [47]. It can be speculated that reduced Kv4.3 expression contributes to reduced Ito currents and loss of the repolarization in the cardiac AP, making Ca2+ release in ventricular myocytes less synchronous and resulting in increased susceptibility to cardiac arrhythmias [48]. These results implied that Kv4.3 acts as a potential substrate that renders the heart susceptible to arrhythmias. Thus, the results of this study indicate that simvastatin may alter electrophysiology in the pathological situation, partially through its effect on Kv4.3. The data obtained in this study demonstrate the effects of simvastatin treatment on Ito levels and Kv4.3 expression and provide support for the existence of cross-talk between simvastatin and electrical remodeling. Furthermore, these data suggest that the anti-arrhythmia effect of simvastatin in cardiac hypertrophy may be associated with its effects on Ito, which are predominantly mediated by Kv4.3.
However, the mechanism underling the simvastatin-mediated effects on Kv4.3 expression and Ito in cardiac hypertrophy is complex. Previous studies have shown the Kv4.3 mRNA reduction observed in hypertrophy results from mRNA destabilization [15]. Furthermore, studies have shown that statins (including simvastatin) reduce mRNA destabilization in several situations [49-52] and it is hypothesized that this is the mechanism responsible for the alleviation in Kv4.3 expression reduction observed in simvastatin-treated hypertrophied rats. It has also been reported that the potassium channel antagonist, 4-aminopyridine, enhanced the activation of extracellular signal-regulated kinases (ERK) [53], which phosphorylate the Kv channel subunit [54-56], resulting in a reduced current [57]. Previous study has shown that simvastatin suppressed ERK phosphorylation and inhibited AngII induced cardiomyocyte hypertrophy [30], indicating that simvastatin may alleviate Ito reduction by suppressing ERK phosphorylation, resulting in alteration in the biophysical properties of these channels. However, Ren et al. showed that reduced expression of Kv channels proteins is accompanied by reduced phosphorylation of extracellular ERK [58]. These contradictory observations may be attributed to different experimental conditions. Furthermore, Martins et al. [59] demonstrated increased rat heart alkaline phosphatase (ALP) activity in response to statins in vitro. Therefore, it can be hypothesized that statin-mediated dephosphorylation of Kv channels in cardiomyocytes occurs through regulation of alkaline phosphatase activity. Thus, simvastatin may also increase alkaline phosphatase activity, resulting in Kv channel dephosphorylation, which are in turn, related to the beneficial effects of simvastatin. However, further studies are required to determine the putative effects of statins on ALP activity in the cardiac tissue.
It should be noted that the reversal in the reduction in Kv4.3 expression was observed in the subepicardial myocardium but not the subendocardial myocardium in simvastatin-treated rats showed in this study. It has been well documented that Kv4.2 mRNA expression varies across the ventricular wall of the rat and Kv4.2 protein expression parallels the regional heterogeneity in Ito density, while Kv4.3 is expressed at equivalent levels across the ventricular wall [60, 12]. However, different potassium channel subunits have been shown to be responsible for the cardiac Ito in higher mammalian species compared with the rodent [12]. It has been reported that Kv4.3, as the pore-forming subunit, appears to be solely responsible for Ito in canine and human heart [61, 62]. Thus, since the distribution of Kv4.3 across the ventricle is homogeneous, blockade of this channel by specific drugs may not alter the normal heterogeneity of Ito current [63]. In our study, with the exception of the AABS group there were no significant differences in Kv4.3 expression between the subendocardial and subepicardial myocardium in the sham, SS and AAB groups. Compared with the AAB group, the AABS group exhibited significantly higher Kv4.3 expression in subepicardial myocardium. These data strongly support the hypothesis that simvastatin altered transmural Kv4.3 expression and may modulate the electrophysiology in hypertrophied heart.
In this study, it was demonstrated that calcineurin activity in subepicardial myocardium was lower than that in subendocardial myocardium in the sham and SS groups. Furthermore, the calcineurin activity was increased in subepicardium myocardium in the AAB group. These data are consistent with the previous reports [64, 65]. Furthermore, in the AABS group, calcineurin activity was partially reversed by simvastatin in subepicardial myocardium. This was accompanied by recovery of transmural Kv4.3 expression in the AABS group. However, it has been reported that simvastatin induced PC3 prostate cancer cell necrosis is mediated by calcineurin [66]. Thus, although it is speculated that calcineurin is involved in the simvastatin induced recovery of Kv4.3, the effects of simvastatin on calcineurin activity remain to be elucidated and experimental discrepancies may result from differences in experimental conditions and cells.
In summary, this study demonstrated that simvastatin modulated Kv4.3 expression and Ito currents in hypertrophied rat hearts. However, it should be noted that interpretation of these results is subject to a number of limitations. Firstly, these results are applicable only to rats due to species differences in Kv4.3 expression and confirmation of similar changes in human hypertrophied cardiomyocytes requires further investigation. Secondly, although the Kv4.3 expression reported here are from the two extreme surfaces of the left ventricular myocardium, it should be noted that the small size of the rat left ventricle may have resulted in inaccuracies in tissue sampling defined as” subepicardial” and “subendocardial” myocardium, which might have significantly influenced the results. Thirdly, due to the limitation of research conditions, monophasic APs were not tested in the whole heart. Furthermore, in this study, expression of another Ito alpha subunit, Kv4.2, was not investigated due to the lack of availability of a specifically reactive detection antibody. Therefore, although Kv4.3 and Ito may contribute to repolarization of the early plateau phase in that even small currents can have significant effects on the balance of currents during the plateau of the AP, there are numerous other ion channels and currents known to be active in the whole heart. Thus, the effect of simvastatin on the electrophysiologic parameters of the whole heart and the surface ECG on the development of pathological electrical stimulation requires further investigation.
Acknowledgements
The authors are grateful to Yunyan Duan of the Department of Ultrasonography and Yan Xu of Department of Clinical Laboratory in Xijing Hospital for their kind help and suggestions.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Dartsch T, Fischer R, Gapelyuk A. et al. Aldosterone induces electrical remodeling independent of hypertension. Int J Cardiol. 2011 [Epub ahead of print]
2. Jin H, Chemaly ER, Lee A. et al. Mechanoelectrical remodeling and arrhythmias during progression of hypertrophy. FASEB J. 2010;24:451-63
3. Wickenden AD, Kaprielian R, Kassiri Z. et al. The role of action potential prolongation and altered intracellular calcium handling in the pathogenesis of heart failure. Cardiovasc Res. 1998;37:312-23
4. Marionneau C, Brunet S, Flagg TP. et al. Distinct cellular and molecular mechanisms underlie functional remodeling of repolarizing K+ currents with left ventricular hypertrophy. Circ Res. 2008;102:1406-15
5. Deal KK, England SK, Tamkun MM. Molecular physiology of cardiac potassium channels. Physiol Rev. 1996;76:49-67
6. Panama BK, Latour-Villamil D, Farman GP. et al. Nuclear factor kappaB downregulates the transient outward potassium current I(to,f) through control of KChIP2 expression. Circ Res. 2011;108:537-43
7. Delpon E, Cordeiro JM, Nunez L. et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol. 2008;1:209-18
8. Kassiri Z, Zobel C, Nguyen TT. et al. Reduction of I(to) causes hypertrophy in neonatal rat ventricular myocytes. Circ Res. 2002;90:578-85
9. Niwa N, Nerbonne JM. Molecular determinants of cardiac transient outward potassium current (I(to)) expression and regulation. J Mol Cell Cardiol. 2010;48:12-25
10. Sah R, Ramirez RJ, Oudit GY. et al. Regulation of cardiac excitation-contraction coupling by action potential repolarization: role of the transient outward potassium current (I(to)). J Physiol. 2003;546:5-18
11. Swynghedauw B, Baillard C, Milliez P. The long QT interval is not only inherited but is also linked to cardiac hypertrophy. J Mol Med (Berl). 2003;81:336-45
12. Guo W, Li H, Aimond F. et al. Role of heteromultimers in the generation of myocardial transient outward K+ currents. Circ Res. 2002;90:586-93
13. Dixon JE, Shi W, Wang HS. et al. Role of the Kv4.3 K+ channel in ventricular muscle. A molecular correlate for the transient outward current. Circ Res. 1996;79:659-68
14. Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270-83
15. Zhou C, Vignere CZ, Levitan ES. AUF1 is upregulated by angiotensin II to destabilize cardiac Kv4.3 channel mRNA. J Mol Cell Cardiol. 2008;45:832-8
16. Pedersen TR, Kjekshus J, Berg K. et al. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). 1994. Atheroscler Suppl. 2004;5:81-7
17. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet. 1994;344:1383-9
18. Guo WG, Su FF, Yuan LJ. et al. Simvastatin inhibits angiotensin II-induced cardiac cell hypertrophy: role of Homer 1a. Clin Exp Pharmacol Physiol. 2010;37:40-5
19. Liu J, Shen Q, Wu Y. Simvastatin prevents cardiac hypertrophy in vitro and in vivo via JAK/STAT pathway. Life Sci. 2008;82:991-6
20. Patel R, Nagueh SF, Tsybouleva N. et al. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 2001;104:317-24
21. Ravingerova T, Adameova A, Kelly T. et al. Changes in PPAR gene expression and myocardial tolerance to ischaemia: relevance to pleiotropic effects of statins. Can J Physiol Pharmacol. 2009;87:1028-36
22. Mithani S, Akbar MS, Johnson DJ. et al. Dose dependent effect of statins on postoperative atrial fibrillation after cardiac surgery among patients treated with beta blockers. J Cardiothorac Surg. 2009;4:61
23. Adameova A, Harcarova A, Matejikova J. et al. Simvastatin alleviates myocardial contractile dysfunction and lethal ischemic injury in rat heart independent of cholesterol-lowering effects. Physiol Res. 2009;58:449-54
24. Liu YB, Lee YT, Pak HN. et al. Effects of simvastatin on cardiac neural and electrophysiologic remodeling in rabbits with hypercholesterolemia. Heart Rhythm. 2009;6:69-75
25. Berni R, Savi M, Bocchi L. et al. Modulation of actin isoform expression before the transition from experimental compensated pressure-overload cardiac hypertrophy to decompensation. Am J Physiol Heart Circ Physiol. 2009;296:H1625-32
26. Panek AN, Posch MG, Alenina N. et al. Connective tissue growth factor overexpression in cardiomyocytes promotes cardiac hypertrophy and protection against pressure overload. PLoS One. 2009;4:e6743
27. Weinberg EO, Schoen FJ, George D. et al. Angiotensin-converting enzyme inhibition prolongs survival and modifies the transition to heart failure in rats with pressure overload hypertrophy due to ascending aortic stenosis. Circulation. 1994;90:1410-22
28. Berni R, Savi M, Bocchi L. et al. Modulation of actin isoform expression before the transition from experimental compensated pressure-overload cardiac hypertrophy to decompensation. Am J Physiol Heart Circ Physiol. 2009;296:H1625-32
29. Guo WG, Yu ZB, Xie MJ. Protein kinase Cdelta is possibly involved in the transition from hypertrophy to apoptosis of myocardiocytes. Sheng Li Xue Bao. 2006;58:269-74
30. Guo WG, Su FF, Yuan LJ. et al. Simvastatin inhibits angiotensin II-induced cardiac cell hypertrophy: role of Homer 1a. Clin Exp Pharmacol Physiol. 2010;37:40-5
31. Yao JA, Jiang M, Fan JS. et al. Heterogeneous changes in K currents in rat ventricles three days after myocardial infarction. Cardiovasc Res. 1999;44:132-45
32. Liu W, Deng J, Xu J. et al. High-mobility group box 1 (HMGB1) downregulates cardiac transient outward potassium current (Ito) through downregulation of Kv4.2 and Kv4.3 channel transcripts and proteins. J Mol Cell Cardiol. 2010;49:438-48
33. Wang C, Li L, Zhang ZG. et al. Globular adiponectin inhibits angiotensin II-induced nuclear factor kappaB activation through AMP-activated protein kinase in cardiac hypertrophy. J Cell Physiol. 2010;222:149-55
34. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101-8
35. Indolfi C, Di Lorenzo E, Perrino C. et al. Hydroxymethylglutaryl coenzyme A reductase inhibitor simvastatin prevents cardiac hypertrophy induced by pressure overload and inhibits p21ras activation. Circulation. 2002;106:2118-24
36. Liu HB, Yang BF, Dong DL. Calcineurin and electrical remodeling in pathologic cardiac hypertrophy. Trends Cardiovasc Med. 2010;20:148-53
37. Willey CD, Palanisamy AP, Johnston RK. et al. STAT3 activation in pressure-overloaded feline myocardium: role for integrins and the tyrosine kinase BMX. Int J Biol Sci. 2008;4:184-99
38. Zhao YH, Cui CC, Li Y. et al. Eukaryotic expression vector pcDNA3-HERG transfection inhibits angiotensin II induced neonatal rabbit ventricular myocyte hypertrophy in vitro. Zhonghua Xin Xue Guan Bing Za Zhi. 2009;37:931-5
39. Correale M, Fanigliulo AM, Ieva R. et al. Anti-hypertensive drugs and left ventricular hypertrophy: a clinical update. Intern Emerg Med. 2011 epub
40. Milan A, Caserta MA, Avenatti E. et al. Anti-hypertensive drugs and left ventricular hypertrophy: a clinical update. Intern Emerg Med. 2010;5:469-79
41. Sundstrom J, Lind L, Arnlov J. et al. Echocardiographic and electrocardiographic diagnoses of left ventricular hypertrophy predict mortality independently of each other in a population of elderly men. Circulation. 2001;103:2346-51
42. Verdecchia P, Schillaci G, Borgioni C. et al. Prognostic significance of serial changes in left ventricular mass in essential hypertension. Circulation. 1998;97:48-54
43. Gao S, Long CL, Wang RH. et al. K(ATP) activation prevents progression of cardiac hypertrophy to failure induced by pressure overload via protecting endothelial function. Cardiovasc Res. 2009;83:444-56
44. Stones R, Billeter R, Zhang H. et al. The role of transient outward K+ current in electrical remodelling induced by voluntary exercise in female rat hearts. Basic Res Cardiol. 2009;104:643-52
45. Shipsey SJ, Bryant SM, Hart G. Effects of hypertrophy on regional action potential characteristics in the rat left ventricle: a cellular basis for T-wave inversion? Circulation. 1997;96:2061-8
46. Volk T, Nguyen TH, Schultz JH. et al. Regional alterations of repolarizing K+ currents among the left ventricular free wall of rats with ascending aortic stenosis. J Physiol. 2001;530:443-55
47. Kuo HC, Cheng CF, Clark RB. et al. A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of I(to) and confers susceptibility to ventricular tachycardia. Cell. 2001;107:801-13
48. Harris DM, Mills GD, Chen X. et al. Alterations in early action potential repolarization causes localized failure of sarcoplasmic reticulum Ca2+ release. Circ Res. 2005;96:543-50
49. Tai SC, Robb GB, Marsden PA. Endothelial nitric oxide synthase: a new paradigm for gene regulation in the injured blood vessel. Arterioscler Thromb Vasc Biol. 2004;24:405-12
50. Jimenez AM, Millas I, Farre J. et al. Effect of HMG-CoA reductase inhibition on endothelial dysfunction-inducing protein in hypercholesterolemic rabbits. Rev Esp Cardiol. 2002;55:1151-8
51. de Frutos, Sanchez DML, Farre J. et al. Expression of an endothelial-type nitric oxide synthase isoform in human neutrophils: modification by tumor necrosis factor-alpha and during acute myocardial infarction. J Am Coll Cardiol. 2001;37:800-7
52. Jimenez A, Arriero MM, Lopez-Blaya A. et al. Regulation of endothelial nitric oxide synthase expression in the vascular wall and in mononuclear cells from hypercholesterolemic rabbits. Circulation. 2001;104:1822-30
53. Liu J, Feng S, Zhang L. et al. Expression and properties of potassium channels in human mammary epithelial cell line MCF10A and its possible role in proliferation. Sheng Li Xue Bao. 2010;62:203-9
54. Sutton GM, Patterson LM, Berthoud HR. Extracellular signal-regulated kinase 1/2 signaling pathway in solitary nucleus mediates cholecystokinin-induced suppression of food intake in rats. J Neurosci. 2004;24:10240-7
55. Varga AW, Anderson AE, Adams JP. et al. Input-specific immunolocalization of differentially phosphorylated Kv4.2 in the mouse brain. Learn Mem. 2000;7:321-32
56. Hu HJ, Alter BJ, Carrasquillo Y. et al. Metabotropic glutamate receptor 5 modulates nociceptive plasticity via extracellular signal-regulated kinase-Kv4.2 signaling in spinal cord dorsal horn neurons. J Neurosci. 2007;27:13181-91
57. Schrader LA, Ren Y, Cheng F. et al. Kv4.2 is a locus for PKC and ERK/MAPK cross-talk. Biochem J. 2009;417:705-15
58. Ren C, Wang F, Li G. et al. Nerve sprouting suppresses myocardial I(to) and I(K1) channels and increases severity to ventricular fibrillation in rat. Auton Neurosci. 2008;144:22-9
59. Negrao MR, Mota A, Azevedo I. et al. Statins and tissue mineralization: putative involvement of alkaline phosphatase. Med Hypotheses. 2006;67:524-8
60. Birnbaum SG, Varga AW, Yuan LL. et al. Structure and function of Kv4-family transient potassium channels. Physiol Rev. 2004;84:803-33
61. Bertaso F, Sharpe CC, Hendry BM. et al. Expression of voltage-gated K+ channels in human atrium. Basic Res Cardiol. 2002;97:424-33
62. Dixon JE, Shi W, Wang HS. et al. Role of the Kv4.3 K+ channel in ventricular muscle. A molecular correlate for the transient outward current. Circ Res. 1996;79:659-68
63. Faivre JF, Calmels TP, Rouanet S. et al. Characterisation of Kv4.3 in HEK293 cells: comparison with the rat ventricular transient outward potassium current. Cardiovasc Res. 1999;41:188-99
64. Rossow CF, Dilly KW, Yuan C. et al. NFATc3-dependent loss of I(to) gradient across the left ventricular wall during chronic beta adrenergic stimulation. J Mol Cell Cardiol. 2009;46:249-56
65. Tan X, Li J, Wang X. et al. Tanshinone IIA protects against cardiac hypertrophy via inhibiting calcineurin/NFATc3 pathway. Int J Biol Sci. 2011;7:383-9
66. Oliveira KA, Zecchin KG, Alberici LC. et al. Simvastatin inducing PC3 prostate cancer cell necrosis mediated by calcineurin and mitochondrial dysfunction. J Bioenerg Biomembr. 2008;40:307-14
Author contact
![]() Corresponding author: Qiangsun Zheng, M.D., Ph.D., Department of Cardiology and Arrhythmologic Center, Tangdu Hospital, Fourth Military Medical University, Xi'an 710038, China. Telephone: +86 29 84777422; Fax: +86 29 84777422; Email: sufeifeiedu.cn
Corresponding author: Qiangsun Zheng, M.D., Ph.D., Department of Cardiology and Arrhythmologic Center, Tangdu Hospital, Fourth Military Medical University, Xi'an 710038, China. Telephone: +86 29 84777422; Fax: +86 29 84777422; Email: sufeifeiedu.cn