Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(4):430-441. doi:10.7150/ijbs.3632 This issue Cite
Research Paper
Hydrogen Sulfide Mitigates Cardiac Remodeling During Myocardial Infarction via Improvement of Angiogenesis
Natia Qipshidze ![]() , Naira Metreveli, Paras K. Mishra, David Lominadze, Suresh C. Tyagi
, Naira Metreveli, Paras K. Mishra, David Lominadze, Suresh C. Tyagi
Departments of Physiology and Biophysics, University of Louisville School of Medicine, Louisville, Kentucky- 40202.
Received 2011-10-12; Accepted 2012-1-12; Published 2012-2-28
Abstract
Exogenous hydrogen sulfide (H2S) leads to down-regulation of inflammatory responses and provides myocardial protection during acute ischemia/reperfusion injury; however its role during chronic heart failure (CHF) due to myocardial infarction (MI) is yet to be unveiled. We previously reported that H2S inhibits antiangiogenic factors such, as endostatin and angiostatin, but a little is known about its effect on parstatin (a fragment of proteinase-activated receptor-1, PAR-1). We hypothesize that H2S inhibits parstatin formation and promotes VEGF activation, thus promoting angiogenesis and significantly limiting the extent of MI injury. To verify this hypothesis MI was created in 12 week-old male mice by ligation of left anterior descending artery (LAD). Sham surgery was performed except LAD ligation. After the surgery mice were treated with sodium hydrogen sulfide (30 μmol/l NaHS, a donor for H2S, in drinking water) for 4 weeks. The LV tissue was analyzed for VEGF, flk-1 and flt-1, endostatin, angiostatin and parstatin. The expression of VEGF, flk-1 and flt-1 were significantly increased in treated mice while the level of endostatin, angiostatin and parstatin were decreased compared to in untreated mice. The echocardiography in mice treated with H2S showed the improvement of heart function compared to in untreated mice. The X-ray and Doppler blood flow measurements showed enhancement of cardiac-angiogenesis in mice treated with H2S. This observed cytoprotection was associated with an inhibition of anti-angiogenic proteins and stimulation of angiogenic factors. We established that administration of H2S at the time of MI ameliorated infarct size and preserved LV function during development of MI in mice. These results suggest that H2S is cytoprotective and angioprotective during evolution of MI.
Keywords: Hydrogen sulfide, VEGF, flk-1, flt-1, endostatin, angiostatin, parstatin Myocardial infarction
Introduction
Cardiovascular disease (CVD) is the leading cause of death for human population. Myocardial infarction (MI), one of the most devastating consequences of CVD, leads to an upregulation of various growth factors [1, 2] and tissue remodeling. Vascular endothelial growth factor (VEGF), the most prominent member of a family of growth factors is strongly associated with angiogenic stimuli in different pathologies [3-8], thus most likely plays a role in left ventricular (LV) remodeling after MI. Recent observations showed that VEGF expression is also substantially increased in chronically ischemic myocardium [9, 10] as well as it is increased instantly after myocardial ischemia [11]. VEGF is thought to stimulate endothelial cell proliferation and to increase permeability by binding with high-affinity to two receptors expressed predominantly on endothelial cells such as the tyrosine kinase receptor - flk-1 [12] and the fms-like tyrosine kinase - flt-1[6]. Flt-1 may mediate vascular organization, and flk-1 mediates endothelial differentiation and proliferation [12, 13]. These receptors interact and modify biological effects of VEGF either positively or negatively, depending on the specific vascular bed, the experimental condition, and disease state [14]. Interestingly, impairment of endogenous angiogenesis in several pathologic and physiologic conditions, such as diabetes, hypercholesterolemia, and aging, has been associated with reduced production of VEGF [15-17]. In previous studies we demonstrated that MI causes hyperhomocysteinemia (HHcy) that is promoted by downregulation of 5-Methyltetrahydrofolate (5-MTHFR), cystathionine-β-synthase (CBS) and cystathionine-γ-lyase (CSE) [18]. Inhibition of angiogenesis recently has been described as a new deleterious effect of HHcy [19, 20] H2S is known to be produced in the vasculature by CSE and to mediate smooth muscle relaxation and subsequent vasodilatation [21]. Previous studies suggest that the heart is one of the major sources of the H2S [22]. Exogenous H2S provides myocardial protection during myocardial ischemia/reperfusion by opening K+ATP channels in cardiomyocytes and down-regulating of inflammatory responses [23]. Recent studies demonstrated that H2S decreased apoptosis, level of inflammation, and size of ischemia in the setting of myocardial ischemia reperfusion [24]. Although previous studies reported that endostatin and angiostatin expression and activity were significantly elevated in hypertrophied hearts [25], only a few studies have examined the role of parstatin during heart failure [26-29] in post-myocardial remodeling and dysfunction. We previously showed that MI causes upregulation of MMP-9, that increases levels of anti-angiogenic factors and contributes LV heart failure [18, 30]. In this study, we tested whether exogenous H2S reduced heart injury after stimulation of VEGF and inhibition of anti-angiogenic factors in MI mice.
Materials and Methods
Animals: Ten weeks old wild type (WT) C57BL6/J male mice were obtained from Jackson Laboratories (Bar Harbor, ME.,) and housed in the animal care facility at the University of Louisville with access to standard chow and tap water. At the age of 12 weeks with an approximate weight of 25-29 grams, animals underwent MI surgery or sham surgery. After surgery mice were divided in two groups: one group was given plain tap water and another were given NaHS (H2S donor; Sigma), which in aqueous solution releases H2S, in drinking water for 4 wk. Although there are differences in the physiological range of H2S and some reports say that it is much lower than the micromolar level, a 30 μmol/l concentration was supplemented to keep the plasma H2S in the physiological range, which is widely variable from 5 to 300 μmol/l. Diffusion of H2S into room air is minimal since its density is 18% higher than that of air. Furthermore, the drinking water was changed daily with fresh NaHS solution to provide adequate levels of H2S to the mice. To estimate daily intake of NaHS, previous studies from our lab reported that there was no difference in the consumption of water among the treated and untreated groups and also that there was an increase in plasma H2S concentration with exogenous supplementation. It was reported that oxidation of H2S due to air occurs. This yields slightly yellow colored water. Since we changed the drinking solution daily and did not see changes in water coloring (it was always yellow to the same extent) we presumed that the content of NaHS in drinking water was changing significantly [25]. To supplement H2S continuously water supply was changed every 24 h. At the end of the study, animals were euthanized, deeply anesthetized with thribromoethanol (confirmed by tail/toe pinch). For heart excision, mice were injected with 20 % KCl solution (0.2 ml/100 g body weight that causes heart arrest in diastole). Then hearts were excised. This was consistent with a veterinarian view of euthanasia. All experiments using mice were approved by and performed according to the Guidelines for the care and Use of laboratory Animals in University of Louisville, which strictly conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Mouse model of Myocardial Infarction: Animals were anesthetized with sodium pentobarbital (70mg/kg), intubated and ventilated with Harvard mini ventilator. Body temperature was maintained with a heating pad (TR 200, Fine Science Tools, Foster City, CA). A left thoracotomy was performed via the fourth intercostal space and the lungs retracted to expose the heart. After opening the pericardium, left anterior descending artery (LAD) was ligated with an 8-0 silk suture near its origin between the pulmonary outflow tract and the edge of the atrium. Ligation was deemed successful when the anterior wall of the left ventricle turned pale. The lungs were inflated by increasing positive end-expiratory pressure and the thoracotomy side closed in layers. Animals were kept on a heating pad until they recovered. Another group of mice underwent a sham ligation surgery. They had a similar surgical procedure done without tightening the suture around the LAD. The lungs were re-expanded and chest was closed. The animals were removed from the ventilator and allowed to recover on a heating pad. After surgery animals allowed to recover in the lab and monitored until their full recovery. Then animals were transferred to the animal care unit. Animals were checked daily for signs of pain or distress (hunch back, restricted movement, etc.) and carprofen at 5mg/kg subcutaneous was given after surgery once a day during 48 hours. Records of observations and procedures were kept in the lab.
Echocardiography: Before and 4 weeks after the surgery, two-dimensional (2-D) echocardiography was performed on mice using a Hewlett-Packard Sonos 5500 ultrasonography with a 15-MHz transducer. The mice were sedated with fresh made 2,2,2 tribromethanol ((100 mg/kg IP)TBE, Sigma T48 402; 240 mg/kg of Bd. wt.), and the chest was shaved. The mice were placed in a custom-made cradle on a heated platform in the supine or the left lateral decubitus position to facilitate echocardiography. For quantification of LV dimensions and wall thickness. LV short- and long-axis loops and LV 2-D image-guided M-mode traces at the level that yielded the largest diastolic dimension were digitally recorded. LV dimensions (LVDs) at diastole and systole (LVDd and LVDs, respectively) were measured from five cycles and averaged. Fractional shortening (FS) was calculated as [(LVDd−LVDs)/LVDd] ×100%. Fractional area change (FAC) was derived from end-diastolic and end-systolic areas of short-axis loops. According to the single-plane Simpson method, LV volumes at end-diastole (EDV) and end-systole (ESV) were derived from long-axis loops. Ejection fraction (EF) was calculated as [(EDV−ESV)/EDV] ×100%.
Western Blot analysis: Changes in protein content of CBS, CSE, endostatin, and angiostatin, induced by MI were assessed by Western blot analyses according to the method described earlier [16]. Briefly, frozen heart tissue washed twice with ice-cold PBS and lysed with ice-cold RIPA buffer (containing 5 mM of ethylenediamine-tetraacetic acid), which was supplemented with phenylmethylsulfonyl fluoride (1 mM) and protease inhibitor cocktail (1 ml/ml of lysis buffer). Protein content of the lysate was determined using the Bicinchronic Acid protein assay kit (Pierce, Rockford, IL). Equal amounts of protein (30 mg) were resolved on 12% SDS-PAGE and transferred onto a polyvinylidene difluoride membrane as described [31, 32]. The blots were incubated with monoclonal anti-mouse CBS (1:1500 dilution, C# H00000875-M01), and anti-mouse CSE (1:1500 dilution, C# H00001491-M02) antibodies (Novus Biologicals), anti -mouse Endostatin (1:1500), and anti-rat Angiostatin (1:1500) for 1 h at room temperature. After incubation, the proteins on blots were detected as described [31]. Membranes were stripped and re-probed for β-actin as a loading control. The blots were analyzed with Gel-Pro Analyzer software (Media Cybernetics, Silver Spring, MD) as described earlier [33]. The protein expression intensity was assessed by the integrated optical density (IOD) of the area of the band in the lane profile. To account for possible differences in the protein load, the measurements presented are the IOD of each band under study (protein of interest) divided by the IOD of the respective β-actin band.
Histology and confocal microscopy: Hearts were collected from experimental animals and thoroughly washed in PBS. For fixation, hearts were perfused with 4% paraformaldehyde and preserved in Peel-A-Way disposable plastic tissue embedding molds (Polysciens Inc, Washington, PA) filled with tissue freezing media (triangle Biomedical Sciences, Durham, NC) and stored at -700 C until analysis. Tissue sections (5 µm in thickness) were made using Leica CM 1850 Cryocut (Bannockburn, IL, USA). Sections were placed on Super frost plus glass slides, air-dried, and processed for histological and Immunohistochemistry (IHC) staining.
Histology: Masson's trichrome staining was performed on frozen tissue sections using a Masson's trichrome kit (Richard-Allan Scintific, Kalamazoo, MI) according to the manufacturer's recommendations. The heart muscle and vascular smooth muscle were stained a pink while the collagen was blue. The level of subcellular matrix collagen was assessed by measuring the optical density of blue color. For quantitation we choose the same distance from septal wall. For example we choose 2-3 mm from apex-septal wall. Because of that in MI we have whole part of scar and in MI+H2S we have border-zone. Ischemic zone itself in MI+H2S is very small compare to MI.
Immunohistochemistry: Immunohistochemistry was performed on frozen tissue sections using a standard IHC protocol. Primary antibodies applied overnight included anti-endostatin, anti-angiostatin, anti-parstatin, anti-Hcy, and anti- MTHFR antibodies (all from Abcam, Cambridge, MA). Secondary antibodies labeled with Alexa Fluor 488 and Texas Red (Invitrogen, Carlsbad, CA) were applied for immunodetection of these proteins. Stained slides were analyzed for fluorescence using a laser scanning confocal microscope (Olympus FluoView-1000, objective 60 xs) set at the appropriate filter settings. The total fluorescence (green or red) intensity in 5 random fields (for each experimental sample) was measured with image analysis software (Image-Pro Plus, Media Cybernetics). Fluorescence intensity values for each experimental group were averaged and presented as fluorescent intensity units (FIU).
Coronary angiography: We used barium sulfate for post mortem imaging of mice vasculature [30]. The size of barium particles range from 1-100 µm whereas most of the mice micro vasculature is less than 30 µm. Moreover barium sulfate is insoluble in water. To overcome this problem, we dissolved barium sulfate in acidic pH buffer and the mixture was used for intravascular infusion. All images were taken with Kodak 4000 MM image station. Dissected animals were placed in the X-ray chamber and angiogram images were captured with high penetrative phosphorous screen by 31 KVP X-ray exposure for 3 minutes using high resolution phosphorous screen and aperture settings of approximately 4.0, f-stop-12 and zoom of 40 mm.
RNA extraction: RNA from LV was extracted using Trizol method. The purity of RNA was estimate by NanoDrop (ND-1000) and only highly pure quality RNA (260/280-2.00 and 260/230-2.0) was used for RT-PCR.
Reverse transcription-polymerase chain reaction: The RT-PCR was performed using Promega kit, following their protocol (Promega Corporation, Madison, WI, USA,)[34]. The RT-PCR amplification program was [95 °C-0.50 min, 55 °C-1.00 min, 72 °C-1.00 min] × 34, 72 °C-5.00 min, 4 °C-∞.
The primers for RT-PCR were:
VEGF- F- 5' GGA CCC TGG CTT TAG TGC 3'
R- 5'CCG GCT TGG CGA TTT AG 3'
Flk-1- F- 5' GGT GCC CGC TCT TTG 3'
R- 5'TGT CTC AGT GGG GAT TGC 3'
Flt-1- F- 5' CCT GGC TA CCC GAT TCC 3'
R- 5' TCC CGC TTT GTT GAT GGC 3'
In vivo blood flow measurement: Mice were anesthetized with Pentobarbital (70mg/kg) were intubated, thoracic cavity was opened and heart was exposed for blood flow measurement. Body temperature was maintained between 37- 39 ºC using a heating pad. The part for measurement was chosen to be 1cm2. The probe was always placed 1cm below the ligation or (in Sham animals) the place where the ligation would have been if we performed it. Carprofen (5mg/kg subcutaneous) was administered after induction of anesthesia. Blood flow on the heart was measured by using LASER DOPPLER IMAGING flow meter.
Statistical analysis: Values are reported as mean ± SEM. Differences between groups were tested by two-way ANOVA. If ANOVA indicated a significant difference (P < 0.05), Tukey's multiple comparison test was used to compare group means and were considered significant if P < 0.05.
Results
To determine cardiac function, echocardiography measurements were obtained immediately before and 4 weeks after the surgery. Results are shown in Table 1. All variables were similar in animals of all groups immediately before the surgery and remained unaltered in the Sham group throughout the study period (Table 1). Supplementation with H2S did not alter fraction shortening (FS), LVDd, and LVDs in Sham mice (Table 1). Induction of MI resulted in decrease of FS and increase of LVDd and LVDs (Table 1). Treatment with H2S decreased dilation of heart and improved FS, LVDd and LVDs in mice with MI (Table 1).
Comparison of LV chamber dimension and fraction shortening (FS) in sham-operated (Sham), myocardial infarction-induced (MI), sham-operated and treated with H2S (Sham+H2S), and myocardial infarction-induced and treated with H2S (MI+H2S) mice.
| Experimental groups | Sham | Sham+H2S | MI | MI+ H2S |
|---|---|---|---|---|
| FS,% | 64±0.4 | 65±0.8 | 28±0.4* | 47±0.3*# |
| LVDd, mm | 2.17±0.5 | 2.16±0.3 | 3.2±0.5* | 2.85±0.5*# |
| LVDs, mm | 0.87±0.2 | 0.98±0.5 | 2.15±0.4* | 1.6±0.9*# |
Presented results are calculated from the following equation [(LVDd−LVDs)/LVDd] ×100%.
* P < 0.05 vs. Sham and Sham+H2S, # P<0.05 vs. MI; n=9 for all groups.
To determine the levels of fibrosis, histological analysis of collagen was performed on the LV. The intensity of trichrome blue stain demonstrated development of significant fibrosis on the LV in MI hearts as compared to Sham (Figure1 A and B). Treatment with H2S mitigated the formation of fibrosis on the left ventricle in MI+H2S group (Figure1 A and B).
Role of H2S in Hcy / MTHFR axis: The confocal image analyses of Hcy and MTHFR indicated that induction of MI increased HCY expression and decreased MTHFR expression in heart tissue compared to Sham (Figure 2 A and B). Supplementation with H2S restored this effect in MI+H2S group (Figure 2). Treatment of Sham mice with H2S (Sham+H2S) did not change expression of Hcy and MTHFR in their hearts compared to those in Sham group (Figure 2 A and B).
Role of H2S in expression of CBS and CSE: Induction of MI significantly decreases expression of CBS, and CSE in mice heart (Figure 2 C and D). Interestingly supplementation with H2S restored expression of CSE in hearts from mice with MI (Figure 2 C and D). Treatment of H2S did not change expression of CBS in mice with MI. Supplementation with H2S did not affect expression of CBS and CSE in Sham+ H2S mice compared to those in Sham group (Figure 2 C and D).
Angiogenic role of H2S: A total RNA Expression of VEGF and its receptors: flk-1 and flt-1 were quantified by PCR of RNAs extracted from the entire LV (including both infarcted and non-infarcted regions at various time intervals after coronary ligation). To facilitate comparison of absolute changes in expression between VEGF and its receptors, baseline expression of each gene in the normal LV was arbitrarily set at 100, and results from animals with infarctions were scaled in proportion. Coronary ligation induced a profound increase in VEGF expression over the time course of the study.
VEGF expression was increased as early as 1 d in MI heart (Figure 3B). It was dramatically (more than twice) increased in MI+H2S mice. Treatment with H2S did not have an effect in sham group. One week after MI, expression of VEGF in H2S-treated MI mice was similar to that in untreated MI mice, but it was still greater than in sham group (Figure 3B). Four weeks after MI expression of VEGF decreased to the level found in sham group (Figure 3B). Expression of flk-1 in MI mice treated with H2S after 1 day of surgery was greatly increased compared to Sham operated mice (Figure 3C). The expression of flk-1 was significantly increased in MI mice without H2S treatment compared to sham group, but it was less than in MI mice treated with H2S (Figure 3C). Treatment with H2S did not have an effect on sham group (Figure 3C). Flk-1 expression in MI mice treated with H2S decreased to the level of MI mice without H2S treatment after 1 week of surgery, but it was still greater than in sham group (Figure 3C). From 1 to 4 weeks after surgery, there were no changes in expression of flk-1 in MI mice treated with H2S (Figure 3C). In mice without H2S treatment, expression of flk-1 decreased to the level of sham group 4 weeks after surgery (Figure 3C). During the first day all MI groups experienced a sharp increase in Flt-1 expression (Figure 3D). After one week of surgery H2S-treated MI mice continued to increase expression in flt-1 at a reduced rate (Figure 3D). Untreated MI mice showed a decrease in expression of flt-1, but that was still greater than in sham group (Figure 3D). The expression of flt-1 was same for all the groups after 4 weeks of surgery (Figure 3D).
Anti anti-angiogenic role of H2S: The confocal image analysis indicated that induction of MI increased endostatin, angiostatin, and parstatin expression in heart tissue compared to those in Sham (Figure 1C, 1D, and 4). Supplementation with H2S decreased this effect in MI+H2S group (Figure 1C, 1D, and 4), although they were still greater than in Sham animals. Treatment of Sham mice with H2S (Sham+H2S) did not change expression of endostatin, angiostatin, and parstatin in heart compared to those in untreated Sham group (Figure 1C, 1D, and 4).
Heart wall anatomical changes in sham-operated (Sham), myocardial infarction-induced (MI), sham-operated and treated with H2S (Sham+H2S), and myocardial infarction-induced and treated with H2S (MI+H2S) animals. A: Examples of cross-sectional view of the left ventricular (LV) walls of Sham, Sham+H2S, MI and MI+H2S hearts. Note: No visible necrosis was found in the LV wall of hearts from Sham and Sham+H2S mice. B: Collagen-associated (blue) intensity changes in hearts from experimental animals. C: Changes in expression of parstatin protein contents, by immunohistochemistry analysis, in hearts from Sham, myocardial infarction-induced (MI), sham-operated and treated with H2S (Sham+ H2S), and myocardial infarction-induced and treated with H2S (MI+ H2S) mice. D Bar graph of changes in Sham, myocardial infarction-induced (MI), sham-operated and treated with H2S (Sham+H2S), and myocardial infarction-induced and treated with H2S (MI+H2S) mice heart tissue. The micrographs were taken under the identical set of conditions for all groups. * P < 0.05 vs. Sham, Sham+H2S, and MI+H2S. # p<0.05 vs. MI. n=6 for all group.
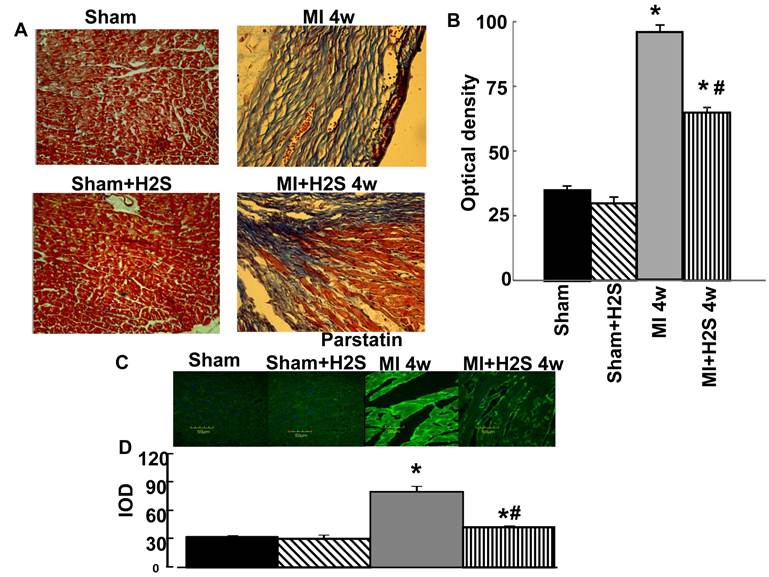
A. Changes in expression of MTHFR (green) and HCY (red) protein contents, by immunohistochemistry analysis, in hearts from Sham, myocardial infarction-induced (MI), sham-operated and treated with H2S (Sham+ H2S), and myocardial infarction-induced and treated with H2S (MI+ H2S) mice. B. Bar graph of changes in integrated optical density (IOD) in expression of MTHFR and Hcy in Sham, MI, Sham+H2S, and MI+ H2S mice heart tissues. The micrographs were taken under the identical set of conditions for all groups. C and D. Changes in expression of CSE and CBS proteins contents, by Western blot analysis, in hearts from Sham, myocardial infarction-induced (MI), sham-operated and treated with H2S (Sham+ H2S), and myocardial infarction-induced and treated with H2S (MI+ H2S) mice. Examples of Western blot images of the proteins studied and contents of β-actin in the respective samples and results of the Western blot analysis, relative protein expression is reported as ratio of integrated optical density (IOD) of each band to the IOD of the respective β-actin band. * P < 0.05 vs. Sham, Sham+H2S, and MI+H2S. # p<0.05 vs. MI. n=6 for all group.
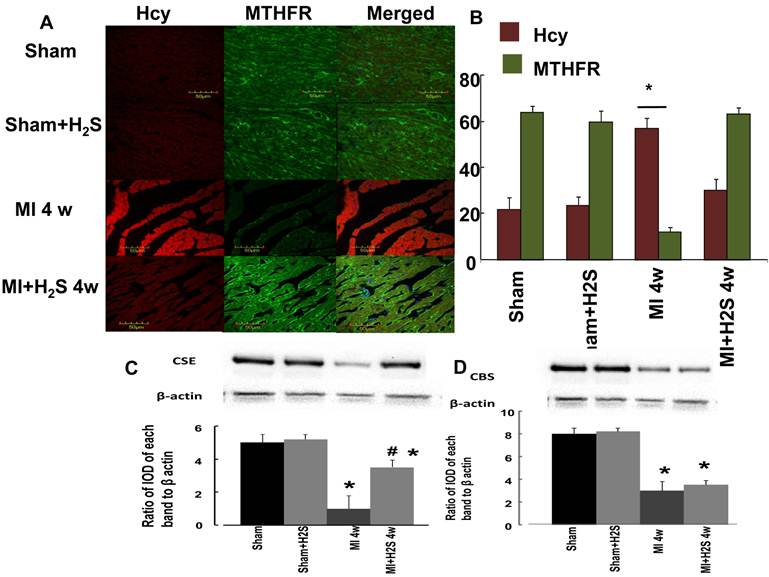
Changes in expression of VEGF, flk-1 and flt-1 RNA contents, by RT-PCR analysis, in hearts from Sham, myocardial infarction-induced (MI), sham-operated and treated with H2S (Sham+ H2S), and myocardial infarction-induced and treated with H2S (MI+ H2S) mice. A: Examples of PCR images of the RNA studied and contents of GAPDH in the respective samples. B: Results of the VEGF RT-PCR analysis. Relative RNA expression is reported as ratio of integrated optical density (IOD) of each band to the IOD of the respective GAPDH. C: Results of the flk-1 RT-PCR analysis. Relative RNA expression is reported as ratio of integrated optical density (IOD) of each band to the IOD of the respective GAPDH, D: Results of the flt-1 RT-PCR analysis. Relative RNA expression is reported as ratio of integrated optical density (IOD) of each band to the IOD of the respective GAPDH; * P < 0.05 vs. Sham, Sham+ H2S, and MI+ H2S. # P < 0.05 vs. MI. n=7 for all groups.
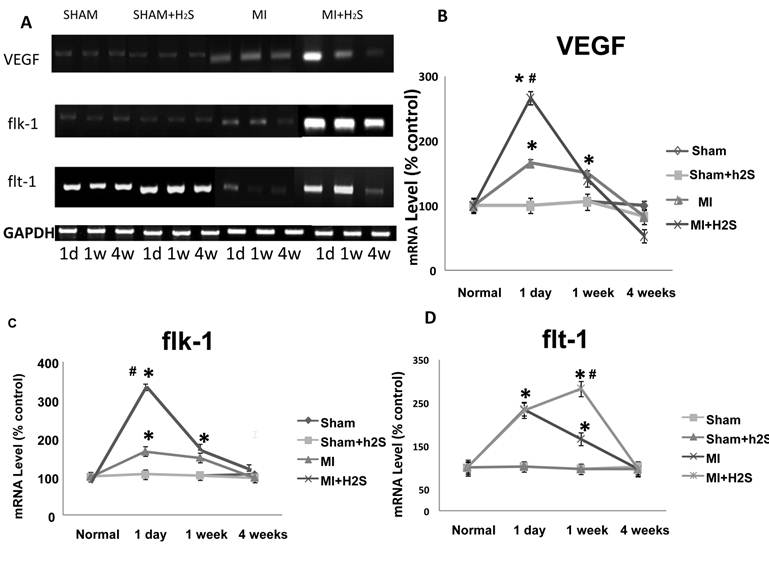
A. Changes in expression of endostatin (green) and angiostatin (red) protein contents, by immunohistochemistry analysis, in hearts from Sham, myocardial infarction-induced (MI), sham-operated and treated with H2S (Sham+ H2S), and myocardial infarction-induced and treated with H2S (MI+ H2S) mice. B. Bar graph of changes in integrated optical density (IOD) in expression of endostatin and angiostatin for all groups. C and D. Examples of Western blot images of the endostatin and angiostatin proteins studied and contents of β-actin in the respective samples. Relative protein expression is reported as ratio of integrated optical density (IOD) of each band to the IOD of the respective β-actin band. * P < 0.05 vs. Sham, Sham+H2S, and MI+H2S. # P<0.05 vs. MI. n=6 for all group.
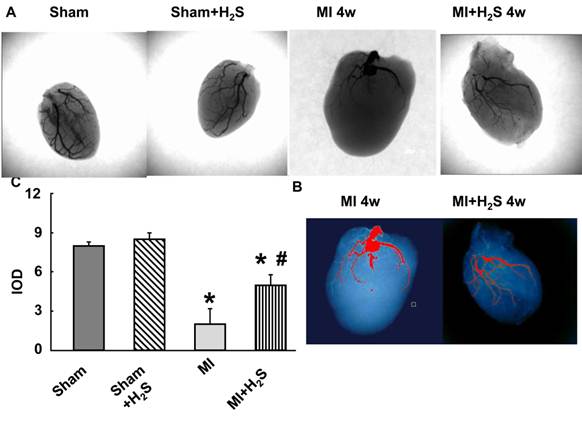
Collateralization: The X-ray vascular density data showed increase in angiogenic vessels at 4 weeks after MI in H2S-treated mice. Capillary rarefaction was observed at 4th weeks post MI without the treatment with H2S (Figure 5).
Barium-contrast X-ray images of the hearts from sham, myocardial infarction-induced (MI), sham-operated and treated with H2S (Sham+H2S), and myocardial infarction-induced and treated with H2S (MI+H2S) mice: Note: There is an increase in collateral vessels in the MI heart after H2S treatment compared to MI. A.X-ray images from experimental groups. B. Bar graph of changes in integrated optical density (IOD) in expression of Barium. C. analyzing x-ray pictures in image-pro system.
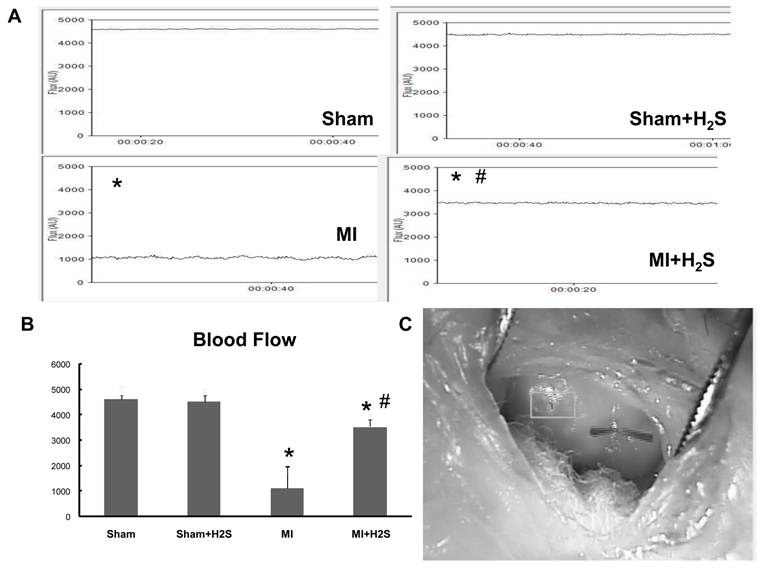
Blood flow measurement: To corroborate the vascular density data with the blood flow we measured blood flow by Laser Doppler. Induction of MI significantly impaired blood flow in MI mice (Figure 6). Supplementation with H2S increased blood flow in mice with MI (Figure 6). Supplementation with H2S did not affect blood flow in Sham+ H2S mice compared to those in Sham group (Figure 6).
Heart blood flow A: Examples of blood flow of the left ventricular (LV) walls of Sham, Sham+H2S, MI and MI+H2S hearts. Note: No visible changes were found in the LV wall's blood flow of hearts from Sham and Sham+H2S mice. B: Bar graph of changes in Flux (AU) in MI, Sham+H2S, and MI+ H2S mice heart tissues. C. Example of the blood flow measurement on the heart * P < 0.05 vs. sham group, # P < 0.05 vs. MI; n=6 for all groups.
Discussion
The principal findings of this study were that an exogenous administration of NaHS, a H2S donor at the time of MI limits the extent of MI in an in vivo mouse model. A dose-response study revealed that H2S displayed a biphasic reduction in infarct size as has previously been reported by Johansen et al [35, 36]. Importantly, the decrease in infarct size translated into reduced LV dilatation and improved LV function as measured by echocardiography.
H2S is generated from L-cysteine in reactions catalyzed by CBS or CSE. CSE is primarily responsible for most of the H2S production in the vasculature [37-39]. We show that exogenous H2S increased level of CSE in MI heart and limited the extent of injury.
Our data supported the hypothesis that the induction of CSE in heart tissue activates endogenous angiogenic agent VEGF via inhibition of anti-angiogenic factors like angiostatin, endostatin and parstatin. This promotes new vessels in heart and reduced infarct size in MI mice. Further our study demonstrated that VEGF expression was up-regulated in H2S treated mice with MI. VEGF receptors flk-1 and flt-1 were induced maximally in MI+H2S mice. Our results confirmed results of previous studies [40] and suggested that treatment with H2S after MI has beneficial effects on the heart. This may be related to the expression of angiogenic cytokines and their receptors, such as VEGF, flk-1 and flt-1. VEGF is a key growth factor in physiological angiogenesis and induces angiogenesis in myocardial ischemia and MI [2, 41-44]. Many studies have focused on the expression of angiogenic factors after infarction, especially VEGF and its receptors. VEGF was a strong mitogen, highly specific to endothelial cells. Its specific receptors include flt-1 and flk-1, which are receptor tyrosine kinases and predominantly expressed in endothelial cells [45]. In our studies, both temporal and spatial changes in flk-1 expression were observed after MI. Whereas flk-1 RNA was up-regulated at day 1 and down-regulated at 4th week after induction of MI. Expression of flt-1 was up-regulated continuously during first week and down-regulated after 4 weeks of MI induction. It was found that flt-1 played a role in the later stages of angiogenesis [45]. In contrast to flt-1, Flk-1 was expressed earlier. Flk-1 plays a major role in angiogenesis and endothelial cell survival [46].
The findings of this study indicate that myocardial levels of the antiangiogenic proteins angiostatin, endostatin and parstatin are increased in MI mice. The myocardial levels of angiostatin, endostatin and parstatin have a strong negative correlation to coronary collateralization in MI mice, which mitigates its reduction of endostatin, angiostatin and parstatin and is positively correlated with more collateralization in MI mice treated with H2S. Endostatin, a powerful endogenous inhibitor of angiogenesis, has been shown to reduce blood flow locally [47]. Endostatin, angiostatin and parstatin inhibits endothelial cell proliferation, migration and tube formation in vitro [47-49], and possesses potent inhibitory effects on tumor growth in vivo [50]. The inhibitory effects of anti-angiogenic factors on the expression of VEGF in tumor cells and on vascular permeability have been reported [50, 51].
In conclusion, we have shown that exogenously given H2S at the time of MI limits the extent of infarction. This protection is accompanied by a decrease in myocardial anti-angiogenic factors, such as endostatin, angiostatin and parstatin, and a preservation of growth factors such as VEGF, flk-1 and flt-1. These results suggest that H2S therapy may be a promising candidate for the treatment of MI.
Limitation: To confirm the theory exposure of CSE in heart tissue activates endogenous angiogenic agent VEGF via inhibition of anti-angiogennic factors, the investigation using anti VEGF antibody and/or CSE knockout mice is necessary. These investigations are in progress.
To quantify the presumed augmented angiogenesis, it may appear that there is lack of clarity between ischaemic zone and infarct size, and the lack of CD31 data from the IHC study. We substantiated this by measuring VEGF gene expression this may lead to increase in VEGF protein levels. The message stabilization is a common way of increasing VEGF protein there are some reports of the opposite trend.
The timing of infarct size reduction with H2S treatment and improved LV function is related to angiogenesis is supported by the evidences, in part by blood flow measurement.
The CD31 data and barium contrast X-ray analysis only reveal the macrovascular expansion, and does not provide any direct evidence for angiogenesis (capillary growth). Indeed, Fig 6 is consistent with collateral dilatation.
Acknowledgements
The study was supported in part by NIH grants: HL-71010; HL-74185; and HL-88012 to SCT and HL-80394 to DL.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Lee SH, Wolf PL, Escudero R, Deutsch R, Jamieson SW, Thistlethwaite PA. Early Expression of Angiogenesis Factors in Acute Myocardial Ischemia and Infarction. New England Journal of Medicine. 2000;342:626-633
2. Li J, Brown LF, Hibberd MG, Grossman JD, Morgan JP, Simons M. VEGF, flk-1, and flt-1 expression in a rat myocardial infarction model of angiogenesis. American Journal of Physiology - Heart and Circulatory Physiology. 1996;270:H1803-H1811
3. Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69:4-10
4. Ferrara N. The role of VEGF in the regulation of physiological and pathological angiogenesis. EXS. 2005:209-231
5. Goodsell DS. The Molecular Perspective: VEGF and Angiogenesis. Oncologist. 2002;7:569-570
6. LeCouter J LR, Ferrara N. The role of EG-VEGF in the regulation of angiogenesis in endocrine glands. Cold Spring Harbor Symposia on Quantitative Biology. 2002;67:217-221
7. McColl BK, Stacker SA, Achen MG. Molecular regulation of the VEGF family - inducers of angiogenesis and lymphangiogenesis. APMIS. 2004;112:463-480
8. McMahon G. VEGF Receptor Signaling in Tumor Angiogenesis. Oncologist. 2000;5:3-10
9. Banai S SD, Pinson A, Chandra M, Lazarovici G, Keshet E. Upregulation of vascular endothelial growth factor expression induced by myocardial ischaemia: implications for coronary angiogenesis. Cardiovascular research. 1994;28:1176-1179
10. Sharma HS WM, Schmidt M, Schott RJ, Kandolf R, Schaper W. Expression of angiogenic growth factors in the collateralized swine myocardium. EXS. 1992;61:255-260
11. Hashimoto E, Ogita T, Nakaoka T, Matsuoka R, Takao A, Kira Y. Rapid induction of vascular endothelial growth factor expression by transient ischemia in rat heart. American Journal of Physiology - Heart and Circulatory Physiology. 1994;267:H1948-H1954
12. Terman BI, Dougher-Vermazen M, Carrion ME, Dimitrov D, Armellino DC, Gospodarowicz D. et al. Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochemical and Biophysical Research Communications. 1992;187:1579-1586
13. Patterson C, Perrella MA, Endege WO, Yoshizumi M, Lee ME, Haber E. Downregulation of vascular endothelial growth factor receptors by tumor necrosis factor-alpha in cultured human vascular endothelial cells. The Journal of Clinical Investigation. 1996;98:490-496
14. Voelkel NF, Vandivier RW, Tuder RM. Vascular endothelial growth factor in the lung. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2006;290:L209-L221
15. Couffinhal T SM, Kearney M, Sullivan A, Witzenbichler B, Magner M, Annex B, Peters K, Isner JM. Impaired Collateral Vessel Development Associated With Reduced Expression of Vascular Endothelial Growth Factor in ApoE-/- Mice. Circulation. 1999;99:3188-3198
16. Rivard A, Berthou-Soulie L, Principe N, Kearney M, Curry C, Branellec D. et al. Age-dependent Defect in Vascular Endothelial Growth Factor Expression Is Associated with Reduced Hypoxia-inducible Factor 1 Activity. Journal of Biological Chemistry. 2000;275:29643-29647
17. Rivard A, Silver M, Chen D, Kearney M, Magner M, Annex B. et al. Rescue of Diabetes-Related Impairment of Angiogenesis by Intramuscular Gene Therapy with Adeno-VEGF. The American Journal of Pathology. 1999;154:355-363
18. Qipshidze N, Tyagi N, Sen U, Givvimani S, Metreveli N, Lominadze D. et al. Folic acid mitigated cardiac dysfunction by normalizing the levels of tissue inhibitor of metalloproteinase and homocysteine-metabolizing enzymes postmyocardial infarction in mice. American Journal of Physiology - Heart and Circulatory Physiology. 2010;299:H1484-H1493
19. Duan J MT, Ikeda H, Katoh A, Shintani S, Sasaki K, Kawata H, Yamamoto N, Imaizumi T. Hypercholesterolemia Inhibits Angiogenesis in Response to Hindlimb Ischemia: Nitric Oxide-Dependent Mechanism. Circulation. 2000;102:III-370 -376
20. Nagai Y, Tasaki H, Takatsu H, Nihei S-i, Yamashita K, Toyokawa T. et al. Homocysteine Inhibits Angiogenesis in Vitro and in Vivo. Biochemical and Biophysical Research Communications. 2001;281:726-731
21. Zhao W, Wang R. H2S-induced vasorelaxation and underlying cellular and molecular mechanisms. American Journal of Physiology - Heart and Circulatory Physiology. 2002;283:H474-H480
22. Geng B, Yang J, Qi Y, Zhao J, Pang Y, Du J. et al. H2S generated by heart in rat and its effects on cardiac function. Biochemical and Biophysical Research Communications. 2004;313:362-368
23. Zhang Z HH, Liu P, Tang C, Wang J. Hydrogen sulfide contributes to cardioprotection during ischemia-reperfusion injury by opening K ATP channels. Canadian Journal of Physiology and Pharmacology. 2007;85:1248-1253
24. Sodha NR, Clements RT, Feng J, Liu Y, Bianchi C, Horvath EM. et al. The effects of therapeutic sulfide on myocardial apoptosis in response to ischemia-reperfusion injury. European Journal of Cardio-Thoracic Surgery. 2008;33:906-913
25. Givvimani S, Munjal C, Gargoum R, Sen U, Tyagi N, Vacek JC. et al. Hydrogen sulfide mitigates transition from compensatory hypertrophy to heart failure. Journal of Applied Physiology. 2011;110:1093-1100
26. Duncan MB, Kalluri R. Parstatin, a Novel Protease-Activated Receptor 1-Derived Inhibitor of Angiogenesis. Molecular Interventions. 2009;9:168-170
27. Frank KF, Müller-Ehmsen J. Angiostatin: drying out the roots in cardiac muscle. Heart. 2009;95:269-270
28. Jing Q, Xinyu Q, Rougcheng L. Recombinant Human Endostatin-associated Acute Left Heart Failure. Clinical Oncology. 2008;20:268-268
29. Yamahara K, Min KD, Tomoike H, Kangawa K, Kitamura S, Nagaya N. Pathological role of angiostatin in heart failure: an endogenous inhibitor of mesenchymal stem-cell activation. Heart. 2009;95:283-289
30. Givvimani S, Tyagi N, Sen U, Mishra PK, Qipshidze N, Munjal C. et al. MMP-2/TIMP-2/TIMP-4 versus MMP-9/TIMP-3 in transition from compensatory hypertrophy and angiogenesis to decompensatory heart failure*. Archives Of Physiology And Biochemistry. 2010;116:63-72
31. Sen U, Tyagi N, Patibandla PK, Dean WL, Tyagi SC, Roberts AM. et al. Fibrinogen-induced endothelin-1 production from endothelial cells. American Journal of Physiology - Cell Physiology. 2009;296:C840-C847
32. Wilkinson IB, MacCallum H, Cockcroft JR, Webb DJ. Inhibition of basal nitric oxide synthesis increases aortic augmentation index and pulse wave velocity in vivo. British Journal of Clinical Pharmacology. 2002;53:189-192
33. Lominadze D, Schuschke DA, Joshua IG, Dean WL. Increased ability of erythrocyrtes to aggregate in spontaneously hypertensive rats. Clinical and Experimental Hypertension. 2002;24:397-406
34. Tyagi N, Ovechkin AV, Lominadze D, Moshal KS, Tyagi SC. Mitochondrial mechanism of microvascular endothelial cells apoptosis in hyperhomocysteinemia. Journal of Cellular Biochemistry. 2006;98:1150-1162
35. Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L. et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proceedings of the National Academy of Sciences. 2007;104:15560-15565
36. Johansen D, Ytrehus K, Baxter G. Exogenous hydrogen sulfide (H2S) protects against regional myocardial ischemia-reperfusion injury--Evidence for a role of K ATP channels. Basic Research in Cardiology. 2006;101:53-60
37. Li L, Moore PK. Putative biological roles of hydrogen sulfide in health and disease: a breath of not so fresh air? Trends in Pharmacological Sciences. 2008;29:84-90
38. Sivarajah A, McDonald MC, Thiemermann C. The Production of Hydrogen Sulfide Limits Myocardial Ischemia and Reperfusion Injury and Contributes To the Cardioprotective Effects of Preconditioning With Endotoxin, But Not Ischemia in the Rat. Shock. 2006;26:154-161
39. Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917-935
40. Sen U MC, Qipshidze N, Abe O, Gargoum R, Tyagi SC. Hydrogen sulfide regulates homocysteine-mediated glomerulosclerosis. American Journal of Nephrology. 2010;31:442-455
41. Laham RJ LJ, Tofukuji M, Post M, Simons M, Sellke FW. Spatial heterogeneity in VEGF-induced vasodilation: VEGF dilates microvessels but not epicardial and systemic arteries and veins. Annals of vascular surgery. 2003;17:245-252
42. Lopez JJ, Laham R, Stamler A, Pearlman JD, Bunting S, Kaplan A. et al. VEGF administration in chronic myocardial ischemia in pigs. Cardiovascular research. 1998;40:272-281
43. Sato K, Wu T, Laham RJ, Johnson RB, Douglas P, Li J. et al. Efficacy of intracoronary or intravenous VEGF165 in a pig model of chronic myocardial ischemia. Journal of the American College of Cardiology. 2001;37:616-623
44. Wu G, Luo J, Rana JS, Laham R, Sellke FW, Li J. Involvement of COX-2 in VEGF-induced angiogenesis via P38 and JNK pathways in vascular endothelial cells. Cardiovascular research. 2006;69:512-519
45. Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671-674
46. House SL, Bolte C, Zhou M, Doetschman T, Klevitsky R, Newman G. et al. Cardiac-Specific Overexpression of Fibroblast Growth Factor-2 Protects Against Myocardial Dysfunction and Infarction in a Murine Model of Low-Flow Ischemia. Circulation. 2003;108:3140-3148
47. Sorensen Dag R. R, Tracy-Ann, Porwol, Torsten, Olsen, Bjorn R, Timpl, Rupert, Sasaki, Takako, Iversen, Per O, Benestad, Haakon B, Lee Sim, Bjerkvig, Rolf. Endostatin reduces vascularization, blood flow, and growth in a rat gliosarcoma. Neuro-Oncology. 2002;4:1-8
48. Moser TL, Stack MS, Asplin I, Enghild JJ, Højrup P, Everitt L. et al. Angiostatin binds ATP synthase on the surface of human endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:2811-2816
49. Yamaguchi N, Anand-Apte B, Lee M, Sasaki T, Fukai N, Shapiro R. et al. Endostatin inhibits VEGF-induced endothelial cell migration and tumor growth independently of zinc binding. EMBO J. 1999;18:4414-4423
50. Hajitou A, Grignet-Debrus C, Devy L, Berndt S, Blacher S, Deroanne CF. et al. The antitumoral effect of endostatin and angiostatin is associated with a down-regulation of vascular endothelial growth factor expression in tumor cells. The FASEB Journal. 2002;16:1802-1804
51. Claesson-Welsh L, Welsh M, Ito N, Anand-Apte B, Soker S, Zetter B. et al. Angiostatin induces endothelial cell apoptosis and activation of focal adhesion kinase independently of the integrin-binding motif RGD. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:5579-5583
Author contact
![]() Corresponding author: Natia Qipshidze MD, University of Louisville, Dept. of Physiology & Biophysics, School of Medicine, Bldg. A, Room 1209, 500 South Preston Street, Louisville, KY 40202, Phone: (502) 852-3627, Fax: (502) 852-6239, E-mail: n0qips01edu.
Corresponding author: Natia Qipshidze MD, University of Louisville, Dept. of Physiology & Biophysics, School of Medicine, Bldg. A, Room 1209, 500 South Preston Street, Louisville, KY 40202, Phone: (502) 852-3627, Fax: (502) 852-6239, E-mail: n0qips01edu.