Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(4):442-450. doi:10.7150/ijbs.4155 This issue Cite
Research Paper
Prognostic Values of Filamin-A Status for Topoisomerase II Poison Chemotherapy
Jingyin Yue2,*, Shijie Lan1,2,*, Changji Yuan1, Zhiyuan Shen2 ![]()
1. The Cancer Center, the First Hospital of Jilin University, Changchun 130021, China.
2. Department of Radiation Oncology, The Cancer Institute of New Jersey, Robert Wood Johnson Medical School, New Brunswick, NJ 08903, USA.
* Authors with equal contribution.
Received 2012-1-26; Accepted 2012-2-15; Published 2012-2-29
Abstract
Filamin-A, also called Actin Binding Protein-280, is not only an essential component of the cytoskeleton networks, but also serves as the scaffold in various signaling networks. It has been shown that filamin-A facilitates DNA repair and filamin-A proficient cells are more resistant to ionizing radiation, bleomycin, and cisplatin. In this study, we assessed the role of filamin-A in modulating cancer cell sensitivity to Topo II poisons, including etoposide and doxorubicin. Intriguingly, we found that cells with filamin-A expression are more sensitive to Topo II poisons than those with defective filamin-A, and filamin-A proficient xenograft melanomas have better response to etoposide treatment than the filamin-A deficient tumors. This is associated with more potent induction of DNA double strand breaks (DSBs) by Topo II poisons in filamin-A proficient cells than the deficient cells. Although the expression of filamin-A enables cells a slightly stronger capability to repair DSB, the net outcome is that filamin-A proficient cells bear more DSBs due to the significantly enhanced DSB induction by Topo II poisons in these cells. We further found that filamin-A proficient cells have increased drug influx and decreased drug efflux, suggesting that filamin-A modulates the intra-cellular drug kinetics of Topo II poisons to facilitate the generation of DSB after Topo II poison exposure. These data suggest a novel function of filamin-A in regulating the pharmacokinetics of Topo II poisons, and that the status of filamin-A may be used as a prognostic marker for Topo II poisons based cancer treatments.
Keywords: Filamin-A, ABP-280, topoisomerase II poisons, chemotherapy, sensitization.
Introduction
Human topoisomerase II (Topo II or Top2) has been used as a therapeutic target for the development of anticancer drugs [1, 2]. Two classes of Topo II poisons, anthracyclines and epipodophyllotoxins, have been extensively studied [3, 4]. Among them, doxorubicin and etoposide are commonly used in clinical practice. These drugs trap Topo II on cleaved DNA to form stable cleavage complex, which results in irreversible double-strand breaks (DSB), leading to cell death [5-8]. Thus the efficacy of Topo II poisons can be modulated by both the ability of cells to repair DNA damage and the drug transport and/or metabolism. Identification of biomarkers that modulate these processes will greatly facilitate individually tailored cancer treatment using Topo II poisons.
Filamin-A (FLNa) is also called Actin Binding Protein-280 (ABP-280) [9-11]. It carries out versatile biological functions in the cell, including organizing cortical actin filament networks and acting as a scaffold for signal transduction [9, 12, 13]. Filamin-A interacts with caveolin-1 to promote caveolae-mediated vesicle internalization, clustering, and trafficking [14-16]. It has also been shown that filamin-A regulates intracellular sorting and redistribution of proteins including epidermal growth factor receptors (EGFR) and furin [17, 18]. We and others have reported that filamin-A plays a role in the regulation of homologous recombination (HR) and non-homologous end joining (NHEJ) through interactions with BRCA1 and BRCA2 [19, 20]. In this context, filamin-A is required for efficient repair of a variety of DNA damages, including single strand breaks (SSBs), double strand breaks (DSBs) and inter-strand cross links (ICLs) [19, 20]. In this study, we tested the role of filamin-A in the modulation of the cellular sensitivity to Topo II poisons. Interestingly, we found that loss of filamin-A expression did not sensitize cells to etoposide and doxorubicin treatments, rather, filamin-A deficient cells are more resistant to these Topo II poisons than filamin-A proficient cells. We further discovered that filamin-A plays a role in regulating the influx and efflux of Topo II poisons. Filamin-A proficient cells incurs more Topo II poison-induced DNA damages compared to filamin-A deficient cells, due to elevated intracellular drug level. This result suggests that filamin-A can be used as a prognostic marker for Topo II poisons based cancer treatments.
Materials and Methods
Cell lines, cell cultures, and cellular sensitivity to Topo II poisons
The three pairs of isogenic filamin-A proficient and deficient cell lines have been described in previous publications [19, 21]. These cells are: M2 (filamin-A deficient) and A7 (filamin-A proficient), C8161-KD (filamin-A knockdown) and C8161-Con (filamin-A proficient), and MB231-KD (filamin-A knockdown) and MB231-Con (filamin-A proficient). The characterization and culture condition for these cells have been described in previous reports [19, 21]. Log-phase cells were treated with etoposide or doxorubicin, the cellular sensitivity was measured with colony formation assay and growth inhibition assay as described in a previous report [21].
Immunofluorescent detection of γH2AX nuclear foci
Cells (5 × 104) were plated and grown on glass coverslips for 16 h, then treated with 10 μM of etoposide or 0.25 μg/ml of doxorubicin for 2 hours. Upon the completion of 2 hours drug exposure, cells were washed with PBS and fed with fresh media to allow recovering. At various time points to be specified in the figures, the cells were fixed and stained for γH2AX nuclear foci, using the procedures as described previously [19, 21].
Drug influx measured by flow cytometry
Cells (5 × 105) were treated with 5 μg/ml of doxorubicin at 37°C. The cells were collected by centrifugation at 4°C, after continuous exposure to the drug for 0, 10 and 30 min. After two washes with ice-cold PBS, the cells were re-suspended in 0.5 ml of ice-cold PBS and kept on ice for flow cytometry analysis immediately to determine the relative level of intracellular doxorubicin based on the strength of its auto-fluorescence. The cells were analyzed using a FACS Calibur benchtop flow cytometer (BD Biosciences, San Jose, CA) with excitation at 488 nm, with emission integrated above 530 nm. For each sample, 20,000 cells were analyzed, and experiment was repeated twice.
The efflux of doxorubicin as assessed with fluorescent microscopy
To measure the efflux of doxorubicin, the cells were treated with 1.0 μg/ml of doxorubicin for 10 min, and then fixed with cold 4% paraformaldehyde at 0, 15 and 30 min after incubation with drug-free medium. Fixed cells were stained with DAPI. Images containing red and blue channels were taken with fluorescent microscope. The signal intensities from red channel (doxorubicin) and blue channel (DNA, internal control) were measured with Image J as described before [22] and the ratio of red signal over blue represents the relative intracellular concentration of doxorubicin.
The efflux of etoposide measured with HPLC
Because the excitation and emission wavelengths of etoposide are out of range of conventional flow cytometry and fluorescent microscope, high performance liquid chromatography (HPLC) approach was used to measure the efflux of etoposide as described before with modification [23]. The HPLC (AKTA Purifier, GE Healthcare Biosciences, Pittsburgh, PA) is equipped with UV detector and autosampler. To separate etoposide, the C18 reverse phase column (ZorBax SB-C18, 250mm×4.5mm, Agilent Technologies, Santa Clara, CA) was used, and the separation was obtained with 50% methanol in H2O as mobile phase. To detect etoposide, 220nm of UV wavelength was used. The cells were treated with 50 μM of etoposide for 2 hours, and then harvested immediately or after 15 or 30 min of incubation with drug-free medium. Etoposide within the cells was extracted with diethyl ether and dissolved with mobile phase for HPLC analysis. The absorbance of etoposide at 220 nm of UV was converted into drug concentration using premeasured standard curve. Phenacitin was added into cell pellet before diethyl ether extraction and used as internal tracer control.
Tumor growth inhibition with a xenograft model
Among the cell lines used in this study, melanoma C8161 forms tumors when subcutaneously inoculated, thus was used for the nude mouse xenograft experiments. Human melanoma xenogafts were generated with C8161-Con (filamin-A wild type) and C8161-KD (filamin-A knockdown) cells and the tumor growth was monitored as described previously [21]. When the tumor volume reached 100-300mm3 after 5-7 days of subcutaneous injection, the xenografts were randomly divided into control and treatment groups, 5 mice per group. Drug treatment group was administrated with etoposide at the dosage of 5 mg/Kg, with 3 days interval and 7 administrations total. Control group was injected with same volume of saline.
Results
Filamin-A expression sensitizes cells to Topo II poisons
We have shown in previous reports that loss of filamin-A sensitizes cells to multiple DNA damaging agents, including ionizing radiation, bleomycin and cisplatin [19, 21]. These studies are in agreement with a model that filamin-A is required for efficient DNA repair through HR and NHEJ [19-21]. Since Topo II poisons mainly generate DSBs [6, 8], which can be repaired by HR and NHEJ [24], we speculate that filamin-A deficient cells might also be more sensitive to Topo II poisons than filamin-A proficient cells. To verify this, we treated the same three pairs of isogenic filamin-A proficient or deficient cells with etoposide and doxorubicin, and measured the cell survival by colony formation. To our great surprise, we observed an elevated survival among filamin-A deficient cells after 2 hours of Topo II poison treatments (Figs. 1A and 1B).
Filamin-A expression sensitizes cells to Topo II poisons. The following three pairs of isogenetic filamin-A proficient and deficient cell lines were used (see Materials and Methods for details): M2 (FLNa−) and A7 (FLNa+); C8161-Con (filamin-A control) and C8161-KD (filamin-A knockdown); MB231-Con and MB231-KD. Panels A and B are the survival curves assessed by colony formation assays. Panel C shows the relative IC50 values of the filamin-A deficient cells normalized to that of the isogenic filamin-A proficient cells (* p<0.05, ** p<0.01). The IC50 values were determined by growth inhibition assays (Materials and Methods). Error bars represent the standard deviation of at least three independent experiments.
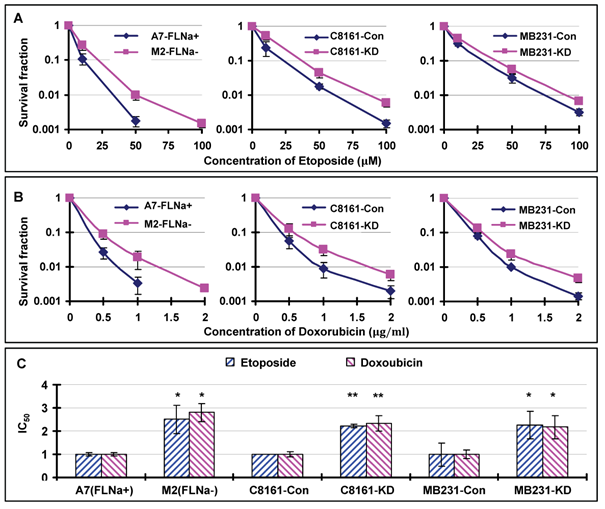
To confirm this result, we employed growth inhibitory assay to measure the IC50s of each cell after continuous treatment of cells with etoposide or doxorubicin (see Materials and Methods for details). As shown in Table 1, filamin-A proficient cells have significantly lower IC50 values to etoposide and doxorubicin than the filamin-A deficient counterparts (p<0.05 or p<0.01). After normalizing the IC50 value to that of the paired filamin-A proficient cells, filamin-A deficient cells showed 2-3 fold increase in IC50 values (Fig. 1C). These data strongly suggest that filamin-A deficient cells are less sensitive to etoposide and doxorubicin than the filamin-A proficient cells, which is in sharp contrast to the sensitivity to ionizing radiation, bleomycin, and cisplatin as reported previously [21].
IC50 for human melanoma cell and breast cancer cells.
| Cell line | Drug Treatment | |
|---|---|---|
| Etoposide (μM) | Doxorubicin (μg/mL) | |
| A7-FLNa+ | 0.35±0.12* | 0.015±0.003* |
| M2-FLNa- | 3.38±0.84 | 0.14±0.02 |
| C8161-Con | 0.87±0.02** | 0.045±0.01** |
| C8161-KD | 4.37±0.15 | 0.11±0.03 |
| MB231-Con | 0.12±0.06* | 0.06±0.01* |
| MB231-KD | 0.27±0.07 | 0.13±0.03 |
* p<0.05; ** p<0.001.
Induction and repair of DSBs
DNA Topo II poisons induce DSB and filamin-A is required for efficient DSB repair through regulating HR and NHEJ. Thus, it was puzzling to us at the moment that filamin-A deficient cells have lesser cellular sensitivity to Topo II poisons than filamin-A proficient cells. To investigate the mechanism of this apparently conflicting phenomenon, we assessed the initial levels of DNA damage and the repair kinetics. Again, we used M2 cells that have spontaneously lost filamin-A expression and the A7 cells that have reinstated filamin-A by stably expressing filamin-A in the M2 cells. After 2 hr exposure to 10 μM of etoposide or 0.25μg/ml of doxorubicin, the drug was washed out. At various time points as shown in Figure 2A, the numbers of DSB in these paired cells were assessed by measuring the number of γH2AX nuclear foci. Significantly more γH2AX nuclear foci were induced in A7 cells than in M2 cells after 2 hours of etoposide incubation (50.7 and 33.3 foci/cell respectively, p<0.01), indicating that there is more initial DNA damage in the filamin-A proficient A7 cells than the deficient M2 cells (Fig.2A). Similarly, higher peak value of γH2AX foci was observed in A7 than M2 after doxorubicin treatment (Fig. 2B). It is notable that the highest level of DSBs induced in doxorubicin treated cells occurs at 6 hours after the drug was removed, instead of right after 2 hour of drug incubation with etoposide (Figs. 2A and B). This observation is in agreement with previous study that the generation of cleaved DNA takes longer for doxorubicin than etoposide [25]. Nevertheless, these data suggest that there was significantly less initial amount of DSB generated in M2 than A7 cells.
Induction and repair of DSBs after 2 hours of etoposide and doxorubicin treatment. The A7 (FLNa+) and M2 (FLNa−) cells were treated with 10 μM etoposide or 0.25 μg/ml doxorubicin for 2 h, and then the drug was removed and the cells were kept in drug-free medium. The cells were fixed at various time points for γH2AX immunostaining. The numbers of γH2AX nuclear foci were counted from 100-150 cells in each slide, and the experiment was repeated twice. Panels A and B show the average numbers of γH2AX nuclear foci after etoposide (Panel A) or doxorubicin (Panel B) treatment (* p<0.05, ** p<0.01). Error bars are standard deviations. Panel C and D show the exponential regression analysis. After DNA damage reached highest level (0 hour for etoposide and 6 hour for doxorubicin), the numbers of γH2AX against the repair time were shown in X-Y scattered plot (Diamond and Square). Solid lines are the exponential regression fitting. The exponential regression equations along with the coefficient of determination, R-squared (R2), are shown in the figure.
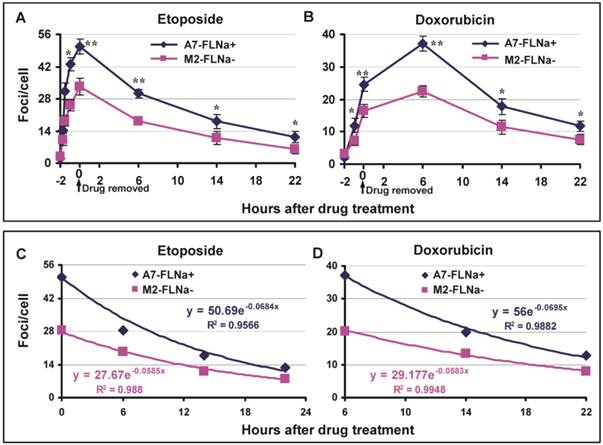
To compare the capability of DSB repair in A7 and M2 cells, the time-points (Figs. 2A and 2B) after the DSB reached the highest level (0 hr for etoposide and 6 hr for doxorubicin) were used for exponential regression analysis. As shown in Figure 2C and 2D, there are strong correlations between the DSB and repair time (R2>0.95) for both drugs. For both etoposide and doxorubicin, the A7 cells have deeper slops than the M2 cells, indicating more efficient repair of DSBs in A7 cells, which is consistent with previous report [19-21]. At 22 hours after drug treatment, the number of γH2AX foci in both cells decreased dramatically, while there was significantly higher number of residual γH2AX foci in A7 cells than in M2 cells (Fig.2A and B).
Collectively, Figure 2 suggests that A7 cells incur more initial DNA damage after Topo II poison treatments. Despite of a faster removal of the DSB, there remains more DSBs in A7 cells than M2 cells due to the excessive level of initial DSB. The net outcome is that A7 cells consistently bear more DNA damage during the first 22 hours after 2 hours of treatment, which likely contributes to the increased cellular sensitivity to Topo II poison.
Filamin-A expression increases drug influx and decreases drug efflux
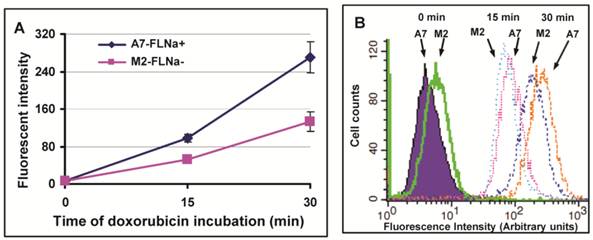
Filamin-A proficient cells showed higher cellular sensitivity to Topo II poisons (Fig. 1), and more initial DSBs induced in filamin-A expressing A7 cells than filamin-A deficient M2 cells (Fig. 2). The extent of cleaved DNA generated in cells is thought to be correlated with intracellular drug concentration. Therefore, we tested drug influx of Topo II poisons in A7 and M2 cells. Since doxorubicin is a natural fluorescent compound, we measured the intracellular concentration of doxorubicin by measuring the fluorescent intensity using flow cytometry. The cells were incubated with 5μg/ml doxorubicin for 0, 15 and 30 min. As shown in Figure 3A, the fluorescent intensity increased with the treatment time. Stronger signals were detected in A7 cells than M2 cells at 15 and 30 min after drug treatment, indicating higher intracellular drug concentration in A7 cells after drug exposure. Figure 3B shows the representative histographs of flow cytometry and about one fold of signal intensity increases was observed in A7 cells compared with M2 cells at 15 and 30 min time points. This result suggests the elevated drug intake in filamin-A expressing cells.
Drug influx of doxorubicin in melanoma A7 and M2 cells. The A7 (FLNa+) and M2 (FLNa−) cells were treated with 5μg/ml doxorubicin for 0, 10min and 30min, the intracellular concentration of doxorubicin was assessed by measuring red fluorescent signal using flow cytometry. Panel A shows the fluorescent intensity of doxorubicin measured in A7 and M2 cells. Shown is the average of three independent experiments, and error bars are standard deviations. Panel B shows the representative histograph of flow cytometry.
Higher intracellular drug concentration may not only reflect increased drug influx, but also decreased drug efflux. To test whether filamin-A affects dug excretion, we measured the efflux of doxorubicin in the paired filamin-A proficient and deficient cells. The cells were treated with 1.0 μg/ml of doxorubicin for 10 min, then were fixed with cold 4% paraformaldehyde immediately or after 15 or 30 min incubation in drug-free medium at 37°C. After staining the cells with DAPI, images containing red and blue channels were taken with fluorescent microscope. Figure 4A shows the representative images of the cells incubated in drug-free medium for 15 min.
Drug efflux in filamin-A proficient and deficient cells. The cells were treated with 1.0 μg/ml of doxorubicin for 10 min, then were fixed with cold 4% paraformaldehyde immediately or after 15 or 30 min incubation in drug-free medium. Fixed cells were stained with DAPI. Images containing red and blue channels were taken with fluorescent microscope. The signal intensities from red channel (doxorubicin) and blue channel (DNA, internal control) were measured with Image J as described before[22]. The ratio of red signal over blue reflects the intracellular concentration of doxorubicin. Panel A shows the representative images taken after 15 min drug efflux. Panel B shows quantitative data of intracellular concentration of doxorubicin with various time drug excretions (* p<0.05, ** p<0.01). 40 cells were analyzed from each slide and shown are the averages from three independent experiments. Error bars are standard errors. To assess the efflux of etoposide, A7 and M2 cells were treated with 50 μM etoposide for 2 hours, and then the cells were harvested immediately or after 15 or 30 min incubation in drug-free medium. The amount of etoposide in the cells was determined with HPLC (Materials and Methods). Panel C shows the intracellular concentration of etoposide of A7 and M2 cells. Shown are the averages from three independent experiments. Error bars are standard errors.
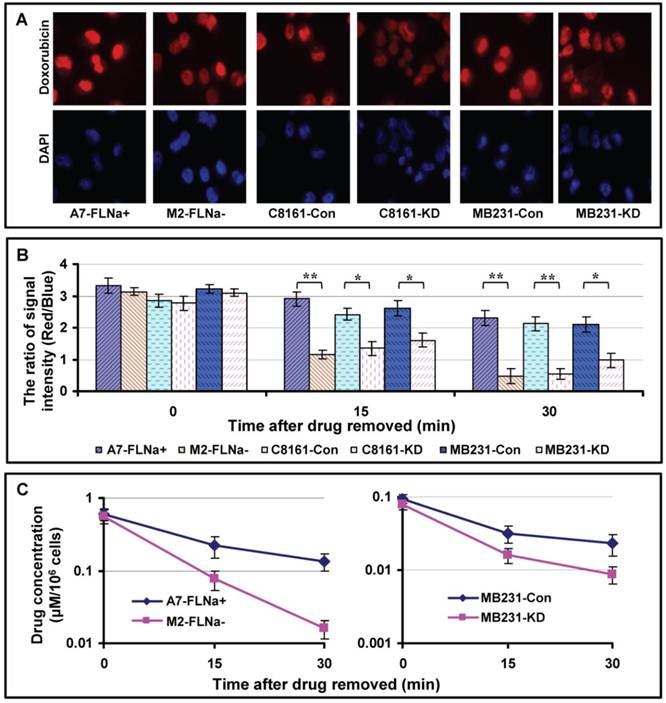
We analyzed signal intensity using Image J software as described before[22]. Stronger red signals (doxorubicin) were observed in filamin-A proficient cells (A7-FLNa+, C8161-Con and MB231-Con) than filamin-A deficient cells (M2, C8161-KD and MB231-KD). After taking the ratio of red signal over blue signal which reflects the intracellular concentration of doxorubicin, significant slow drug efflux was observed in filamin-A expressing cells than filamin-A deficient cells (Fig. 4B).
To assess the efflux of etoposide, we employed HPLC equipped with UV detector to measure intra-cellular levels of etoposide. As shown in Figure 4C, significantly faster etoposide excretion was detected in filamin-A deficient cells (M2 and MB231-KD) than filamin-A proficient cells (A7 and MB231-Con). These data suggest that filamin-A expression affects the cellular drug kinetics of Topo II poisons including etoposide and doxorubicin, by increasing drug influx and decreasing drug efflux (Figs. 3 and 4), which may contribute to the increased toxicity to filamin-A proficient cells.
Filamin-A expression sensitizes the tumor xenografts to Topo II poisons
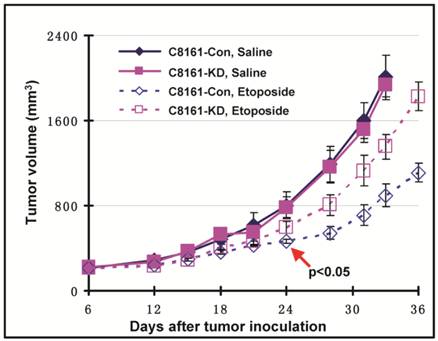
We have shown that filamin-A expression increases the cellular sensitivity to Topo II poisons (Figs. 1 and 2), and this sensitization effect is the consequence of increased drug influx and decreased drug efflux in filamin-A proficient cells (Figs. 3 and 4). These data raise a possibility that filamin-A may be used as a prognostic marker for cancer treatment with Topo II poisons. To verify this, we generated human melanoma tumor xenografts with paired C8161 cell lines (C8161-Con and C8161-KD). The tumor growth was monitored during control (saline) and etoposide treatments. Both control (C8161-Con) and filamin-A knockdown (C8161-KD) cells formed xenograft tumors in nude mice with similar growth rates (Fig. 5). As expected, xenograft tumors treated with etoposide (5 mg/kg) displayed growth delay compared with those treated with saline alone. Furthermore, filamin-A proficient tumors grew significantly slower than the filamin-A deficient tumors (p < 0.05) at 24 days with etoposide treatments, suggesting that filamin-A expression significantly sensitizes melanoma cells to etoposide (Fig. 5). These data indicate that lack of filamin-A expression impairs the efficacy of Topo II poisons in cancer treatment.
Filamin-A expression sensitizes C8161 melanoma cells to etoposide treatment in xenograft mouse model. Human melanoma tumor xenografts were generated as described previously. Shown is the tumor growth in response to control (saline) and etoposide treatment. Treatment group was administrated with etoposide at the dosage of 5 mg/Kg, with 3 days interval and 7 administrations total. Control group was injected with same volume of saline. Error bars represent the standard errors from 5 mice. Red arrow shown in figure indicates the significant growth delay was observed in filamin-A proficient tumors compared with filamin-A deficient tumors (p < 0.05) at 24 days and thereafter with etoposide treatments.
Discussion
Etoposide and doxorubicin are among the most commonly used agents in clinical cancer treatment. They interfere with the rejoining reaction of cleaved DNA by stabilizing the covalent Topo II-DNA complex, which in turn generates DSBs that can be visualized as γH2AX nuclear foci. In this report, we illustrate a novel function of filamin-A in regulating Topo II poison intracellular pharmacokinetics. Lack of filamin-A expression decreases the intracellular drug levels, reduces the amount of DNA damage caused by etoposide and doxorubicin, and subsequently abrogates cell sensitivity to these Topo II poisons.
Filamin-A is depicted as multifunctional protein with variety of interacting partners. It is not only an essential component of the cytoskeleton networks, but also serves as the scaffold in various signaling networks (see reviews [13, 26, 27]). Recently, it has been shown that filamin-A regulates HR and NHEJ and thus modulates DNA damage repair [19, 20]. Consistent with these reports, we observed a faster removal of drug-induced DSBs in filamin-A proficient cells than deficient cells during the period of DSB repair (Fig. 2C and D), confirming the role of filamin-A in DNA repair. However, filamin-A proficient cells also have higher levels of initial amount of DNA damage induced by both etoposide and doxorubicin (Fig. 2A and B). The net outcome of these two opposing effects is the constantly higher levels of DNA damage in the filamin-A proficient cells than the deficient cells, at least within the first 22 hours after the removal of the drugs (Figure 2). We believe that this is likely the main determinant in the increased sensitivity to etoposide and doxorubicin among filamin-A proficient cells.
We further demonstrated that the elevated level of initial DNA damage in filamin-A proficient cells is strongly supported by the increased intracellular drug concentration in these cells. As shown in Figure 3, filamin-A proficient cells have an elevated drug intake compared to deficient cells as soon as 15 min after doxorubicin was added into the medium. At 30min after continuously exposure, A7 cells have about one fold increase of intracellular doxorubicin. At the same time, after the drugs are removed, the filamin-A proficient cells also have slower clearance of intracellular doxorubicin and etoposide (Fig. 4), suggesting that the expression of filamin-A decreases the efflux of Topo II poisons.
The mechanisms of how filamin-A affects the drug influx and efflux remain elusive. It is known that influx of drug into the cells may take place by simple diffusion. In multidrug resistant cells (with P-glycoprotein expression), increased efflux can be accompanied by a decrease in influx [28, 29], which is similar to what we have found in filamin-A deficient cells. However, the expression of P-glycoprotein (MDR-1), the main drug transporter [30], was below the detectable level in the cell lines used in this study, as shown in our previous report[21]. Thus, alternative mechanisms remain to be investigated. It is known that other membrane proteins in addition to MDR-1, such as ATP-binding cassette (ABC) transporters and multidrug resistance-associated protein (MRP), may promote the efflux of etoposide and doxorubicin [31-33]. Filamin-A has been reported to play a role in intracellular trafficking and redistribution of some proteins, such as epidermal growth factor receptors (EGFR) and furin [17, 18]. Thus, it is possible that filamin-A may regulate drug efflux by affecting the cellular trafficking or membrane distribution of these proteins which are responsible for the drug transporting. In addition, filamin-A has been shown to interact with caveolin-1 and promote caveolae-mediated vesicle internalization, clustering, and trafficking [14-16]. Both endocytosis and intracellular vesicle trafficking have been shown to affect the intake of hormone, protein, and lipids. It remains to be determined whether this activity also contributes to the intake of Topo II poisons.
Although the mechanism by which filamin-A regulates the intake and efflux of Topo II poisons requires further investigation, our study raises a possibility that filamin-A can be used as a biomarker for cancer treatment by Topo II poisons. As shown in previous study, about 40-50% of melanoma and breast tumors are filamin-A positive [19, 21]. It is likely that these patients will have better prognosis in terms of response to Topo II poisons based chemotherapy due to higher intracellular drug concentration than filamin-A deficient cancers. However, they will have poorer prognosis to radiation, bleomycin, and cisplatin treatments, due to enhanced ability to repair damaged DNA. Therefore, the status of filamin-A can be used to differentiate the selection of different types of DNA damage based treatments, serving as an important biomarker for individualized treatment.
In summary, our current study greatly expands the spectrum of filamin-A's function, regulating the pharmacokinetics of Topo II poisons. Combining with previous studies, filamin-A can be used as a biomarker for individually tailored therapeutic regimes establishment. The previous reports [19, 21] and this study suggest that, for cancers expressing filamin-A, Topo II poisons based therapy is a better choice than radiation, bleomycin, and cisplatin. For filamin-A deficient cancers, radiation, bleomycin and cisplatin would be a better choice than Topo II poisons.
Acknowledgements
This research is supported by NIH R01CA156706. S. Lan is a recipient of a scholarship under the State Scholarship Fund from China Scholarship Council.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Liu LF. DNA topoisomerase poisons as antitumor drugs. Annu Rev Biochem. 1989;58:351-75
2. Pommier Y. DNA topoisomerase I and II in cancer chemotherapy: update and perspectives. Cancer Chemother Pharmacol. 1993;32:103-8
3. Weiss RB. The anthracyclines: will we ever find a better doxorubicin? Semin Oncol. 1992;19:670-86
4. Damayanthi Y, Lown JW. Podophyllotoxins: current status and recent developments. Curr Med Chem. 1998;5:205-52
5. Corbett AH, Osheroff N. When good enzymes go bad: conversion of topoisomerase II to a cellular toxin by antineoplastic drugs. Chem Res Toxicol. 1993;6:585-97
6. Mao Y, Desai SD, Ting CY, Hwang J, Liu LF. 26 S proteasome-mediated degradation of topoisomerase II cleavable complexes. J Biol Chem. 2001;276:40652-8
7. Caldecott K, Banks G, Jeggo P. DNA double-strand break repair pathways and cellular tolerance to inhibitors of topoisomerase II. Cancer Res. 1990;50:5778-83
8. Wilstermann AM, Osheroff N. Stabilization of eukaryotic topoisomerase II-DNA cleavage complexes. Curr Top Med Chem. 2003;3:321-38
9. Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M. et al. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol. 2001;2:138-45
10. Wang K. Filamin, a new high-molecular-weight protein found in smooth muscle and nonmuscle cells. Purification and properties of chicken gizzard filamin. Biochemistry. 1977;16:1857-65
11. Wang K, Ash JF, Singer SJ. Filamin, a new high-molecular-weight protein found in smooth muscle and non-muscle cells. Proc Natl Acad Sci U S A. 1975;72:4483-6
12. Popowicz GM, Schleicher M, Noegel AA, Holak TA. Filamins: promiscuous organizers of the cytoskeleton. Trends Biochem Sci. 2006;31:411-9
13. Feng Y, Walsh CA. The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat Cell Biol. 2004;6:1034-8
14. Muriel O, Echarri A, Hellriegel C, Pavon DM, Beccari L, Del Pozo MA. Phosphorylated filamin A regulates actin-linked caveolae dynamics. J Cell Sci. 2011;124:2763-76
15. Sverdlov M, Shinin V, Place AT, Castellon M, Minshall RD. Filamin A regulates caveolae internalization and trafficking in endothelial cells. Mol Biol Cell. 2009;20:4531-40
16. Stahlhut M, van Deurs B. Identification of filamin as a novel ligand for caveolin-1: evidence for the organization of caveolin-1-associated membrane domains by the actin cytoskeleton. Mol Biol Cell. 2000;11:325-37
17. Fiori JL, Zhu TN, O'Connell MP, Hoek KS, Indig FE, Frank BP. et al. Filamin A modulates kinase activation and intracellular trafficking of epidermal growth factor receptors in human melanoma cells. Endocrinology. 2009;150:2551-60
18. Liu G, Thomas L, Warren RA, Enns CA, Cunningham CC, Hartwig JH. et al. Cytoskeletal protein ABP-280 directs the intracellular trafficking of furin and modulates proprotein processing in the endocytic pathway. J Cell Biol. 1997;139:1719-33
19. Yue J, Wang Q, Lu H, Brenneman M, Fan F, Shen Z. The cytoskeleton protein filamin-A is required for an efficient recombinational DNA double strand break repair. Cancer Res. 2009;69:7978-85
20. Velkova A, Carvalho MA, Johnson JO, Tavtigian SV, Monteiro AN. Identification of Filamin A as a BRCA1-interacting protein required for efficient DNA repair. Cell cycle (Georgetown, Tex). 2010;9:1421-33
21. Yue J, Lu H, Liu J, Berwick M, Shen Z. Filamin-A as a marker and target for DNA damage based cancer therapy. DNA repair. 2012;11:192-200
22. Lu H, Yue J, Meng X, Nickoloff JA, Shen Z. BCCIP regulates homologous recombination by distinct domains and suppresses spontaneous DNA damage. Nucleic Acids Res. 2007;35:7160-70
23. Shirazi FH, Bahrami G, Stewart DJ, Tomiak E, Delorme F, Noel D. et al. A rapid reversed phase high performance liquid chromatographic method for determination of etoposide (VP-16) in human plasma. J Pharm Biomed Anal. 2001;25:353-6
24. de Campos-Nebel M, Larripa I, Gonzalez-Cid M. Topoisomerase II-mediated DNA damage is differently repaired during the cell cycle by non-homologous end joining and homologous recombination. PloS one. 2010 5. doi:10.1371/journal.pone.0012541
25. Binaschi M, Capranico G, De Isabella P, Mariani M, Supino R, Tinelli S. et al. Comparison of DNA cleavage induced by etoposide and doxorubicin in two human small-cell lung cancer lines with different sensitivities to topoisomerase II inhibitors. Int J Cancer. 1990;45:347-52
26. Zhou AX, Hartwig JH, Akyurek LM. Filamins in cell signaling, transcription and organ development. Trends Cell Biol. 2010;20:113-23
27. Nakamura F, Stossel TP, Hartwig JH. The filamins: organizers of cell structure and function. Cell Adh Migr. 2011;5:160-9
28. Nielsen D, Maare C, Skovsgaard T. Influx of daunorubicin in multidrug resistant Ehrlich ascites tumour cells: correlation to expression of P-glycoprotein and efflux. Influence of verapamil. Biochem Pharmacol. 1995;50:443-50
29. Shen F, Chu S, Bence AK, Bailey B, Xue X, Erickson PA. et al. Quantitation of doxorubicin uptake, efflux, and modulation of multidrug resistance (MDR) in MDR human cancer cells. J Pharmacol Exp Ther. 2008;324:95-102
30. Bosch I, Croop J. P-glycoprotein multidrug resistance and cancer. Biochim Biophys Acta. 1996;1288:F37-54
31. Morjani H, Pignon B, Vilque JP, Millot JM, Lartigue B, Simon G. et al. [Evaluation of multidrug resistance phenotype on medullary specimens from patients with acute leukemia by determination of nuclear efflux of tetrahydropyranyl-doxorubicin. Approach by confocal laser microspectrofluorometry]. Ann Biol Clin (Paris). 1996;54:9-15
32. Benderra Z, Morjani H, Trussardi A, Manfait M. Role of the vacuolar H+-ATPase in daunorubicin distribution in etoposide-resistant MCF7 cells overexpressing the multidrug-resistance associated protein. Int J Oncol. 1998;12:711-5
33. Grant CE, Valdimarsson G, Hipfner DR, Almquist KC, Cole SP, Deeley RG. Overexpression of multidrug resistance-associated protein (MRP) increases resistance to natural product drugs. Cancer Res. 1994;54:357-61
Author contact
![]() Corresponding author: Zhiyuan Shen at email: shenzhedu.
Corresponding author: Zhiyuan Shen at email: shenzhedu.