Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(4):451-458. doi:10.7150/ijbs.4038 This issue Cite
Research Paper
The Silencing of RECK Gene is Associated with Promoter Hypermethylation and Poor Survival in Hepatocellular Carcinoma
Changsong Zhang1,2,4*, Yang Ling1*, Chenghui Zhang3*, Yun Xu3, Lu Gao2, Rong Li2, Jing Zhu1, Lieying Fan4, Lixin Wei2 ![]()
1. Clinical Oncology Laboratory, Changzhou Tumor Hospital, Soochow University, Changzhou, China;
2. Tumor Immunology and Gene Therapy Center, Eastern Hepatobiliary Surgery Hospital, the Second Military Medical University, Shanghai, China;
3. Department of Oncology, Nanyang Center Hospital, Nanyang, China;
4. Department of Clinical Laboratory, Shanghai East Hospital, Tongji University Medical School, Shanghai, China.
* These authors contributed equally to this work.
Received 2012-1-2; Accepted 2012-2-28; Published 2012-3-3
Abstract
Background: To evaluate the promoter methylation status of RECK gene and mRNA expression in patients with hepatocellular carcinoma (HCC).
Methods: We analyzed RECK methylation by MSP, and RECK mRNA by real-time PCR in 74 HCC. The liver cell lines (7721, Chang and Hep-G2) were treated with 5-Aza-CdR and TSA.
Results: RECK mRNA were lower in HCC tissues (Mean -∆Ct = -3.29) than that in Non-Hcc tissues (Mean -∆Ct = -2.42). Expression of RECK was elevated in only 24 (32.43%) of the 74 HCC patients but decreased (-∆∆Ct<0) in 50 (67.57%) of the patients. RECK promoter was hypermethylated in 55.4% (41/74) of HCCs, and in only 17.6% (13/74) of Non-Hcc samples. RECK mRNA were lower in HCC patients with hypermethylation (∆MI>=0.5) (Mean -∆∆Ct = -1.75) than those with demethylation (∆MI<0.5) (Mean -∆∆Ct = 0.05), and there is a decreased tendency for RECK mRNA in HCC patients with promoter hypermethylation (p = 0.002). There was a significantly correlation found between RECK mRNA and poor survival after surgery. After treated by 5-Aza-CdR and TSA, we found that RECK mRNA induced different changes in 7721, Chang and Hep-G2 cells. And RECK demethylation also induced by epigenetic inhibitors.
Conclusion: The results suggested that the hypermethylation may lead to promoter silencing of RECK mRNA and associated with poor survival in HCC.
Keywords: RECK, hepatocellular carcinoma, methylation.
Introduction
The reversion-inducing cysteine-rich protein with Kazal motifs (RECK) is anchored to the cell surface via glycosylphosphatidylinositol, which can suppress tumour invasion and metastasis through involving in breakdown of the extracellular matrix [1]. RECK modulates the function of MT1-MMP in directly inhibiting its proteolytic activity and modulating the endocytic pathway. Hence, RECK could play a key role of tumor suppression by involving the regulation of angiogenesis and maintenance of vascular integrity [2].
RECK appears to down-regulation and have a good prognostic significance across several cancers. Reduced RECK expression was an independent and significant factor to predict a poor prognosis in Non-small-cell lung carcinoma, colorectal cancer, pancreatic cancer, and gastric cancer [3-6]. The level of RECK gene expression is inversely correlated with histological grade, infiltrative depth and lymphatic metastasis in esophageal squamous cell carcinoma (ESCC) [7]. The breast carcinoma patients whose tumors exhibited low levels of RECK had a poor 5-year RFS rate than those with high RECK expression [8]. RECK displays as a metastasis suppressor in cholangiocarcinoma, and upregulation of RECK expression could provide a potential therapy to improve the prognosis [9].
There are almost certainly pathways by which RECK is down-regulated in cancer. Hypoxia induces RECK downregulation through the recruitment of HDAC1 and HIF-1α to the rHRE2 site in the promoter and the inhibition of hypoxic RECK silencing would be a therapeutic and preventive target for early tumorigenesis. However, the CpG island promoter hypermethylation is associated silencing of tumor suppressor genes, which is the most recognized epigenetic disruption in human tumors [10]. RECK downregulation is caused by promoter methylation in metastatic non-small cell lung cancer and promoter methylation was also detected in human lung cancer cell lines [11]. Hypermethylation of RECK promoter is also a common event in human ESCC, which occurs concurrently in tumor-adjacent normal mucosa and is correlated with poor prognosis in ESCC patients [12]. It implied that the promoter methylation of RECK could play a particularly important regulatory role for RECK expression in carcinogenesis.
The molecular mechanisms behind RECK transcriptional regulation are clearly complex, and are still yet to be fully defined. So far the few studies conducted focused on RECK methylation and no data are available regarding RECK in HCC. The purpose of the study was to investigate the relationship between the expression of RECK and promoter methylation in HCC, and determined the effects of Trichostatin A (TSA) treatment on the methylation status and expression level of the RECK gene in liver cell lines.
Materials and methods
Patients and tissue samples
This study was approved by our institutional review board. A total of 74 patients with HCC who underwent complete tumor resection at the Second Military Medical University between January 2008 and December 2009 and whose histological specimens were available for immunohistochemical staining (IHS) were retrospectively reviewed. The patients consisted of 65 men and 9 women, ranging in age from 27 to 70 years (mean ± SD, 49.51 ± 11.12 years). All tumors were histologically diagnosed as HCC according to the Edmondson-Steiner classification system [13]. In all cases, H&E-stained slides were re-examined independently by three experienced pathologists without any knowledge of clinical data. The matched Non-Hcc tissues were obtained from the liver >3 cm away from tumors and were confirmed to be free of tumor by microscopic examination. The frozen tissue samples were flash frozen in liquid nitrogen immediately after surgical resection until analysis.
Cell Culture and Treatment
The liver cell lines (7721, Chang and Hep-G2) were kept in our laboratory. The cells were cultured in RPMI medium plus 10% fetal bovine serum in a humidified 37°C incubator containing 5% CO2. They were plated and treated with final concentration of 10 µM 5-Aza-2'-deoxycytidine (5-Aza-CdR, Sigma) and 400 ng/ml TSA. The fresh medium was changed every 24 hours to maintain the 5-Aza-CdR and TSA concentration. RNA was isolated 3 days after treatment.
Methylation analysis of RECK
Genomic DNA was isolated by Commercial QIAamp DNA Mini kit (QIAGEN, Hilden, Germany). The bisulfite modification of DNA was done according to described previously [14]. RECK methylation was measured using a methylation-specific PCR assay as previously described [15]. Primers used were: unmethylated RECK forward primer, 5'- TAAAGAGTTTTGGTATGGGGTATGT-3', reverse primer, 5'- CTCCA AACCACAAAATACTCAAA-3'; methylated RECK forward primer, 5'- AATAAAGAGTTTTGGTACGGGGTAC-3', reverse primer, 5'- AAAACCGCGAAATACTCGAA-3'. Two microliter of modified genomic DNA samples were PCR amplified in a total volume of 50 μl. The PCR was performed in a thermal cycler for 40 cycles consisting of denaturation at 95°C for 30 sec, annealing at 58°C for 30 sec, and extension at 72°C for 30 sec, followed by a final 10 min extension. The PCR products were separated on 2% agarose gels and visualized using ethidium bromide staining. The methylation index (MI) of RECK was calculated by the following formula: 100 x methylated reaction/(unmethylated reaction + methylated reaction) [16]. ΔMI defined as MIHCC - MINon-Hcc.
Quantitative real-time PCR analysis for RECK
Total RNA was isolated from 74 HCC, adjacent normal tissues, and cultured cells. In brief, total RNA was extracted with TRIzol reagent, according to the protocol provided by the manufacturer. The quantity and quality of the RNA samples were measured carefully by spectrophotometer and electrophoresis. The first-strand cDNA was synthesised from 2 μg of total RNA. Primer sequences of RECK for reverse transcription-PCR (RT-PCR) reaction were forward (5'- CCTCAGTGAGCACAGTTCAGA-3') and reverse (5'- GCAGCACACACACTGCTGTA-3') [15]. Quantitative real-time PCR (qPCR) were carried out by using the Mx3000P QPCR System (Stratagene, USA). The cDNA was then used for qPCR in a 20 μl SYBR Premix Ex Taq. qPCR for RECK mRNA expression was performed under the following conditions: 5 min at 95°C, 40 cycles of 30 seconds at 95°C, 30 seconds at 60°C, and 1min at 72°C. As an internal control for qPCR, β-actin mRNA expression was amplified from the same cDNA samples. All results were normalized to β-actin amplification. CT values for triplicate reactions were averaged and relative RECK expression was determined with the comparative CT method, using average CT values for RECK and β-actin.
Statistical analysis
All data were generated without knowledge of the clinical status of the samples analyzed by SPSS 17.0 software (SPSS, Inc., Chicago, USA). Associations between categorical variables were examined by using the Pearson χ2 and Fisher exact tests. Kaplan-Meier analysis and the log rank test were performed to identify survival differences in HCC. A p value of less than 0.05 was considered statistically significant.
Results
The levels of RECK mRNA in HCC and corresponding Non-Hcc tissues
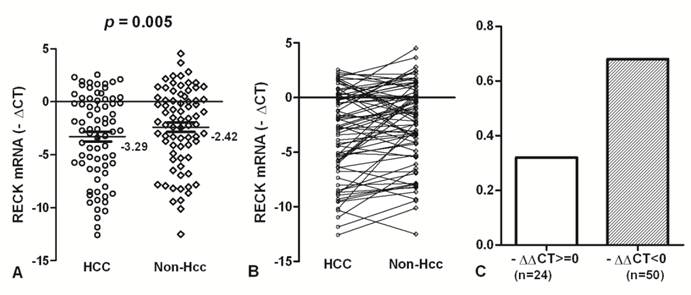
To accurately quantify relatively RECK mRNA levels, we used a real-time PCR assay in 74 HCC and matched Non-Hcc tissues. Overall results of RECK mRNA are summarized in Fig. 1. We found that RECK mRNA expression lower in HCC tissues (Mean -∆Ct = -3.29; 95%CI, -4.23 - -2.36) than that in Non-Hcc tissues (Mean -∆Ct = -2.42; 95%CI, -3.27 - -1.54). There was a significant difference was found in RECK expression between HCC tissues and matching Non-Hcc tissues (p = 0.005; Fig.1A). There was a decreased tendency for RECK expression from Non-Hcc tissues to HCCs, and more HCC samples showed lower RECK expression (Fig. 1B). Expression of RECK was elevated (-∆∆Ct>=0) in only 24 (32.43%) of the 74 HCC patients but decreased (-∆∆Ct<0) in 50 (67.57%) of the patients (Fig. 1C). It suggests that RECK genes expression may be critical for the development of HCC.
The expression of RECK mRNA in HCC tissues. (A)The RECK mRNA was indicated in HCC and Non-Hcc samples. Data are shown by the Mean -∆CT and 95%CI. The RECK mRNA in HCC was lower than that in the matched Non-Hcc tissues (p = 0.005). (B,C) There was a decreased tendency for RECK expression from Non-Hcc tissues to HCC. Expression of RECK was elevated (-∆∆Ct>=0) in only 24 of the 74 HCC patients but decreased (-∆∆Ct<0) in 50 of the patients. Statistical analyses were done using the paired t test.
Association of RECK mRNA with clinicopathologic features
The relationship between RECK mRNA status and known clinicopathologic factors in 74 tumor tissues were examined. Initially analyzed were the associations between mRNA status and available clinical information including age, gender, differentiation of the tumor, presence of hepatitis, presence of cirrhosis, tobacco, alcohol, AFP. These analyses were summarized in Table 1.
Correlation of clinicophthologic variables with RECK mRNA in HCC.
| Variable | No.(74) | Mean-∆ct ( ± SE) | P value* | |
|---|---|---|---|---|
| Gender | Male | 65 | -0.87 ( ± 0.33) | 0.974 |
| Female | 9 | -0.90 ( ± 0.51) | ||
| Age(y) | >= 50 | 34 | -1.04 ( ± 0.37) | 0.616 |
| < 50 | 40 | -0.73 ( ± 0.45) | ||
| HBV history | Yes | 43 | -0.52 ( ± 0.40) | 0.169 |
| No | 31 | -1.36 ( ± 0.44) | ||
| Differentiation | G1 | 62 | -0.64 ( ± 0.33) | 0.071 |
| G2-3 | 12 | -2.10 ( ± 0.55) | ||
| Cirrhosis | Yes | 39 | -0.75 ( ± 0.43) | 0.662 |
| No | 35 | -1.01 ( ± 0.42) | ||
| Tobacco | Yes | 33 | -0.87 ( ± 0.44) | 0.984 |
| No | 41 | -0.88 ( ± 0.41) | ||
| Alcohol | Yes | 14 | -0.38 ( ± 0.75) | 0.431 |
| No | 60 | -0.99 ( ± 0.33) | ||
| HBV (copy/ml) | >= 105 | 25 | -1.09 ( ± 0.55) | 0.601 |
| < 105 | 49 | -0.76 ( ± 0.35) | ||
| AFP (µg/ l) | >= 30 | 45 | -0.40 ( ± 0.37) | 0.072 |
| < 30 | 29 | -1.50 ( ± 0.49) |
* Statistical significance determined using the independent samples test.
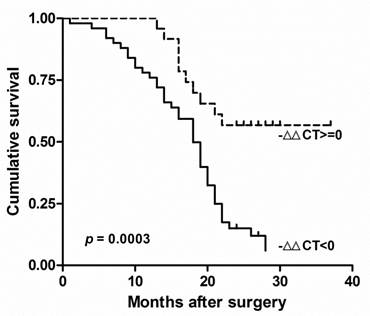
There was no other significant correlation found between clinicopathological factors and RECK mRNA in Chinese HCC. The survival of these patients was compared by the Kaplan-Meier method and the log rank test (Fig. 2). We found the decreased Expression of RECK (-∆∆Ct<0) were significantly correlated with poor overall survival (p = 0.0003; HR=0.336, 95% CI: 0.19-0.60). These results suggested that RECK mRNA level could be prognostic factors in HCC.
RECK mRNA associated with poor prognosis in HCC. Kaplan-Meier analysis of survival months after surgery according to RECK mRNA level. The decreased Expression of RECK (-∆∆Ct<0) were significantly correlated with poor overall survival after surgery.
Methylation status of RECK promoter in HCC and its adjacent tissue
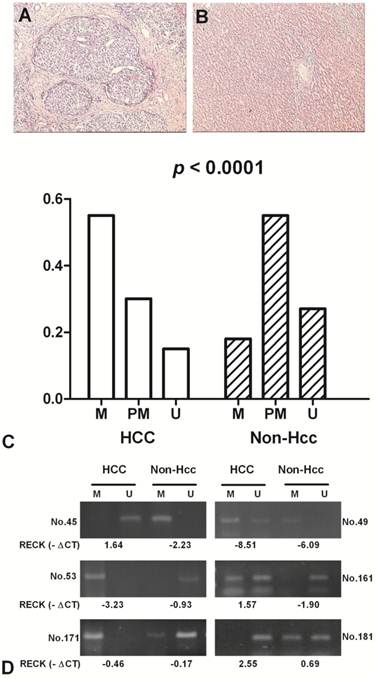
The methylation status of RECK promoter region was analysed as one of the putative regulatory mechanisms of RECK mRNA expression in HCCs and their adjacent normal tissues. The hypermethylation contains only methylated PCR product, the partial methylation contains both methylated and unmethylated PCR products, and the unmethylation contains only unmethylated product [17]. RECK promoter was hypermethylated in 55.4% (41/74) of HCCs, and in 17.6% (13/74) of Non-Hcc samples; partial methylated in 29.7% (22/74) vs 55.4% (41/74); unmethylated in 14.9% (11/74) vs 27.0% (20/74). The difference of RECK methylation between the tumour and Non-Hcc groups was statistically significant (p < 0.0001) (Fig 3).
The methylation frequency of RECK promoter in HCC patients by MSP. Representative H&E staining in HCC (A) and the matched Non-Hcc tissues (B). (C) Methylation of RECK promoter in HCC and corresponding Non-Hcc tissues. M, hypermethylation; PM, partial methylation; U, unmethylation; (D) Representative patterns of RECK promoter methylation. U, reaction specific for unmethylated DNA; M, reaction specific for methylated DNA. -∆Ct, - (CTRECK - CTβ-actin).
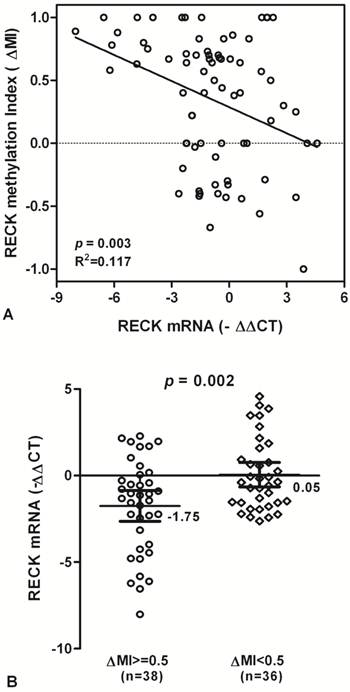
RECK mRNA and promoter methylation in HCC patients. (A) The scatter plots summarized relative expression levels of RECK mRNA (-∆∆CT) associated with relative methylation index (∆MI) in HCC patients. (B) RECK mRNA expression were lower in HCC patients with ∆MI>= 0.5 than those with ∆MI<0.5. Statistical analyses were done using the unpaired t test. Statistical analyses were done using Pearson correlation test. -∆∆CT, - (∆CTHCC-∆CTNon-Hcc); ∆MI, MIHCC-MINon-Hcc.
Association of RECK methylation with RECK mRNA expression in HCC and corresponding Non-Hcc tissues
To test whether RECK promoter methylation in HCC might be correlated with repression of RECK mRNA transcription, qPCR was used for the expression of RECK transcripts in all tissue samples. We found that RECK methylation is correlated significantly with RECK mRNA expression, and there is a decreased tendency for RECK mRNA in HCC patients with promoter hypermethylation (R2 Linear = 0.117, p = 0.003; Fig. 3A).
The levels of RECK mRNA expression were significantly decreased in HCC samples with methylation (∆MI>=0.5) than in those with hypomethylation (∆MI<0.5) (Mean -∆Ct ± SE, -1.75 ± 0.44 and 0.05 ± 0.35, respectively; p = 0.002; Fig. 3B). The results suggested that HCC showing hypermethylation of RECK promoter may lead to silencing of RECK mRNA.
RECK mRNA by 5-Aza-CdR and TSA
To analyze the effects of epigenetic inhibitor on RECK gene expression, Real-time PCR analyses were performed using liver cell lines (7721, Chang and Hep-G2) treated with final concentration of 10 µM 5-Aza-CdR and 400 ng/ml TSA. After normalizing mRNA levels to β-actin, a 1.6-2.4 ∆Ct induction of RECK mRNA was detected after 5-Aza-CdR treatment in Chang and 7721 cells, but decrease for Hep-G2 cells (Fig. 5A). Additionally, qRT-PCR assays found that the expression of RECK gene was induced 3.2-6.9 ∆Ct after TSA treatment in Hep-G2 and Chang cells, and undetectable in 7721 cells. For the 5-Aza-CdR + TSA treatment, we found that a 1.4-4.4 ∆Ct induction of RECK mRNA was detected in three cells. Taken together, the above results suggested that the expression of RECK can be activated by 5-Aza-CdR or TSA, and the effect on RECK expression is very various for different cells.
Effect of 5-aza-CdR and TSA on expression of RECK mRNA in liver cell lines. (A) The RECK mRNA was detected by real-time PCR for 7721, Chang, and Hep-G2 treated with 5-aza-CdR, TSA, alone or combination. (B) It indicated that RECK demethylation induced significantly by TSA or TSA in 7721, Chang and Hep-G2 cells.
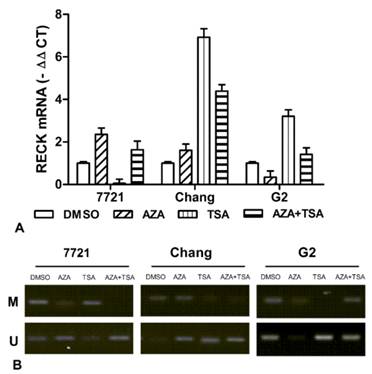
We also examined promoter methylation of RECK in liver cell lines. Meanwhile, we observed the effects of 5-aza-CdR and TSA on promoter methylation of RECK gene by MSP. The RECK promoter was found to be hypermethylated in 7721 and Chang, but partial methylation in HepG2 cells (Figure 5B). The demethylation of RECK was found by 5-aza-CdR or TSA treatment in three cells. However, it showed different effects on RECK methylation. These data suggested the demethylation of RECK promoter by epigenetic inhibitor could play an important role for restoration of RECK expression.
Discussion
The RECK gene is widely expressed in normal human tissues but is downregulated in tumor cell lines. RECK encodes a membrane-anchored glycoprotein that suppresses tumor invasion and angiogenesis by regulating matrix-metalloproteinases [18]. Understanding of the transcriptional regulation of RECK constitutes a potent approach to the molecular basis of liver malignant. DNA methylation of the RECK promoter and histone acetylation/deacetylation have been studied to understand the underlying mechanisms of RECK expression [19].
RECK gene is a common negative target for oncogenic signals and that its downregulation is a step in the pathway towards malignant conversion. Our study confirmed that RECK was lower expression in HCC tissues than corresponding Non-Hcc tissues by Real-time PCR. Meanwhile, 24 of HCC patients were elevated for RECK mRNA, and 50 cases were decreased for RECK mRNA. RECK mRNA was a statistically significant difference between HCC and Non-Hcc tissues. However, the loss of RECK mRNA could play an important role in the pathogenesis of HCC. Recent studies implied RECK expression is closely related to tumor invasion and metastasis, but there is no difference significantly between the level of RECK and clinical factors in our HCC patients. Our findings implied that HCC patients with high RECK mRNA expression tended to show better survival, which support that the feasibility of RECK mRNA as a promising prognostic molecular marker for HCC. These results support the view that RECK is an important tumor suppressor gene.
Aberrant CpG methylation is common in cancer development and may play an important role in the carcinogenic process. Recent studies have also shown that RECK methylation is associated with increase of metastasis and invasion in human cancers. The hypermethylation of RECK is associated with a low level of mRNA expression in oral squamous cell carcinoma cell lines [12]. We found that the frequency of RECK hypermethylation was higher in HCC samples than in non-tumor tissues, and more unmethylation of RECK in corresponding normal tissues. Meanwhile, the RECK mRNA expression were significantly decreased in HCC samples with hypermethylation (∆MI>=0) than in those with hypomethylation (∆MI>=0). The results suggested that HCC showing hypermethylation of RECK promoter may lead to silencing of RECK mRNA. This study implied also that RECK expression could be regulated by DNA methylation in the promoter region in HCC.
DNMT inhibitor could reverse promoter methylation and restores related gene expression in human cancer cell Lines [20]. DNMT inhibitor may restore RECK expression to inhibit cell invasion in colon cancer and lung cancer cell lines [11, 15]. EGCG, a major component of green tea, also may enhance RECK expression by reversal of hypermethylation of RECK promoter and inhibit MMP activities as well as cancer cell invasion in OSCC cell lines [21]. DNMT inhibitors may be useful for the treatment of RAS-induced metastasis by restored RECK expression [22]. And HDAC inhibitors, TSA, up-regulated RECK via transcriptional activation in CL-1 human lung cancer cells or restored the expression of RECK under hypoxic conditions [23, 24]. Our results showed that liver cells treated with 5-Aza-CdR or TSA gave a different change in RECK mRNA. The 5-Aza-CdR alone treatment was more effective in 7721 and Chang than Hep-G2. The TSA alone treatment was more effective in Chang and Hep-G2 than 7721. And the combination treatment has a change on three cells. Meanwhile, we observed the different effects of epigenetic inhibitors on promoter demethylation of RECK gene in three cells.
Taken together, RECK promoter hypermethylation is frequently observed in HCC and is associated with loss of mRNA expression, and decreased RECK mRNA is significantly correlated with worse survival in HCC. Additional work is needed to determine the mechanism of promoter methylation and RECK mRNA in liver tumourigenesis.
Acknowledgements
This project was supported by the Natural Science Foundation of China (NO: 30901722, 81000970, 81030041, 31171321, 81101622); Shanghai Science and Technology Committee (NO: 11ZR1449500, 10411963100, 10ZR1439900, 10ZR1439600); Key Basic Research Project of China (NO: 2010CB945600, 2011CB966200); Science Fund for Creative Research Groups, NSFC, China (NO: 30921006); the Natural Science Foundation of Jiangsu (NO: BK2011251). The Health Talents and “333” Project for Jiangsu (NO: LJ201157, RC2011038, BRA2011038), and the Technology Planning Project of Changzhou (CS2008214, CS20102021, CS20102022); and the Talents Project (“831” and health) of Changzhou.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Clark JC, Thomas DM, Choong PF, Dass CR. RECK--a newly discovered inhibitor of metastasis with prognostic significance in multiple forms of cancer. Cancer Metastasis Rev. 2007;26:675-83 doi:10.1007/s10555-007-9093-8
2. Noda M, Takahashi C, Matsuzaki T, Kitayama H. What we learn from transformation suppressor genes: lessons from RECK. Future Oncol. 2010;6:1105-16 doi:10.2217/fon.10.80
3. Takenaka K, Ishikawa S, Yanagihara K, Miyahara R, Hasegawa S, Otake Y. et al. Prognostic significance of reversion-inducing cysteine-rich protein with Kazal motifs expression in resected pathologic stage IIIA N2 non-small-cell lung cancer. Ann Surg Oncol. 2005;12:817-24 doi:10.1245/ASO.2005.09.018
4. van der Jagt MF, Sweep FC, Waas ET, Hendriks T, Ruers TJ, Merry AH. et al. Correlation of reversion-inducing cysteine-rich protein with kazal motifs (RECK) and extracellular matrix metalloproteinase inducer (EMMPRIN), with MMP-2, MMP-9, and survival in colorectal cancer. Cancer Lett. 2006;237:289-97 doi:S0304-3835(05)00554-9 [pii] 10.1016/j.canlet.2005.06.009
5. Masui T, Doi R, Koshiba T, Fujimoto K, Tsuji S, Nakajima S. et al. RECK expression in pancreatic cancer: its correlation with lower invasiveness and better prognosis. Clin Cancer Res. 2003;9:1779-84
6. Song SY, Son HJ, Nam E, Rhee JC, Park C. Expression of reversion-inducing-cysteine-rich protein with Kazal motifs (RECK) as a prognostic indicator in gastric cancer. Eur J Cancer. 2006;42:101-8 doi:S0959-8049(05)00871-3 [pii] 10.1016/j.ejca.2005.09.016
7. Li SL, Gao DL, Zhao ZH, Liu ZW, Zhao QM, Yu JX. et al. Correlation of matrix metalloproteinase suppressor genes RECK, VEGF, and CD105 with angiogenesis and biological behavior in esophageal squamous cell carcinoma. World J Gastroenterol. 2007;13:6076-81
8. Span PN, Sweep CG, Manders P, Beex LV, Leppert D, Lindberg RL. Matrix metalloproteinase inhibitor reversion-inducing cysteine-rich protein with Kazal motifs: a prognostic marker for good clinical outcome in human breast carcinoma. Cancer. 2003;97:2710-5 doi:10.1002/cncr.11395
9. Namwat N, Puetkasichonpasutha J, Loilome W, Yongvanit P, Techasen A, Puapairoj A. et al. Downregulation of reversion-inducing-cysteine-rich protein with Kazal motifs (RECK) is associated with enhanced expression of matrix metalloproteinases and cholangiocarcinoma metastases. J Gastroenterol. 2011;46:664-75 doi:10.1007/s00535-010-0345-y
10. Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17:330-9 doi:nm.2305 [pii] 10.1038/nm.2305
11. Chang HC, Cho CY, Hung WC. Downregulation of RECK by promoter methylation correlates with lymph node metastasis in non-small cell lung cancer. Cancer Sci. 2007;98:169-73 doi:CAS367 [pii] 10.1111/j.1349-7006.2006.00367.x
12. Long NK, Kato K, Yamashita T, Makita H, Toida M, Hatakeyama D. et al. Hypermethylation of the RECK gene predicts poor prognosis in oral squamous cell carcinomas. Oral Oncol. 2008;44:1052-8 doi:S1368-8375(08)00048-1 [pii] 10.1016/j.oraloncology.2008.02.004
13. Bruix J, Sherman M, Llovet JM, Beaugrand M, Lencioni R, Burroughs AK. et al. Clinical management of hepatocellular carcinoma. Conclusions of the Barcelona-2000 EASL conference. European Association for the Study of the Liver. J Hepatol. 2001;35:421-30
14. Zhang C, Xu Y, Zhao J, Fan L, Jiang G, Li R. et al. Elevated expression of the stem cell marker CD133 associated with Line-1 demethylation in hepatocellular carcinoma. Ann Surg Oncol. 2011;18:2373-80 doi:10.1245/s10434-011-1599-1
15. Cho CY, Wang JH, Chang HC, Chang CK, Hung WC. Epigenetic inactivation of the metastasis suppressor RECK enhances invasion of human colon cancer cells. J Cell Physiol. 2007;213:65-9 doi:10.1002/jcp.21089
16. Bais AJ, Gardner AE, McKenzie OL, Callen DF, Sutherland GR, Kremmidiotis G. Aberrant CBFA2T3B gene promoter methylation in breast tumors. Mol Cancer. 2004;3:22. doi:10.1186/1476-4598-3-22 1476-4598-3-22 [pii]
17. Zhang C, Guo X, Zhang L, Lu Z, Ma N, Cheng Y. et al. Methylation-related silencing of p14ARF gene correlates with telomerase activity and mRNA expression of human telomerase reverse transcriptase in hepatocellular carcinoma. J Surg Oncol. 2008;98:462-8 doi:10.1002/jso.21131
18. Simizu S, Takagi S, Tamura Y, Osada H. RECK-mediated suppression of tumor cell invasion is regulated by glycosylation in human tumor cell lines. Cancer Res. 2005;65:7455-61 doi:65/16/7455 [pii] 10.1158/0008-5472.CAN-04-4446
19. Sasahara RM, Brochado SM, Takahashi C, Oh J, Maria-Engler SS, Granjeiro JM. et al. Transcriptional control of the RECK metastasis/angiogenesis suppressor gene. Cancer Detect Prev. 2002;26:435-43
20. Ferguson LR, Tatham AL, Lin Z, Denny WA. Epigenetic regulation of gene expression as an anticancer drug target. Curr Cancer Drug Targets. 2011;11:199-212 doi:EPub-Abstract-CCDT-102 [pii]
21. Kato K, Long NK, Makita H, Toida M, Yamashita T, Hatakeyama D. et al. Effects of green tea polyphenol on methylation status of RECK gene and cancer cell invasion in oral squamous cell carcinoma cells. Br J Cancer. 2008;99:647-54 doi:6604521 [pii] 10.1038/sj.bjc.6604521
22. Chang HC, Cho CY, Hung WC. Silencing of the metastasis suppressor RECK by RAS oncogene is mediated by DNA methyltransferase 3b-induced promoter methylation. Cancer Res. 2006;66:8413-20 doi:66/17/8413 [pii] 10.1158/0008-5472.CAN-06-0685
23. Liu LT, Chang HC, Chiang LC, Hung WC. Histone deacetylase inhibitor up-regulates RECK to inhibit MMP-2 activation and cancer cell invasion. Cancer Res. 2003;63:3069-72
24. Jeon HW, Lee YM. Inhibition of histone deacetylase attenuates hypoxia-induced migration and invasion of cancer cells via the restoration of RECK expression. Mol Cancer Ther. 2010;9:1361-70 doi:1535-7163.MCT-09-0717 [pii] 10.1158/1535-7163.MCT-09-0717
Author contact
![]() Corresponding author: Lixin Wei, MD., Ph.D.,Tumor Immunology and Gene Therapy Center, Eastern Hepatobiliary Surgery Hospital, The Second Military Medical University, 225 Changhai Road, Shanghai 200438, China. Tel: +86-21-81875331 Fax : +86-21-65566349 E-mail addresse: lixinweiedu.cn or Changsong Zhang, Clinical Oncology Laboratory, Changzhou Tumor Hospital, Soochow University, No.1 Northern huaide Road, 213001, Changzhou, China. Phone: +86-519-86867281; Fax : +86-519-86867830 E-mail: zhangchangsongedu.cn.
Corresponding author: Lixin Wei, MD., Ph.D.,Tumor Immunology and Gene Therapy Center, Eastern Hepatobiliary Surgery Hospital, The Second Military Medical University, 225 Changhai Road, Shanghai 200438, China. Tel: +86-21-81875331 Fax : +86-21-65566349 E-mail addresse: lixinweiedu.cn or Changsong Zhang, Clinical Oncology Laboratory, Changzhou Tumor Hospital, Soochow University, No.1 Northern huaide Road, 213001, Changzhou, China. Phone: +86-519-86867281; Fax : +86-519-86867830 E-mail: zhangchangsongedu.cn.