Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(4):522-532. doi:10.7150/ijbs.4164 This issue Cite
Research Paper
TWIST Represses Estrogen Receptor-alpha Expression by Recruiting the NuRD Protein Complex in Breast Cancer Cells
Junjiang Fu1 ![]() , Lianmei Zhang1, Tao He1, Xiuli Xiao1, Xiaoyan Liu1, Li Wang1, Luquan Yang1, Manman Yang1, Tiandan Zhang1, Rui Chen2, Jianming Xu1,3
, Lianmei Zhang1, Tao He1, Xiuli Xiao1, Xiaoyan Liu1, Li Wang1, Luquan Yang1, Manman Yang1, Tiandan Zhang1, Rui Chen2, Jianming Xu1,3
1. The Research Center for Preclinical Medicine, Luzhou Medical College, Luzhou City, China 646000
2. Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas 77030, USA
3. Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, Texas 77030, USA
Received 2012-1-19; Accepted 2012-3-8; Published 2012-3-22
Abstract
Loss of estrogen receptor α (ERα) expression and gain of TWIST (TWIST1) expression in breast tumors correlate with increased disease recurrence and metastasis and poor disease-free survival. However, the molecular and functional regulatory relationship between TWIST and ERα are unclear. In this study, we found TWIST was associated with a chromatin region in intron 7 of the human ESR1 gene coding for ERα. This association of TWIST efficiently recruited the nucleosome remodeling and deacetylase (NuRD) repressor complex to this region, which subsequently decreased histone H3K9 acetylation, increased histone H3K9 methylation and repressed ESR1 expression in breast cancer cells. In agreement with these molecular events, TWIST expression was inversely correlated with ERα expression in both breast cancer cell lines and human breast ductal carcinomas. Forced expression of TWIST in TWIST-negative and ERα-positive breast cancer cells such as T47D and MCF-7 cells reduced ERα expression, while knockdown of TWIST in TWIST-positive and ERα-negative breast cancer cells such as MDA-MB-435 and 4T1 cells increased ERα expression. Furthermore, inhibition of histone deacetylase (HDAC) activity including the one in NuRD complex significantly increased ERα expression in MDA-MB-435 and 4T1 cells. HDAC inhibition together with TWIST knockdown did not further increase ERα expression in 4T1 and MDA-MB-435 cells. These results demonstrate that TWIST/NuRD represses ERα expression in breast cancer cells. Therefore, TWIST may serve as a potential molecular target for converting ERα-negative breast cancers to ERα-positive breast cancers, allowing these cancers to restore their sensitivity to endocrine therapy with selective ERα antagonists such as tamoxifen and raloxifene.
Keywords: TWIST, ERα, NuRD complex, gene repression, breast cancer
Introduction
TWIST, also known as TWIST1, is a class II member of the basic helix-loop-helix (bHLH) transcription factor family. During embryonic development, TWIST plays an essential role in specification of the mesoderm and differentiation of the mesoderm-derived tissues. During carcinogenesis TWIST expression is activated in a subset of breast, gastric, hepatocellular, prostate and bladder cancers, and its upregulation in these cancers usually correlates with low levels of E-cadherin expression, high levels of cancer aggressiveness and poor patient survival rates (1). TWIST expression in cancer cells is usually associated with cell survival, angiogenesis, epithelial-mesenchymal transition (EMT), metastasis, and drug resistance (2-4). Recent studies also suggest that TWIST expression may increase the subpopulation of breast cancer stem-like cells in culture, which may be relevant to drug resistance and distant metastasis (2, 5, 6). Mechanistically, TWIST has been shown to promote EMT, cell migration, invasion and metastasis by upregulating the expression of AKT2 (7), SNAIL1, SNAIL2 and PDGF receptor-α (4, 8, 9) and downregulating the expression of E-cadherin (2, 10, 11). Our recent study further demonstrated that TWIST directly interacts with multiple components of the nucleosome remodeling and deacetylase (NuRD) gene repression complex. Through direct association with the E-boxes proximate to the E-cadherin gene promoter, TWIST effectively recruits the NuRD complex to repress E-cadherin expression, which significantly promotes EMT, migration, invasion and metastasis of beast cancer cells (10). In addition, it has been reported that coexpression of ErbB2 or H-RasV12 with TWIST could significantly enhance TWIST-mediated EMT and override the Ras oncogene-induced premature cell senescence (12). Our study revealed that the Ras-activated MAPKs phosphorylate TWIST protein at serine 68, leading to stabilization of TWIST protein and enhancement of its function in induction of EMT and cell invasiveness in breast cancer cells (13).
Breast cancer is the most common cancer type in women (14). The estrogen receptor α (ERα) protein coded by the ESR1 gene plays an important role in regulation of mammary epithelial cell proliferation, differentiation and tumorigenesis (15). Since estrogen-activated ERα induces mammary epithelial cell proliferation, elevated estrogen or ERα activity is a risk factor in development of breast cancer and ERα function is a survival factor in estrogen-dependent ERα-positive breast cancer cells. Thus, inhibition of ERα function in breast cancer cells by selective ERα modulators (SERMs) such as tamoxifen and raloxifene serves as an effective endocrine therapy to treat estrogen sensitive breast cancers (16). However, most breast cancers treated with endocrine therapy will develop resistance to anti-ERα treatment. One reason for developing such kind of resistance is attributed to the loss of the expression of ERα and other epithelial marker genes such as E-cadherin through EMT process. Loss of ERα and gain of EMT features are also usually associated with development of resistance to chemotherapy and poor survival rate (17). Genetic deletion or mutation of the ESR1 gene is not common; instead, loss of ERα expression is mostly due to transcriptional repression by histone modifications and DNA methylation (18). EMT can be induced by multiple signaling pathways such as TGF-β/SMADs, Wnt/β-catelin and hypoxia/HIF1 signaling pathways and a group of transcription factors including TWIST, SNAIL, SLUG, Zeb1 and SIP1 that repress E-cadherin expression and enhance tumor cell migration, invasion and metastasis (19, 20). However, the crosstalk mechanisms between the loss of ERα expression and the EMT process are poorly understood.
In this study, we found that TWIST expression is inversely correlated with ERα expression in breast cancer cell lines and human breast invasive ductal carcinomas. TWIST interacts with the NuRD complex and recruits this gene repression complex to a chromatin region of the human ESR1 gene to repress ERα expression in ERα-negative breast cancer cells. Knockdown of TWIST in these cells resulted in ERα expression.
Materials and Methods
Cell lines and plasmids
The doxycycline-inducible HEK293 cell lines with stable Flag-TWIST or Flag expression were generated and maintained as described previously (10, 13). Stable 4T1 and MDA-MB-435 cell lines expressing a non-targeting shRNA or specific shRNAs that target TWIST mRNAs were also described previously (10). A 332-bp DNA fragment spanning the TWIST-binding region in intron 7 of the human ESR1 gene was amplified by PCR from a human genomic DNA sample using primers ESR1-BamH1-F (5'-cgcggatccGGGGGGAGGTTCTTCTTC) and ESR1-SalI-R (5'- acgcgtcgacTTATTGCTCAGAAATAGTCATG). This amplified DNA fragment was subcloned into the pGL3-Pro-Luc vector (Promega) for constructing the pGL-332ESR1-Pro-Luc reporter plasmid. The cloned DNA fragment was validated by DNA sequencing.
Chromatin immunoprecipitation (ChIP) and ChIP-Sequencing (ChIP-Seq) assays
ChIP assay was performed as described previously (10, 21). Briefly, the Flag-Twist and Flag control HEK293 cells were cultured in 15-cm plates to 65% confluence and treated with 0.1 μg/ml of doxycycline for 16 hours. MDA-MB-435 cells with TWIST shRNA or non-targeting shRNA expression were cultured in 15-cm plates to almost 100% confluence. The cells were cross-linked with formalin and lysed in 1 ml of cell lysis buffer containing protease inhibitors. The lysates were centrifuged at 3000g for 5 minutes. The pellets were washed and then re-suspended in 600 µl of solution with protease inhibitors and sonicated. The supernatants were pre-cleaned and diluted with 1.4 ml of solution containing 2 mM EDTA, 100 mM NaCl, 20 mM Tris-HCl (pH 8.0), 0.5% Triton X-100 and protease inhibitors. Each aliquot of 0.4 ml samples was mixed with 6 µl of M2 Flag antibody beads or specific antibodies against TWIST, MTA2, HDAC2, Mi2, acetylated histone H3K9 and methylated histone H3K9. The mixtures were incubated overnight at 4oC with rotation. The beads were sequentially washed in ChIP I, II and III buffers and the TE buffer containing 1 mM DTT. The protein-DNA complexes were eluted and digested with 5 µg of proteinase K overnight at 65oC to release DNA from cross-linked proteins. The DNA was cleaned by phenol and chloroform extraction, precipitated and dissolved in 20 µl of ddH2O for PCR analysis.
For ChIP-re-ChIP assays, the above ChIP III buffer-washed antibody beads were further washed by Tris-buffered saline and eluted using a 3×Flag peptide solution. The eluted materials were diluted and subjected to the re-ChIP step using antibodies against MTA2, HDAC2 or Mi2 and the protein A/G beads. The recovered DNA samples from ChIP and ChIP-re-ChIP were analyzed by quantitative real time PCR using three primer pairs. Primers P3 (5' agctgggctcactcacacat-3') and P4 (5'-gaggcatagagcagagtcacc-3') were used to detect the TWIST binding region mapped in intron 7 of the human ESR1 gene. Primers P1 (5'-tcacaccttgcttcgcatag-3') and P2 (5'-tgagctcagaaatccacttcc-3') were used to detect a negative control region in intron 1 of the ESR1 gene. Primers P5 (5'- acctgtgagcagggtgactt-3') and P6 (5'- acaaaaacccgcaacttcct-3') were used to detect a negative control region located 3' to the ESR1 gene.
For ChIP-Seq analysis, crosslinked DNA-protein samples were harvested from Flag-TWIST and Flag HEK293 cells and sonicated into 200-500 bp in length of DNA fragments. The prepared samples were immunoprecipitated using the M2 Flag antibody beads. The washed beads were eluted with a 3×Flag peptide solution and the yielded DNA fragements were ligated to oligo adapters using T4 DNA ligase for PCR to establish DNA libraries. The libraries were processed for massively parallel sequencing at the Human Genome Sequencing Center at Baylor College of Medicine. The Flag-TWIST binding sites were determined after subtracting the Flag-binding background. Sequencing reads were mapped onto the Human Mar. 2006 Assembly (hg18) using BWA (22) and the peaks are identified using MACS (23) and visualized with the Integrated Genome Browser (IGB) (http://www.affymetrix.com/partners_programs/programs/developer/tools/download_igb.affx).
Breast tumors and total RNA isolation
A total of 28 human breast invasive ductal carcinoma specimens were collected from surgically removed tumor tissues at Luzhou Medical College Affiliated Hospital in China with informed consent. All protocols were approved by Luzhou Medical College. All patients were Chinese women and aged from 33 to 72 years old. No patient survival data were available at this stage. The tumor tissues were immediately frozen in liquid nitrogen and stored at -80oC. Total RNA was isolated from tumor tissues and cultured cells using the TRIZOL reagent and stored at -80oC (Invitrogen).
Analysis of mRNA expression by quantitative and semi-quantitative PCR
The cDNA libraries were generated using a reverse transcriptase kit (Invitrogen) from 1 μg of total RNA. Real-time PCR was performed using the qPCR MasterMix Plus (Eurogentec) and the StepOnePlus Real-Time PCR system (Applied Biosystems) as described previously (10, 21). The primers 5'-tcctaacttgctcttggacagg-3' and 5'-gtagccagcagcatgtcg-3' and the TaqMan probe 22 were used to measure ERα cDNA levels in the libraries. The primers 5'-gggccggagacctagatg-3' and 5'-tttccaagaaaatctttggcata-3' and the TaqMan probe 50 were used to measure TWIST cDNA levels in the libraries.
For semi-quantitative RT-PCR using breast tumor mRNA samples, reverse transcription and PCR were performed using the following mRNA-specific primers: RT-ESR1-5 (5'-gtgcctggctagagatcctg) and RT-ESR1-3 (5'-agagacttcagggtgctgga) for measuring ERα mRNA; RT-TWIST-5 (5'-acgagctggactccaagatg) and RT-TWIST-3 (5'-cacgccctgtttctttgaat) for measuring TWIST mRNA. The GAPDH mRNA was measured in the same reaction for each sample to serve as an endogenous control for normalizing the relative levels of ERα and TWIST mRNAs.
Western Blotting
Western blotting analysis was performed as described previously (10, 21). Antibodies for TWIST1, E-cadherin, β-actin and HA-tag were used previously (10, 13), anti-ERα monoclonal antibody (D-12) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Luciferase reporter assay
Cells were cultured to 40% confluence in 12-well plates and then co-transfected with indicated amount of pGL3-Pro-Luc or pGL-332ESR1-Pro-Luc reporter plasmid, indicated amount of pSG5-HA-TWIST plasmid or pSG5 empty plasmid and 30 ng of β-Gal expression plasmids using Lipofectamin 2000. Luciferase and β-gal activities were measured at 2 days after transfection. The luciferase activity of each sample was normalized to the β-gal activity of the same sample. All experiments were performed in triplicates.
Immunohistochemical staining
Immunohistochemistry was done on breast tumor sections prepared from tumor tissues fixed in 10% buffered formalin and embedded in paraffin. Sections (4 μm thick) were deparaffinized in xylene and rehydrated in a series of diluted alcohol and distilled water. After antigen retrieval, endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide in methanol for 30 minutes, followed by rehydration in 1×PBS and incubation with 5% rabbit serum for 30 minutes to block nonspecific background. The slides were then incubated overnight at 4oC with a TWIST monoclonal antibody (ab 50887, abcam) or an ERα antibody in Tris-buffered saline containing 2% of serum and 1% of bovine serum albumin (BSA). The washed sections were incubated with appropriate secondary antibodies at dilution of 1:100 for 30 minutes at room temperature and then incubated with peroxidase-conjugated avidin-biotin complexes. The immunostaining signals were developed by DAB staining. The sections were counterstained with hematoxylin and analyzed by standard light microscopy. Sections incubated with TBS containing 2% rabbit serum and 1% BSA without primary antibody served as negative controls.
Statistical analysis
Paired Student's t-test was used to determine any significant differences. P-value ≤ 0.05 was considered to be significant.
Results
TWIST protein is associated with an intronic chromatin region of the human ESR1 gene
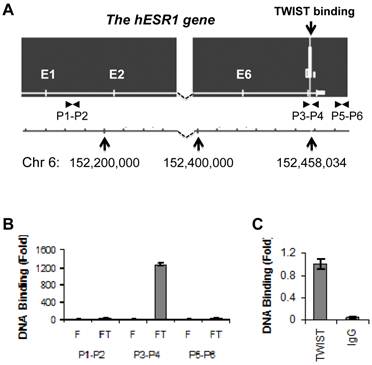
To identify potential TWIST-binding sites in the human genome, we performed ChIP-Seq analysis in previously described HEK293 cell lines that express either Flag-TWIST or Flag control (10). We indentified one TWIST association region within intron 7 of the human ESR1 gene (Fig. 1A). This binding site was centralized at bp 152458103 of chromosome 6, and TWIST bound at this site was enriched highly in Flag-TWIST cells versus Flag control cells. Sequence analysis of a 600 bp region located up and down streams of the binding peak revealed six of the potential TWIST-binding E-boxes (CANNTG). ChIP analysis using a pair of specific primers P3 and P4 spanning the mapped TWIST binding region demonstrated more than 1200-fold enrichment of TWIST in Flag-TWIST cells versus Flag control cells. However, TWIST was not enriched with the two regions used as negative controls, one in the first intron amplified by primers P1 and P2 and the other in a 3' non-coding region amplified by primers P5 and P6 (Fig. 1A and B). Furthermore, ChIP assay performed with primers P3 and P4 also proved that the endogenously expressed TWIST protein in MDA-MB-435 cells was specifically and efficiently recruited to the TWIST binding site located in the intron 7 of the human ESR1 gene (Fig. 1C).
TWIST is associated with a chromatin region in intron 7 of the ESR1 gene. A. The ChIP-Seq map showed one TWIST binding site located in intron 7 of the ESR1 gene in Flag-Twist HEK293 cells. The peak value was obtained by normalizing to the background, if any, in Flag HEK293 control cells. The locations of the ESR1 gene in chromosome 6 and the TWIST binding sites are indicated. The locations of primer pairs used for ChIP assays are also indicated. B. ChIP assays were performed using Flag-TWIST (FT) and Flag (F) HEK293 cells and Flag antibody beads. Precipitated DNA was assayed by PCR using three pairs of primers indicated in panel A. Data obtained from Flag-TWIST cells were normalized with that from Flag control cells. C. ChIP assay in MDA-MB-435 cells with endogenously expressed TWIST. ChIP assay was performed using TWIST antibody or using non-immune IgG as a negative control. The precipitated DNA was assayed by PCR using primers P3 and P4 shown in panel A.
The NuRD repression complex is recruited to the TWIST-binding region of the human ESR1 gene
We have previously shown that TWIST interacts and recruits the NuRD complex to a promoter region of the E-cadherin gene in breast cancer cells (10). To examine whether the components of NuRD complex are also recruited to the TWSIT binding site of the ESR1 gene in HEK293 cells expressing Flag-TWIST, we performed sequential ChIP-re-ChIP assays using the Flag antibody in the first step ChIP, followed by the second step ChIP using antibodies specific to individual NuRD complex components, including Mi-2, MTA2 and HDAC2. HEK293 cells expressing Flag were side-by-side assayed as a background control. This experiment revealed a 100-fold enrichment of TWIST-associated Mi-2, MTA2 and HDAC2 at the TWIST binding site of the ESR1 gene in Flag-TWIST cells over the Flag control cells (Fig. 2A). This indicates that TWIST can efficiently recruit the NuRD complex to the TWIST-binding site of the ESR1 gene in HEK293 cells.
TWIST recruits NuRD complex to the TWIST-binding site of the ESR1 gene. A. ChIP-re-ChIP assays were performed with Flag HEK293 (F) and Flag-TWIST HEK293 (FT) cells using the Flag antibody beads in the first step ChIP and the antibodies against Mi2, MTA2 or HDAC2 in the second step ChIP. The precipitated DNA from the second step ChIP was assayed by real time PCR using primers P3 and P4 indicated in Fig. 1A. B. MDA-MB-435 cells expressing non-targeting shRNA (shCtrl) or shRNA targeting TWIST mRNA (shT) were used in ChIP assays with antibodies against TWIST, MTA2, HDAC2 or RbAp46 as indicated. Real-time PCR was performed as panel A. C. MDA-MB-435 cells with shCtrl or shT were used in ChIP assays with antibodies against acetylated or methylated histone H3K9 as indicated. Real-time PCR was performed as panel A.
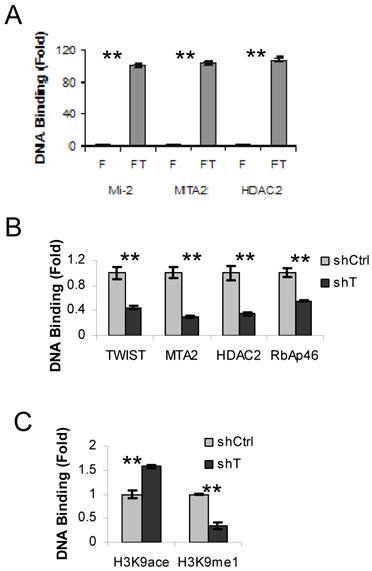
Next, we used ERα-negative MDA-MB-435 cells to check the recruitment of NuRD complex by the endogenous TWIST to the TWIST-binding site of the ESR1 gene. Indeed, TWIST and multiple NuRD complex components including MTA2, HDAC2 and RbAp46 were found to be associated with the TWIST-binding site of the ESR1 gene in MDA-MB-435 cells that express non-targeting shRNA, while these recruitments were significantly reduced in MDA-MB-435 cells with shRNA-mediated stable knockdown of TWIST (Fig. 2B). These MDA-MB-435 cell lines expressing either non-targeting shRNA or TWIST mRNA-targeting shRNA were described previously (10). These results demonstrate that the core components of NuRD complex are indeed recruited to the TWIST-binding site of the ESR1 gene by interaction with TWIST.
To evaluate the effects of recruited NuRD complex on the epigenetic changes of chromatin around the TWIST-binding site of the ESR1 gene, we examined the levels of histone acetylation and methylation by ChIP assays. Although both chromatin-associated histone H3K9 acetylation and mono-methylation were detected in the MDA-MB-435 cells expressing non-targeting shRNA, the acetylated histone H3K9 and the mono-methylated histone H3K9 were increased and decreased, respectively, in the MDA-MB-435 cells with TWIST knockdown (Fig. 2C). These results demonstrate that TWIST-mediated recruitment of NuRD complex is consistent with a repressed epigenetic chromatin modification in the TWIST-binding chromatin region of the ESR1 gene.
TWIST represses ERα expression
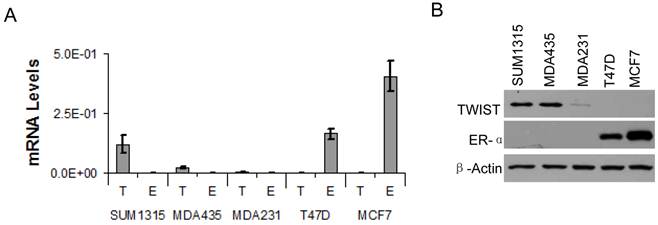
Based on the association of TWIST with the ESR1 gene and TWIST-mediated recruitment of the NuRD complex for gene repression, we hypothesized that TWIST plays an important role in suppression of ERα expression in breast cancer cells. To test this hypothesis, we first measured ERα mRNA and protein levels in several breast cancer cell lines by real-time RT-PCR and Western blot analysis. High levels of TWIST mRNA and protein were detected in the ERα-negative SUM1315 and MDA-MB-435 cell lines, and a low level of TWIST protein was also detected in the ERα-negative MDA-MB-231 cell line. In contrast, TWIST mRNA and protein were undetectable in T47D and MCF-7 cells that express high levels of ERα (Fig. 3A and B). These results indicate that TWIST expression is negatively correlated with ERα expression in these cell lines. Since the three TWIST-positive cell lines are poorly differentiated metastatic cell lines and the two TWIST-negative cell lines are much more differentiated non-metastatic cell lines as assayed in xenograft mouse models, repression of ERα expression by TWIST may also contribute to the progression of breast cancer to an EMT and metastatic stage.
TWIST expression is inversely correlated with ERα expression in human breast cancer cell lines. A. Real-time RT-PCR analysis for the mRNA levels of TWIST (T) and ERα (E) in SUM1315, MDA-MB-435 (MDA435), MDA-MB-231 (MDA231), T47D and MCF-7 human breast cancer cell lines. The data were normalized to the mRNA levels of GAPDH and presented as average ± standard deviation. B. Western blot analysis for TWIST and ERα in the above human breast cancer cell lines. β-actin was assayed as a control for the loaded total cellular proteins.
Next, we amplified a 332 bp DNA fragment containing the six E-Boxes within the TWIST-binding region from human ESR1 gene, cloned this fragment into the pGL3-Pro plasmid with a basal promoter and a luciferase reporter coding sequences. After this pGL3-332ESR1-Pro-Luc reporter plasmid was co-transfected with a TWIST expression vector or a parent control vector into HEK293T cells, we observed that overexpression of TWIST significantly repressed the activity of this reporter. In contrast, overexpression of TWIST did not repress the activity of the pGL3-Pro-Luc parent reporter without the 332ESR1 sequence (Fig. 4A). These results suggest that the TWIST/NuRD-associated region of the ESR1 gene has a transcriptional repression function.
TWIST represses ERα expression. A. 293T cells were transiently co-transfected with 120 ng of HA-TWIST expression plasmid or parent control plasmid DNA and 50 ng of pGL3-332ESR1-Pro-Luc or pGL-Pro-Luc reporter plasmid DNA as indicated. Western blot analysis using antibodies against HA and β-actin (lower panel) and luciferase activity assay (upper panel) were performed in triplicates. **, p < 0.001. B. T47D cell lines with control parent vector (T47D/V) and TWIST expression vector (T47D/T) were established. The levels of TWIST and ERα mRNAs were measured by real time RT-PCR. **, p < 0.001. C. Western blot analysis for ERα, E-cadherin, TWIST and β-actin in MCF-7 cells transfected with a control parent vector or a TWIST expression vector. D. Measurement of ERα mRNA levels by real-time RT-PCR in 4T1 and MDA-MB-435 (MDA435) cells expressing a non-targeting shRNA (shCtrl) or an shRNA targeting TWIST mRNA (shT). **, p < 0.001. E. The 4T1 and MDA-MB-435 cells with TWIST knockdown (shTWIST) or control shRNA (shCtrl) were treated with vehicle (-) or 330 nM of TSA (+) as indicated. Western blot analysis was performed using antibodies against ERα and β-actin.
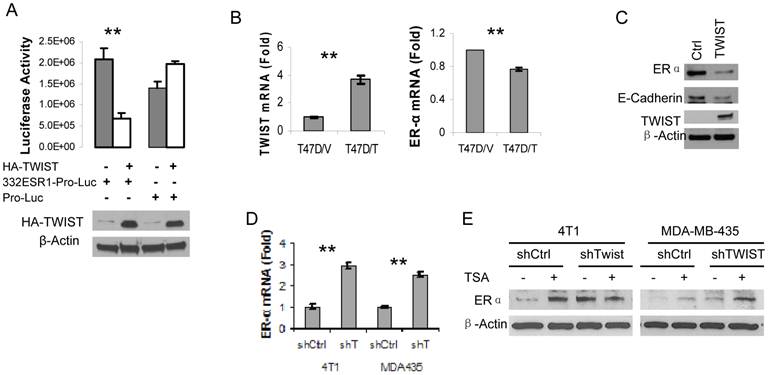
Furthermore, we addressed whether TWIST expression would repress endogenous ERα expression in TWIST-negative and ERα-positive breast cancer cells such as T47D and MCF-7 cell lines. Our experiments demonstrated that forced expression of TWIST indeed repressed ERα mRNA expression in T47D cells and reduced ERα protein level in MCF-7 cells (Fig. 4B and C). In agreement with our previous report (10), forced expression of TWIST in MCF-7 cells significantly reduced the level of E-cadherin protein (Fig. 4C). Conversely, we also tested whether knockdown of TWIST expression would result in ERα expression in TWIST-positive and ERα-negative breast cancer cells, including MDA-MB-435 and 4T1 cell lines. Our analysis revealed that knockdown of TWIST in both of these cell lines increased 2.5-3 folds of ERα mRNA expression (Fig. 4D). Accordingly, the ERα protein levels in 4T1 and MDA-MB-435 cells with non-targeting shRNA expression were extremely low, while knockdown of TWIST expression in these cells markedly increased ERα protein in these cells (Fig. 4E, lanes 1, 3, 5 and 7). Since the NuRD complex recruited by TWIST contains histone deacytalases such as HDAC2 (10, 24), we tested whether Trichostain A (TSA), a HDAC inhibitor, could inhibit TWIST/NuRD-mediated repression of ERα expression. Importantly, both TSA-treated 4T1 and MDA-MB-435 cells with non-targeting shRNA expressed much higher levels of ERα protein compared with their respective vehicle-treated control groups (Fig. 4E, compare lane 1 with lane 2 and lane 5 with lane 6). In the TSA-treated 4T1 and MDA-MB-435 cells with shRNA-mediated TWIST knockdown, ERα protein showed no significant further increase (Fig. 4E, compare the ratios of ERα to β-actin in lane 4 with lanes 2 and 3 and in lane 8 with lanes 6 and 7), suggesting that either knockdown TWIST or inhibition of NuRD HDAC activity can effectively relieve the transcriptional repression of the ESR1 gene.
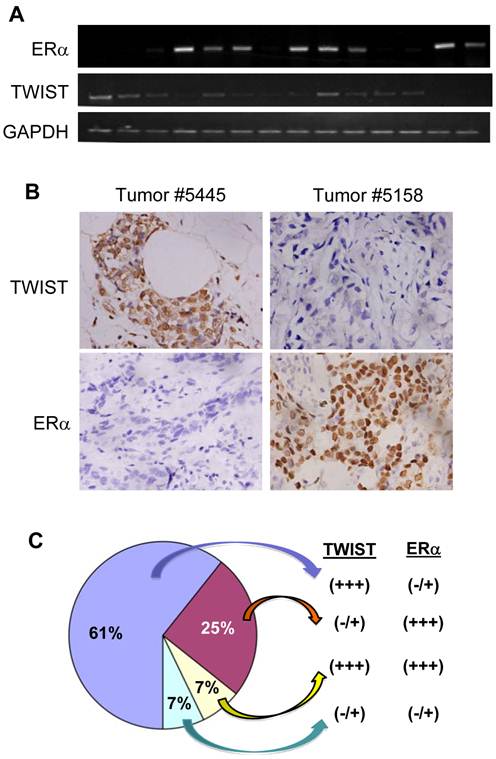
TWIST expression is negatively associated with ERα protein expression in human breast tumors
To examine the effect of TWIST on ERα expression in human breast tumors, we performed semi-quantitative RT-PCR and immunohistochemistry to measure TWIST and ERα mRNA and proteins in a total of 28 invasive breast ductal carcinomas. Representative results are shown in Fig. 5A and B. By analyzing data obtained from these assays we identified 17 (61%) tumors with high levels of TWIST mRNA and protein and very low or undetectable levels of ERα mRNA and protein, 7 (25%) tumors with very low or undetectable levels of TWIST mRNA and protein and high levels of ERα, 2 (7%) tumors with high levels of both TWIST and ERα mRNA and proteins and 2 (7%) tumor with very low or undetectable levels of both TWIST and ERα mRNA and proteins (Fig. 5C). These results indicate that TWIST expression is inversely correlated with ERα expression in most (85.7%) of the invasive breast carcinomas.
TWIST expression inversely correlates with the levels of ERα protein in human breast invasive ductal carcinomas. A. Semi-quantitative analysis of ERα, TWIST and GAPDH mRNA expression by RT-PCR in human breast invasive ductal carcinomas. A total of 28 independent tumor mRNA samples were assayed with three repeat experiments and a part of the representative results is shown. B. Immunohistochemistry analysis (brown color) for TWIST and ERα in human breast invasive ductal carcinomas. Tissue sections prepared from the 28 human invasive breast ductal carcinomas were immunostained with TWIST and ERα antibodies. Representative results obtained from two tumors are shown. Photos were taken under a microscope set at 400×magnification. C. A summary of the data according to TWIST and ERα immunohistochemistry in the 28 human invasive breast ductal carcinomas.
Discussion
Most of the breast cancers initially express ERα and depend on estrogen to grow. However, some of these ERα-positive breast cancers lose ERα and acquire an estrogen-independent feature during progression (16, 17). Because ERα-positive breast cancers have a better prognosis and are more likely to respond to endocrine therapy with SERMs such as tamoxifen and raloxifene compared with ERα-negative breast cancers, the loss of ERα and the gain of resistance to endocrine therapy represent a daunting challenge to breast cancer treatment. Therefore, understanding the molecular mechanisms responsible for repression of ERα expression may help to restore ERα expression and endocrine therapy sensitivity in ERα-negative breast cancers.
In this study, we demonstrated that TWIST expression is inversely correlated with ERα expression in both breast cancer cell lines and human breast invasive ductal carcinomas, suggesting a potential regulatory relationship between these two proteins. More importantly, expression of TWIST in ERα-positive breast cancer cells significantly reduced ERα levels, while knockdown of TWIST in ERα-negative breast cancer cells caused re-expression of endogenous ERα. These results further supported the notion that TWIST may directly repress ERα expression and contribute to breast cancer progression not only in the induction of EMT but also in the conversion of breast cancer from an ERα-positive and endocrine therapy-sensitive status to an ERα-negative and endocrine therapy-resistent status.
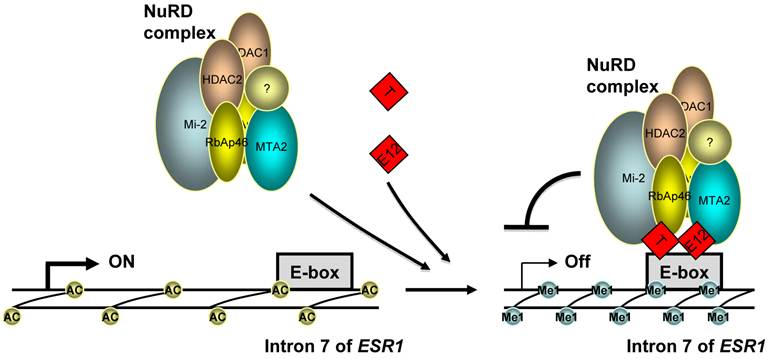
Indeed, our molecular analyses provided compelling evidence for direct repression of ERα expression by TWIST. First, we identified a functional TWIST binding region in intron 7 of the ESR1 gene by an unbiased ChIP-Seq approach and validated the association of TWIST with this site by ChIP assay in breast cancer cells with endogenous TWIST expression. Furthermore, the DNA region associated with TWIST contains multiple E-Boxes for TWIST binding and it indeed mediates TWIST-dependent repression of the transcriptional activity of a heterologous promoter linked with a luciferase reporter. Moreover, through binding to the chromatin region of the ESR1 gene and interacting with the components of the NuRD complex, TWIST effectively recruits NuRD complex to repress ERα expression. This result is consistent with our previous finding that TWIST can recruit NuRD complex to the E-cadherin promoter and repress E-cadherin expression to promote EMT and breast cancer metastasis (10). Finally, we demonstrated that the recruitment of NuRD complex decreased histone H3K9 acetylation and increased H3K9 methylation; inhibition of HDAC activity resulted in ERα expression in ERα-negative breast cancer cells. These findings are consistent with the TWIST-mediated gene repression function. Taken together, these results demonstrate that ESR1 gene is a direct target gene of TWIST and TWIST can recruit NuRD complex to directly repress ERα expression in breast cancer cells (Fig. 6).
A model for the repression of ERα expression by TWIST. TWIST protein (T) binds to E-boxes in the 7th intron of ESR1 gene as homodimer or heterodimer with E12 or TWIST2, which recruits NuRD complex to decrease H3K9 acetylation (Ac) and increase H3K9 methylation (Me1) levels, resulting in transcriptional repression of ERα expression.
Both SUM1315 and MDA-MB-435 cells express relatively high-level TWIST protein, while other types of cells examined in this study express either a low level or no TWIST. Because SUM1315 breast cancer cells grow slowly, most experiments were performed in MDA-MB-435 cells. We are fully aware of that the origin of the MDA-MB-435 cell line was somewhat controversial. It was initially described and used as a metastatic breast cancer cell line by many studies, but later was considered as a melanoma cell line due to its karyotype and microsatellite similar to those of the M14 melanoma cell line (25). However, multiple lines of evidence suggest that the MDA-MB-435 cell line is still a poorly differentiated, metastatic breast cancer cell line. Firstly, this cell line has a female origin with two X chromosomes and without Y chromosome (25), while the initial M14 melanoma cell line had a male origin (26). Furthermore, MDA-MB-435 cells can express epithelial markers and secrete milk proteins and lipids under certain experimental conditions. When the nm23 metastasis suppressor is ectopically expressed in MDA-MB-435 cells, these cells exhibit a similar morphology to normal breast epithelial cells, including acinus formation in three-dimensional culture and production of milk proteins (27, 28). In this study, we showed that knockdown of TWIST in MDA-MB-435 cells resulted in ERα expression. This finding should serve as an additional evidence to support the breast cancer origin of MDA-MB-435 cells.
During the preparation of this paper, Vesuna et al. reported that TWIST also bound a different DNA region adjacent to the ESR1 gene promoter and recruited the HDAC1-DNMT3B repressor complex to repress ERα expression (29). This study demonstrated that TWIST expression in ERα-positive cells induced tamoxifen resistance, while TWIST knockdown in ERα-negative breast cancer cells could restore ERα expression and the inhibitory effect of tamoxifen on estrogen-stimulated cell growth. Therefore, our independent findings are conceptually consistent with this recent study but provide another completely different molecular mechanism responsible for TWIST to repress ERα expression.
In conclusion, this study demonstrated that the expression and repression of the ESR1 gene in breast cancer cells are reversible processes. TWIST-NuRD complex-mediated repression is, at least in part, responsible for silencing ERα expression in TWIST-positive and ERα-negative breast cancer cells. Therefore, targeting TWIST function may facilitate restoration of ERα expression and endocrine therapy sensitivity in these breast cancer cells.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81172049, 30371493), the Scientific Research Foundation of Education Bureau of Sichuan Province (10ZA038), a Key Research Project of Luzhou City (2011-S-30) and a Starting Package of Luzhou Medical College to J.F., and the National Institutes of Health grant R01 CA112403 and the Cancer Prevention and Research Institute of Texas grant RP101251-P4 to J.X. We thank Drs. Jiemin Wong, Zhihui Yang, Mr. Jun Liu, and members of the Research Center for Preclinical Medicine of Luzhou Medical College for suggestions, experimental materials and technical assistance.
Abbreviations
ERα: estrogen receptor alpha; NuRD: The nucleosome remodelling and histone deacetylase; HDAC2: histone deacetylase 2; MTA2: metastasis associated 1 family, member 2; SERMs: Selective estrogen receptor modulators; ChIP: Chromatin immunoprecipitation; ChIP-Seq: ChIP-Sequencing; RT-PCR: Reverse transcription-PCR; IHC: immunohistochemical staining
Competing Interests
The authors have declared that no competing interest exists.
References
1. Qin Q, Xu Y, He T, Qin C, Xu J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012;22:90-106
2. Yang J, Mani SA, Donaher JL. et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927-39
3. Cheng GZ, Zhang W, Wang LH. Regulation of cancer cell survival, migration, and invasion by Twist: AKT2 comes to interplay. Cancer Res. 2008;68:957-60
4. Smit MA, Geiger TR, Song JY, Gitelman I, Peeper DS. A Twist-Snail axis critical for TrkB-induced epithelial-mesenchymal transition-like transformation, anoikis resistance, and metastasis. Mol Cell Biol. 2009;29:3722-37
5. Mani SA, Guo W, Liao MJ. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704-15
6. Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia. 2010;15:117-34
7. Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007;67:1979-87
8. Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res. 2011;71:245-54
9. Eckert MA, Lwin TM, Chang AT. et al. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell. 2011;19:372-86
10. Fu J, Qin L, He T. et al. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 2011;21:275-89
11. Vesuna F, van Diest P, Chen JH, Raman V. Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochem Biophys Res Commun. 2008;367:235-41
12. Ansieau S, Bastid J, Doreau A. et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell. 2008;14:79-89
13. Hong J, Zhou J, Fu J. et al. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011;71:3980-90
14. Komen J, Wolbers F, Franke HR, Andersson H, Vermes I, van den Berg A. Viability analysis and apoptosis induction of breast cancer cells in a microfluidic device: effect of cytostatic drugs. Biomed Microdevices. 2008;10:727-37
15. Russo J, Russo IH. The role of estrogen in the initiation of breast cancer. J Steroid Biochem Mol Biol. 2006;102:89-96
16. Duffy MJ. Estrogen receptors: role in breast cancer. Crit Rev Clin Lab Sci. 2006;43:325-47
17. Bardin A, Boulle N, Lazennec G, Vignon F, Pujol P. Loss of ERbeta expression as a common step in estrogen-dependent tumor progression. Endocr Relat Cancer. 2004;11:537-51
18. Tempfer CB, Schneeberger C, Huber JC. Applications of polymorphisms and pharmacogenomics in obstetrics and gynecology. Pharmacogenomics. 2004;5:57-65
19. Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548-58
20. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442-54
21. Fu J, Jiang J, Li J. et al. Deleted in breast cancer 1, a novel androgen receptor (AR) coactivator that promotes AR DNA-binding activity. J Biol Chem. 2009;284:6832-40
22. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754-60
23. Zhang Y, Liu T, Meyer CA. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137
24. Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999;13:1924-35
25. Rae JM, Ramus SJ, Waltham M. et al. Common origins of MDA-MB-435 cells from various sources with those shown to have melanoma properties. Clin Exp Metastasis. 2004;21:543-52
26. Tuna M, Knuutila S, Mills GB. Uniparental disomy in cancer. Trends Mol Med. 2009;15:120-8
27. Sellappan S, Grijalva R, Zhou X. et al. Lineage infidelity of MDA-MB-435 cells: expression of melanocyte proteins in a breast cancer cell line. Cancer Res. 2004;64:3479-85
28. Howlett AR, Petersen OW, Steeg PS, Bissell MJ. A novel function for the nm23-H1 gene: overexpression in human breast carcinoma cells leads to the formation of basement membrane and growth arrest. J Natl Cancer Inst. 1994;86:1838-44
29. Vesuna F, Lisok A, Kimble B. et al. Twist contributes to hormone resistance in breast cancer by downregulating estrogen receptor-alpha. Oncogene. 2012 [Epub ahead of print]
30. Denslow SA, Wade PA. The human Mi-2/NuRD complex and gene regulation. Oncogene. 2007;26:5433-8
Author contact
![]() Corresponding author: Junjiang Fu, The Research Center for Preclinical Medicine, Luzhou Medical College, 3-319 Zhongshan Road, Luzhou, Sichuan, China 646000. Telephone: (+) 86-830-3160283; Fax: (+) 86-830-3160283. E-mail: fujunjiangcom
Corresponding author: Junjiang Fu, The Research Center for Preclinical Medicine, Luzhou Medical College, 3-319 Zhongshan Road, Luzhou, Sichuan, China 646000. Telephone: (+) 86-830-3160283; Fax: (+) 86-830-3160283. E-mail: fujunjiangcom