Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(5):630-639. doi:10.7150/ijbs.3684 This issue Cite
Research Paper
Glycosylation of the Sodium Channel β4 Subunit is Developmentally Regulated and Involves in Neuritic Degeneration
Ting-ting Zhou1, Zhen-wei Zhang1, Jun Liu2, Jian-peng Zhang1 ![]() , Bing-hua Jiao1
, Bing-hua Jiao1 ![]()
1. Department of Biochemistry and Molecular Biology, Second Military Medical University, Shanghai, 200433, China;
2. Department of Neurology and Institute of Neurology, Ruijin Hospital affiliated to Shanghai Jiaotong University School of Medicine, Shanghai, 200032, China.
Received 2011-10-21; Accepted 2012-4-20; Published 2012-4-27
Abstract
Aberrant protein glycosylation plays major roles in neurodegenerative diseases, including Parkinson's disease (PD). Glycoproteomics showed that the glycosylation of sodium channel β4 was significantly increased in human brain tissue. β4-specific antibodies reacted in immunoblot assays with the 35- and 38-kDa bands from the membrane fractions isolated from neonatal PD transgenic mice but only with the 35-kDa band of the neonatal wild-type mice. The size of the 38-kDa immunoreactive protein is in close agreement with previously reported, suggesting heavy glycosylation of this protein in adult wild-type and neonatal PD transgenic brain tissues. However, the neonatal wild-type mice membrane fractions only contained the 35-kDa immunoreactive protein, and the additional 38-kDa band was not shown until postnatal day 7. Enzymatic deglycosylation of the membrane preparations only converted the 38-kDa band into a faster migrating protein, which was consistent with heavy glycosylation of this protein. The glycosylated state of β4 was developmentally regulated and was altered in disease state. Neurite outgrowth assay demonstrated that overexpression of deglycosylated mutant β4-MUT accelerated neurite extension and increased the number of filopodia-like protrusions, when compared with β4-WT and the vector. These results suggest that extensive glycosylation of β4 subunit play roles in morphological changes, and the altered glycosylation may be involved in the pathogenesis of PD.
Keywords: Voltage-gated sodium channel β4 subunit, Glycosylation, Filopodia-like protrusion, Neurite outgrowth, Parkinson's disease.
Introduction
Voltage-gated sodium channel (VGSC) β subunits are required for normal kinetics and also participate in invasion, aggregation, migration, axonal fasciculation, and neurite outgrowth [1-6]. β4 is an auxiliary subunit similar to β2 subunit and is associated with α subunits by covalent linkages [7]. Structurally, β4 consists of an N-terminal cleaved signal sequence, an extracellular V-type Ig-like fold, a transmembrane domain, and an intracellular region. Co-expression of β4 with Nav1.2 or Nav1.4 in the human embryonic cell line tsA-201 resulted in a negative shift and increased membrane excitability [7]. It has been shown that β4 subunit was the first endogenous blocking protein responsible for resurgent kinetics. The peptide of β4 was necessary for generating a resurgent current and influenced the Na+ channel gating and action potential firing when β4 subunit was knocked down in cultured cerebellar granule neurons [8, 9]. β4 subunit was down-regulated in Huntington's disease transgenic mice, overexpression of β4 induced neurite extension and increased the spine density [10]. Furthermore, β4 subunit regulated neurite length and filopodia-like protrusion density in cultured cells [6].
All subunits of the VGSCs are extensively glycosylated, with 15-40% of their total mass estimated to consist of the carbohydrates [11, 12]. The β4 is predicted to have four potential N-linked glycosylation sites in the extracellular region. The molecular mass of β4 subunit is greater than the predicted 22 kDa by its amino acid sequence; thus, β4 is thought to be glycosylated in the mature protein [7]. Aberrant protein glycosylation has been shown to play major roles in human disorders, including neurodegenerative diseases [13-15]. Parkinson's disease (PD) is associated with α-synuclein pathology and is accompanied by the formation of intraneuronal inclusions called Lewy bodies [16, 17]. A connection has been demonstrated between dysfunction of the parkin and the formation of α-synuclein inclusions; the mechanism of this process may be due to parkin's function as an E3 ligase, which is to mediate the ubiquitination of an O-linked glycosylated form of α-synuclein [18, 19]. However, very little has been elucidated about the role of glycoproteins in PD progression until now.
Our previous research was an ongoing study where the glycoproteins were isolated from human cerebrospinal fluid (CSF) [20]. The brain tissues were also characterized in different stages of PD and in controls. Herein, we quantitatively compared the protein profiles of brain tissues with PD at different pathological stages of disease with age-matched controls. The study revealed many novel proteins with quantitative expression differences as the disease progressed and provided additional insight into the molecular mechanism of one of these proteins. The glycosylation of β4 may be involved in PD progression.
Materials and methods
Animals
Brain tissues were collected from adult and postnatal [postnatal day 0 (P0), P2, P4, and P28] C57BL/6J mice (SLAC Laboratory Animal Company, Shanghai, China). The A53T transgenic mice used in the present study [21] were obtained from Ruijin Hospital affiliated to Shanghai Jiaotong University School of Medicine. All of the animal experiments were performed in compliance with the institutional guidelines.
Quantitative real-time reverse transcriptase polymerase chain reaction (qRT-PCR)
All RNA and cDNA were prepared according to the protocol provided by Invitrogen (Carlsbad, USA), and RT-PCR was performed on a real-time PCR machine (ABI7300, USA) using a standard SYBR Green PCR kit (Toyobo, Japan). The reactions were run for 40 cycles (90 °C for 15 s and 60 °C for 60 s). All of the samples were examined in duplicate. The following primers were used: mouse β4 gene (GenBankTM Accession No.BK001031) (5′-TTGGAGGTATCTGTGGGAAAGG-3′and5′-CGCTGTTATTGTAGGACCACTTG-3′) and GAPDH (5′-AGAAACCTGCCAAGTATGATGACA-3′and5′-GGAAGAGTGGGAGTTGCTGTTG-3′). The relative expression of the β4 gene compared with GAPDH was calculated using the 2-ΔΔCt method [22].
Preparation of membrane fractions
Four brain and liver tissues were dissected from WT and A53T neonatal transgenic mice. The tissues were either immediately processed or snap frozen in liquid nitrogen and kept at -80°C for future processing. The membrane fractions were obtained as previously described [23]. The protein concentrations of high-detergent samples were determined using bicinchoninic acid assays (Beyotime Biotechnology, China).
Antibodies
The rabbit polyclonal antibody specific for the mouse VGSC β4 subunit was generated as previously described [7, 10, 24] with minor modifications. Amino acids 80-228 of the mouse β4 gene were selected and then cloned into the constructed P3 expression vector for expression of the mβ4 protein in a prokaryotic system. The specificity of the β4 antibodies was confirmed by immunoblot assay. The other antibody used in the present study was actin (Abmart, Shanghai, China). The peroxidase-conjugated AffiniPure goat anti-mouse IgG and goat anti-rabbit IgG (Jackson ImmunoResearch, USA) were used as secondary antibodies in immunoblot assay. The CyTM 3-conjugated AffiniPure goat anti-rabbit IgG (H+L) (Jackson ImmunoResearch Laboratories, USA) was utilized as secondary antibody in immunufluorescence experiments.
Immunoblot assay
The samples (20-30 μg) were denatured in 5× sample buffer for 5 min at 100 °C. The proteins were fractionated using SDS-PAGE, using either a 5% or a 12% gradient. The proteins were excised from the Tris-HCl gels and electrotransferred onto Nitrocellulose (NC) membrane (Millipore Corporation, Billerica, MA) for 1 h at 300 mA and 4 °C. The blots were blocked with 5% dried milk in TBST (10 mM Tris, pH 7.5, 50 mM NaCl and 0.1% Tween-20, Sigma) for 2 h at room temperature before their incubation overnight at 4°C with the primary antibody (1:10,000) diluted in 5% dried milk in TBST. The blots were washed extensively in TBST. The immunoreactive proteins were detected by incubation with a 1:20,000 dilution of the horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG secondary antibodies (Jackson ImmunoResearch, USA) in 5% dried milk in TBST for 2 h at room temperature. The signal was detected by chemiluminescence of the HRP substrate, according to recommendations of the manufacturer (Millipore Corporation, Billerica, MA).
Deglycosylation of membrane fractions
The membrane fraction of the brain and liver tissues from four mice at different postnatal ages were prepared as previously described [23] with minor modifications. The membrane pellet was suspended in 100 μl of 0.2 M KCl and 10 mM HEPES at pH 7.4. Sodium dodecyl sulfate (SDS) was added to a final concentration of 0.3%, and then, 10 μl of a 10× glycoprotein denaturation buffer (5% SDS, 0.4 M DTT) was added. The reaction solution was boiled at 100 °C for 10 min, and then, 1/10 volume of 10× G7 buffer (0.5 M sodium phosphate) and 10% NP-40 were added. This was followed by the addition of 1 μl (500 U) N-glucosidase PNGase F (New England Biolabs, Beverly, MA). The mixture was then incubated at 37°C for 1 h. The enzyme cleaves the glycosidic band between the N-acetylglucosamine (GluNAc) group and the asparagine residues of N-linked glycoproteins. A control sample was prepared without the addition of PNGase F. Up to 33 μl of 5× sample buffer was added to the reaction. The proteins were fractionated on a gradient gel as described above.
Plasmid construction
The full-length mouse β4 gene was synthesized and cloned into pUC57 (Invitrogen, USA). The mouse β4 mutant (β4-MUT) was created similarly to that previously described for β1 and β2 subunit mutagenesis [25, 26]. Each asparagine that initiated a potential N-glycosylation site (Asn45, Asn71, Asn113, and Asn145) was changed to a glutamine residue to create the deglycosylated mutant β4-MUT. The mouse sodium channel β1, β2, and β3 cDNAs were amplified from mouse brain cDNAs by PCR using the primers listed in Supplementary Material: Table S2 as previously described [10] with minor modifications. The PCR products were then subcloned into the bicistronic vector pEGFP-N1 (Invitrogen, USA). All of the constructs were verified by sequencing.
Cell culture, transient transfection and treatments
The HEK-293 and Neuro2A cell lines were purchased from the cell bank of the Shanghai Institute of Cell Biology (Shanghai, China). Cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (GIBCO Laboratories, Grand Island, USA). The day before transfection, cells were seeded and left to adhere overnight. The cells were then transfected with the β4-WT and the β4-MUT plasmids using Lipofectamine 2000 (Invitrogen, USA) in accordance with the manufacturer's protocol. 48 hours after transfection, cells were collected for expression analysis. For neurite growth analysis, cells were differentiated by treatment with 5 mM dbcAMP (N6, 2'-O-dibutyryladenosine-3',5'-cyclic monophosphate sodium salt) (Sigma Chemical Co, St. Louis, USA).
Immunofluorescence technique
The cells that were grown on glass cover slips (Corning, USA) were transfected and differentiated for 1 day. Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min, permeabilized with 0.2% Triton X-100 in PBS for 10 min, washed extensively with PBS and then blocked with 10% bovine serum albumin (BSA) (Beyotime, Shanghai, China) in PBS for 1 h. The primary antibody incubation was performed overnight at 4°C. After several washings with PBS, cells were incubated with appropriate secondary antibody for 1 h and washed several times. The CyTM 3-conjugated AffiniPure Goat Anti-Rabbit IgG (H+L) (Jackson ImmunoResearch laboratories, UAS) was used to visualize expression. The samples were observed using a confocal microscope (Olympus FV1000). In some experiments, nuclei were counterstained with DAPI (Roche, Germany).
Quantitative measurement of neurite outgrowth
Neurite outgrowth activity induced by the β4-WT and the β4-MUT in Neuro2A cells was quantified using a confocal microscope (Olympus FV1000; UPLSAPO 60XW NA 1.20). This application utilizes three fluorescent channels, for nuclei (DAPI), green fluorescent protein (GFP+) and anti-β4 antibody (visualized by CyTM 3-conjugated AffiniPure Goat Anti-Rabbit IgG (H+L)), to identify and count the Neuro2a cells. An observer blind to the experimental settings then analyzes the images by quantitating the total length of the neurites of 86-150 cells from three to five independent experiments using the Image-Pro Plus 6.0. The number of filopodia-like protrusions (of 30 μm) on any neurite was counted from at least 20 randomly selected cells.
Statistical analyses
All of the data are presented as the means ± SEM of at least three independent experiments. The statistically significant values (*P<0.05 or **P<0.01) were determined by Student's t-test or ANOVA followed by the Student-Newman-Keuls test using SPSS13.0.
Results
Expression of β4 subunit is higher in PD patient brain and neonatal A53T transgenic mice
The glycoproteins from PD brain tissues were isolated using the methods described for CSF [20]. Using a robust shotgun proteomic approach in conjunction with an isotope-labeling technique, isobaric tagging for relative and absolute quantification (iTRAQ), we generated a list of proteins from top-ranked GO categories that were identified in four independent experiments, which are listed in Table 1. Several of these proteins, including alpha-1-antichymotrypsin [27, 28] and sodium channel β4 subunit [6, 10], are known to have the potential in PD or other neurodegenerative disease pathogenesis. In addition to these proteins, many other proteins are known to be important in other disease pathogenesis but have yet to be linked to PD pathogenesis. Thus, we generated a list of proteins that may yield insight to novel mechanisms underlying PD progression. Among the top-ranked proteins, β4 was selected as our first candidate for confirmation.
Top-ranked proteins (Gene Ontology Analysis) with changes between Parkinson's disease and control brains tissues (Glycoprotein Capture).
| IPI Number | Protein Name | Peptides Found | PD-B/Ctrl* | PD-L/Ctrl* | PD-I/Ctrl* |
|---|---|---|---|---|---|
| IPI00793751.1 | MFAP4 uncharacterized protein MFAP4 | 7 | 0.46 ± 0.13 | 0.49 ± 0.06 | 1.03 ± 0.06 |
| IPI00328113.2 | FBN1 Fibrillin-1 precursor | 6 | 0.78 ± 0.03 | 1.17 ± 0.11 | 1.72 ± 0.13 |
| IPI00329573.9 | COL12A1 Isoform1of Collagen alpha-1(XII) chain precursor | 6 | 0.93 ± 0.14 | 1.82 ± 0.07 | 1.55 ± 0.12 |
| IPI00292732.3 | FMOD Fibromodulin precursor | 4 | 1.20 ± 0.08 | 2.11 ± 0.21 | 1.44 ± 0.16 |
| IPI00009111.1 | TPBG Trophoblast glycoprotein precursor | 8 | 1.48 ± 0.37 | 1.83 ± 0.22 | 1.29 ± 0.13 |
| IPI00847635.1 | SERPINA3 Isoform1of Alpha-1-antichymotrypsin precursor | 15 | 1.62 ± 0.14 | 1.38 ± 0.06 | 1.45 ± 0.09 |
| IPI00015872.3 | TSPAN8 Tetraspanin-8 | 4 | 2.13 ± 0.25 | 1.37 ± 0.13 | 1.44 ± 0.13 |
| IPI00514804.1 | SCN4B Isoform2 of Sodium channel subunit beta-4 precursor | 4 | 2.48 ± 0.30 | 4.44 ± 0.80 | 2.12 ± 0.28 |
Ctrl, Control; PD-B, PD-brainstem; PD-L, PD-limbic; PD-I, PD-isocortex. The protein names are from the IPI human protein database (Version 3.18).
*Peptides found denotes the sum of all the different peptides found and is relatively quantified from the proteins shown in four replicate experiments.
The ratio shown is the means ± SEM of four independent experiments.
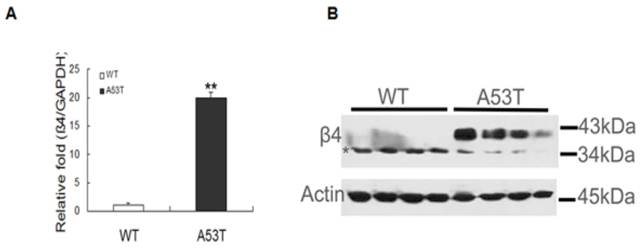
The expression of β4 was significantly increased in PD patient brain tissues and validated through RT-PCR and western blot analyses. In neonatal A53T mice, β4 mRNA expression increased (Fig. 1A), and in the western blot, β4 was expressed at molecular masses of ~38 kDa and 35 kDa (Fig. 1B). The 38 kDa band appeared as a strong, slightly smeared band when compared with the 35 kDa band. The multiple protein bands were expected due to the N-linked glycosylation at multiple sites; the expression of β4 was significantly higher than in neonatal WT mice. The expression of β4 was also compared between the postnatal day 7 WT mice and the adults; the results revealed two different molecules of different masses in immunoblot assays. There was no change in total protein between the two groups (Supplementary Material: Fig S2). These results indicate that β4 protein may exist in two different glycosylated states under normal physiological circumstances; however, the heavily glycosylated state, the 38 kDa band, appears prematurely early during pathogenesis.
Expression level of the sodium channel β4 subunit in neonatal mice. (A) Real-time PCR analysis was performed, and all of the values obtained were normalized with respect to the level of GAPDH. The level of β4 mRNA markedly increased in neonatal A53T mice (**p < 0.01). The values are expressed as the means ± SEM (**p < 0.01 compared with the controls). (B) Confirmation of β4 expression through western blot analysis. Equal amounts (30 μg) of neonatal WT and A53T mice brain tissue samples (four per group, two groups in total) were sequentially analyzed with anti-β 4 antibodies, and actin was used as the loading control. β4 expression increased under disease progression. WT: neonatal wild-type mice; A53T: neonatal PD transgenic mice.
Immunoblot analysis of β4 reveals developmental regulation of the glycosylation of this subunit
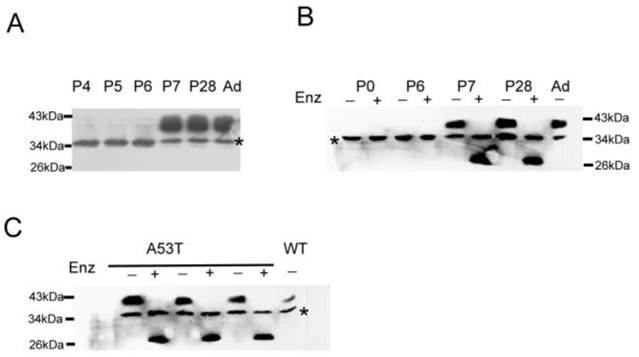
Immunoblot analysis of the membrane fractions showed that WT mice only contained the ~35 kDa band until postnatal day 7 (Fig 2A). The Na+ channels are heavily glycosylated [12, 29]; therefore, the developmentally regulated N-linked glycosylation of β4 is likely the source of the 38 kDa molecular weight band in adult tissues. The ~35 kDa band was attributed to an expression state that is either glycosylated or a nonspecific band. To test this hypothesis, membrane fractions were treated with the glycosidase enzyme PNGase F and analyzed by immunoblot assay. The PNGase F cleaved the glycosidic bond of the N-acetylglucosamine linked to the aspargine (N) side chain of the N-glycosylated proteins. Fig. 2B showed the effect of the enzymatic treatment on the size of the β4 protein of the membrane fractions from P0, P6, P7, P28, and adult WT mice. The enzymatic treatment converted the 38 kDa protein to a faster migrating band in P7, P28 and adult WT mice compared with the untreated samples. There was no change observed between the enzymatically treated and untreated samples in P0-P6 tissues, and there was no expression in liver and lung tissues (data not shown). These findings clearly showed that β4 was heavily glycosylated in brain tissues and was developmentally regulated. Fig. 2C showed the mobility shift of the immunoreactive protein after PNGase F treatment and also indicated that the ~38 kDa protein appeared prematurely in the neonatal A53T mice brain fractions and was heavily glycosylated.
Immunoblot assays show the developmental regulation and glycosylation of β4 subunit. (A) The ~38 kDa band was not expressed until postnatal day 7 in wild-type mice. (B) The treatment of the membrane fractions from the P7, P28 and adult brain tissues with glycosidase PNase F (Enz) converted the immunoreactive proteins into a faster migrating species. P0 and P6 show the immunoreactive proteins without changes between the treated and untreated samples. (C) The similar treatment of the membrane fractions from neonatal A53T mice brain tissues converted the ~38 kDa protein into a faster migrating species, but there is no change in the 35 kDa band with or without PNGase F treatment. The + and - signs indicate the treated and untreated samples, respectively. WT: wild-type mice; A53T: PD transgenic mice; Ad: adult wild-type mice.
Immunoblot analysis of the heterologous expression system reveals that β4 subunit was glycosylated in the mature protein
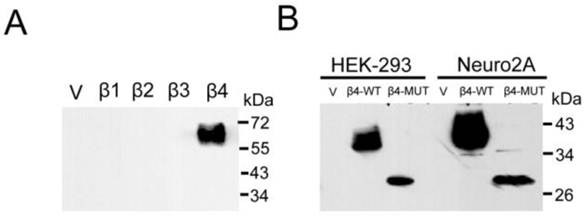
To examine β4 subunit in mouse brains, we generated a polyclonal antibody. To check the specificity of the antibody, we expressed the mouse sodium channels β1-β4 individually in HEK-293 cell line and tested for their immunoreactivities by western blot. As shown in Fig. 3A, the β4 antibody gave no signal against the recombinant β1, β2, and β3 subunits. In contrast, this antibody strongly recognized the β4 subunit and appeared as strong, slightly smeared bands, as described in the brain tissue above. To examine whether the 35 kDa protein exists in another glycosylated state, we transiently expressed the β4-WT and the β4-MUT plasmids (especially when the expression of GFP was removed) in cultured cells. The anti-β4 antibodies were highly specific to the β4-WT and the β4-MUT proteins in membrane fractions isolated from cells in immunoblot analysis. Two immunoreactive proteins of different molecular weight were observed, and the molecular weight difference was consistent with the results observed in the enzymatically treated and untreated samples above (Fig. 3B). These results indicated that in the brain tissues, the β4 protein that was heavily glycosylated was expressed as the 38 kDa band, and the 35 kDa band was demonstrated to be a nonspecific protein.
Immunoblot analysis reveals the expression of β4 subunit. Different cell lines were individually transfected with vector, β1~β4, and β4-MUT expression plasmids. (A) To analyze the specificity of the anti-β4 antibody, HEK-293 cells were transfected individually with the β1~β4 expression plasmids. Purified membrane proteins (20 μg) were fractionated by SDS-PAGE, immunoblotted, and probed using an anti-β4 antibody. (B) The western blot analysis of β4-WT and β4-MUT protein expression minus the expression of GFP after transfection in cultured cell lines. The molecular weight difference was consistent with the results observed in the enzymatically treated and untreated brain samples. β4 protein is heavily glycosylated, and the ~35 kDa band expressed in the brain membrane fractions is demonstrated to be a nonspecific band. β4-WT: β4-wild-type plasmid; β4-MUT: β4 deglycosylated mutant plasmid.
Overexpression of β4-WT and β4-MUT extends neurite length and increases the number of filopodia-like protrusions
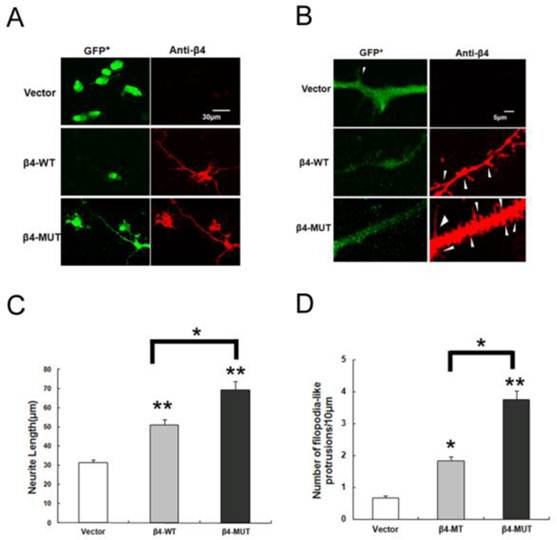
Voltage-gated sodium channel β subunits serve as cell adhesion molecules, based on structural and amino acid homologies [30, 31]. In addition, β4 contains four potential N-linked glycosylation sites in the extracellular region, and the expression of β4 increased under disease progression. Thus, we expressed the β4-WT and β4-MUT plasmids in Neuro2a cells to gain additional insight into the function of the N-glycosylation of β4 subunit. Overexpression of β4-WT induced neurite extension and increased the number of filopodia-like protrusions [6, 10]. To investigate the morphological changes between the β4-WT and β4-MUT expressing cells, Neuro2a cells were transiently transfected with the β4-WT, β4-MUT and vector plasmids. Quantification of the neurites was significantly longer in cells expressing β4-MUT (69.34±4.2μm) when compared with cells expressing β4-WT (50.97±2.7μm) and vector (31.43±1.5μm) (Fig. 4 A, B). In addition to the extension of neurites, overexpression of β4-MUT also increased the number of filopodia-like protrusions (3.7±0.3 per 10 μm of neurite) when compared with cells expressing β4-WT (1.8±0.1 per 10 μm) and vector (0.7±0.1 per 10 μm) (Fig. 4 C, D). The same features of β4-MUT and β4-WT induced protrusions, which are similar to the small, thin, and headless dendritic filopodia, were observed as previously reported [6, 32]. These results indicated that the β4-MUT modulated neurite outgrowth activities and spine formation in cultured cells; in other words, the N-linked glycosylation of the β4 protein may be involved in neurite degeneration.
Overexpression of β4-WT and β4-MUT induces neurite extension and increases the formation of filopodia-like protrusions in Neuro2a cells. (A, B) Overexpression of β4-WT and β4-MUT in Neuro2a cells displayed a marked alteration in cell morphology. High-magnification images of Neuro2a cells overexpressing vector, β4-WT and β4-MUT immunostained by anti-β4 (red) and GFP+ (green) are shown. Scale bar 30 μm for A; Scale bar 5 μm for B. (C) The quantification of the average neurite extension induced by β4-WT and β4-MUT. The average neurite length for cells overexpressing β4-WT and β4-MUT were significantly greater than those for the vector control transfected cells (**p < 0.01); the average neurite length for the β4-MUT overexpressing cells were higher than the β4-WT transfected cells (*p < 0.05). (D) The quantification of the number of filopodia-like protrusions. The cells overexpressing β4-WT and β4-MUT had a significantly increased number of filopodia-like protrusions (*p < 0.05, **p < 0.01). The number of filopodia-like protrusions in the β4-MUT transfected cells was significantly higher than the β4-WT cells (*p < 0.05). The values in the histograms are means ± SEM.
Discussion
Glycosylation represents the most common and complicated post-translational modifications (PTMs) and may contribute to the progression of disease [14, 15]. In the present study, quantitative data of aberrant glycoproteins in PD patient brain tissues are listed in Table 1 from four independent experiments. The expression of β4 was significantly higher in disease tissues than in controls. One PD transgenic mouse line, A53T, was used in this study. Initially, we identified that β4 was significantly up-regulated in the brain tissue of A53T mice. There were two molecular weight bands in neonatal A53T mice, but only the smaller one existed in neonatal WT mice. We first thought that there would be two β4 protein bands that would indicate different glycosylation patterns, but the ~35 kDa band was shown to be a nonspecific band by its changes in mobility after PNGase F treatment and was expressed with β4-MUT plasmid in heterologous expression systems. As we know, there are four potential glycosylation sites in the extracellular region, and the ~38 kDa band is expressed until postnatal day 7; these results showed that the glycosylation of β4 subunit underwent developmental regulation after birth. The extra mass is estimated to be attributed to carbohydrates.
Immunocytochemical analysis shows that filopodia-like protrusions contain F-actin and are similar to dendritic filopodia, which are highly motile structures that play important roles in the process of axon-dendrite contact and thought to be a precursor of the spine [6, 32]. The β4 subunit also served as CAMs can promote neurite outgrowth and is the only endogenous open-channel blocker responsible for resurgent current [8, 9]. However, we noticed that the effect of β4-WT overexpression on neurite morphological changes observed in our study is somewhat different from that reported in a previous study [10]. This could be due to the different culture conditions and the level of protein expression from transfection. Because we observed similar effects in cultured cells, it is worth noting that overexpression of β4-MUT could promote neurite outgrowth in Neuro2a cells and increase the number of filopodia-like protrusions when compared with the cells expressing β4-WT and vector plasmids. These results suggested that the extracellular domain of β4, especially the N-linked oligosaccharides, may play important roles in neuritic degeneration.
Glycosylation alters many properties of membrane proteins, such as targeting, folding, affinity, and cell surface expression [33-35]. Furthermore, the carbohydrate structures of proteins on the cell surface have been shown to correlate with disease, for example, the N-linked oligosaccharides of the 5-HT2A receptor are important for JC virus infection in PML [36]. We examined that the expression of β4 markedly increased in the neonatal PD transgenic mice. We also suggested that the β4 protein underwent developmental regulation and was involved in morphological changes in Neuro2a cells. Taken together, these data suggest that the sialic acid component of β4 protein is required for proper physiological function, but its abnormal glycosylation process may be involved in neuritic degeneration.
The β4 subunit has an important effect on neurons and other excitable cells, regardless of whether the cells express other β subunits or not [7]. The β4 subunit caused negative shifts in the voltage dependence of activation and overrode other subunits' effects; the resulting action potentials increased neuronal sensitivity to excitatory inputs in cells expressing the β4 subunit. PD-linked parkin is involved in regulating mitochondrial dynamics and the stability of excitatory glutamatergic synapses [37, 38]. The excitatory response produced by the disruption of parkin and an increased β4 expression may contribute to the pathophysiology of PD. Sodium channel subunits are glycosylated [39], thus, the extent and type of glycosylation during different stages of development, in different cell types or during different disease progressions may be important in their ability to modulate channel activities [40]; glycosylation of the different β subunits has different mechanisms by which sodium channel activity is modulated [25, 26], so different glycosylations of auxiliary subunits may interfere with neuronal function at multiple levels, heavy glycosylation of β4 in adult tissues may be essential for the shifts in sodium channel gating. However, β4 appears prematurely in the disease state, and increased expression in neonatal PD transgenic mice may imply that N-linked oligosaccharides play roles in neuritic degeneration. Its sialic acid component may regulate cell adhesion, migration and sodium channel activities.
In conclusion, this study provided a new insight into the role of protein glycosylation under disease progression. We demonstrated that the expression of β4 was up-regulated in human PD brain tissue and in neonatal transgenic mice. We also proved that the glycosylated β4 is developmentally regulated and that the N-linked oligosaccharides are involved in neurite degeneration in cultured cells. To confirm the role of the sialic acid component of β4 during PD progression, further studies will focus on the mechanisms of neuritic degeneration and the effects of the oligosaccharides on the sodium channel activities.
Supplementary Material
Table S1: Summary of control and PD cases. Table S2: The nucleotide sequence for amplifying β subunit cDNAs. Fig S1: MS, MS/MS and iTRAQ of sodium channel β4 subunit. Fig S2: Developmental expression of the sodium channel β4 subunit in WT mice.
Acknowledgements
The authors are deeply grateful to Dr. Wu for the image captures. We also thank Shanghai ImmunoGen Bio-Tech Co. Ltd. for antibody preparation.
This work was supported by a grant from the Shanghai Natural Science Foundation (Project No. 09ZR1439600).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Brackenbury WJ, Davis TH, Chen C. et al. Voltage-gated Na+ channel beta1 subunit-mediated neurite outgrowth requires Fyn kinase and contributes to postnatal CNS development in vivo. J Neurosci. 2008;28:3246-56
2. Isom LL, Ragsdale DS, De Jongh KS. et al. Structure and function of the beta 2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell. 1995;83:433-42
3. Kim DY, Ingamo LA, Carey BW. et al. Presenilin/gamma-secretase-mediated cleavage of the voltage-gated sodium channel beta2-subunit regulates cell adhesion and migration. J Biol Chem. 2005;280:23251-61
4. McEwen DP, Isom LL. Heterophilic interactions of sodium channel beta1 subunits with axonal and glial cell adhesion molecules. J Biol Chem. 2004;279:52744-52
5. Ratcliffe CF, Westenbroek RE, Curtis R. et al. Sodium channel beta1 and beta3 subunits associate with neurofascin through their extracellular immunoglobulin-like domain. J Cell Biol. 2001;154:427-34
6. Miyazaki H, Oyama F, Wong HK. et al. BACE1 modulates filopodia-like protrusions induced by sodium channel beta4 subunit. Biochem Biophys Res Commun. 2007;361:43-8
7. Yu FH, Westenbroek RE, Silos-Santiago I. et al. Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2. J Neurosci. 2003;23:7577-85
8. Grieco TM, Malhotra JD, Chen C. et al. Open-channel block by the cytoplasmic tail of sodium channel beta4 as a mechanism for resurgent sodium current. Neuron. 2005;45:233-44
9. Bant JS, Raman IM. Control of transient, resurgent, and persistent current by open-channel block by Na channel beta4 in cultured cerebellar granule neurons. Proc Natl Acad Sci U S A. 2010;107:12357-62
10. Oyama F, Miyazaki H, Sakamoto N. et al. Sodium channel beta4 subunit: down-regulation and possible involvement in neuritic degeneration in Huntington's disease transgenic mice. J Neurochem. 2006;98:518-29
11. Roberts RH, Barchi RL. The voltage-sensitive sodium channel from rabbit skeletal muscle. Chemical characterization of subunits. J Biol Chem. 1987;262:2298-303
12. Schmidt JW, Catterall WA. Palmitylation, sulfation, and glycosylation of the alpha subunit of the sodium channel. Role of post-translational modifications in channel assembly. J Biol Chem. 1987;262:13713-23
13. Liu F, Zaidi T, Iqbal K. et al. Role of glycosylation in hyperphosphorylation of tau in Alzheimer's disease. FEBS Lett. 2002;512:101-6
14. Saez-Valero J, Fodero LR, Sjogren M, Andreasen N. et al. Glycosylation of acetylcholinesterase and butyrylcholinesterase changes as a function of the duration of Alzheimer's disease. J Neurosci Res. 2003;72:520-6
15. Silveyra MX, Cuadrado-Corrales N, Marcos A. et al. Altered glycosylation of acetylcholinesterase in Creutzfeldt-Jakob disease. J Neurochem. 2006;96:97-104
16. Spillantini MG, Schmidt ML, Lee VM. et al. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839-40
17. Licker V, Kovaei E, Hochstrasser DF. et al. Proteomics in human Parkinson's disease research. J Proteomics. 2009;73:10-29
18. Farrer M, Chan P, Chen R, Tan L. et al. Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol. 2001;50:293-300
19. Shimura H. et al. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson's disease. Science. 2001;293:263-9
20. Hwang H, Zhang J, Chung KA. et al. Glycoproteomics in neurodegenerative diseases. Mass Spectrom Rev. 2009;29:79-125
21. Giasson BI, Duda JE, Quinn SM. et al. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521-33
22. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-8
23. Tyrrell L, Renganathan M, Dib-hajj SD, Waxman SG. et al. Glycosylation alters steady-state inactivation of sodium channel Nav1.9/NaN in dorsal root ganglion neurons and is developmentally regulated. J Neurosci. 2001;21:9629-37
24. Wong HK, Sakurai T, Oyama F, Kaneko K. et al. beta Subunits of voltage-gated sodium channels are novel substrates of beta-site amyloid precursor protein-cleaving enzyme (BACE1) and gamma-secretase. J Biol Chem. 2005;280:23009-17
25. Johnson D, Montpetie ML, Stocker PJ, Bennett ES. et al. The sialic acid component of the beta1 subunit modulates voltage-gated sodium channel function. J Biol Chem. 2004;279:44303-10
26. Johnson D, Bennett ES. Isoform-specific effects of the beta2 subunit on voltage-gated sodium channel gating. J Biol Chem. 2006;281:25875-81
27. Yamamoto M, Kondo I, Ogawa N, Asanuma M. et al. Genetic association between susceptibility to Parkinson's disease and alpha1-antichymotrypsin polymorphism. Brain Res. 1997;759:153-5
28. Munoz E, Obach V, Oliva R. et al. Alpha1-antichymotrypsin gene polymorphism and susceptibility to Parkinson's disease. Neurology. 1999;52:297-301
29. Barchi RL, Cohen SA, Murphy LE. Purification from rat sarcolemma of the saxitoxin-binding component of the excitable membrane sodium channel. Proc Natl Acad Sci U S A. 1980;77:1306-10
30. Vaughn DE, Bjorkman PJ. The (Greek) key to structures of neural adhesion molecules. Neuron. 1996;16:261-73
31. Kaczmarek LK. Non-conducting functions of voltage-gated ion channels. Nat Rev Neurosci. 2006;7:761-71
32. Yuste R, Bonhoeffer T. Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat Rev Neurosci. 2004;5:24-34
33. Lanctot PM, Leclerc PC, Clement M. et al. Importance of N-glycosylation positioning for cell-surface expression, targeting, affinity and quality control of the human AT1 receptor. Biochem J. 2005;390:367-76
34. Drescher B, Witte T, Schmidt RE. et al. Glycosylation of FcgammaRIII in N163 as mechanism of regulating receptor affinity. Immunology. 2003;110:335-40
35. Niu L, Heaney ML, Vera JC. et al. High-affinity binding to the GM-CSF receptor requires intact N-glycosylation sites in the extracellular domain of the beta subunit. Blood. 2000;95:3357-62
36. Maginnis MS, Haley SA, Gee GV. et al. Role of N-linked glycosylation of the 5-HT2A receptor in JC virus infection. J Virol. 2010;84:9677-84
37. Yu W, Sun Y, Guo S. et al. The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum Mol Genet. 2011;20:3227-40
38. Helton TD, Otsuka T, Lee MC. et al. Pruning and loss of excitatory synapses by the parkin ubiquitin ligase. Proc Natl Acad Sci U S A. 2008;105:19492-7
39. Messner DJ, Catterall WA. The sodium channel from rat brain. Separation and characterization of subunits. J Biol Chem. 1985;260:10597-604
40. Davis TH, Chen C, Isom LL. et al. Sodium channel beta1 subunits promote neurite outgrowth in cerebellar granule neurons. J Biol Chem. 2004;279:51424-32
Author contact
![]() Corresponding author: Department of Biochemistry and Molecular Biology, Second Military Medical University, Shanghai 200433, China. Tel: +86-21-81870970-8017; Fax: +86-21-65334344, E-mail address: zjp8007202com (Jian-peng Zhang), jiaobhuacn (Bing-hua Jiao).
Corresponding author: Department of Biochemistry and Molecular Biology, Second Military Medical University, Shanghai 200433, China. Tel: +86-21-81870970-8017; Fax: +86-21-65334344, E-mail address: zjp8007202com (Jian-peng Zhang), jiaobhuacn (Bing-hua Jiao).