Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Vitamin D status in chronic...
Biphasic cardiovascular effects...
Molecular structure and...
Metabolism of FGF-23 in CKD
FGF-23 and vascular calcification
FGF-23 and SHPT
FGF-23 and LVH
FGF-23 and endothelial...
Perspectives
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(5):663-671. doi:10.7150/ijbs.3886 This issue Cite
Review
Fibroblast Growth Factor-23 Helps Explain the Biphasic Cardiovascular Effects of Vitamin D in Chronic Kidney Disease
Peng Hu1 ![]() , Qiang Xuan2, Bo Hu1, Ling Lu1, Jing Wang1, Yuan Han Qin3
, Qiang Xuan2, Bo Hu1, Ling Lu1, Jing Wang1, Yuan Han Qin3
1. Department of Pediatrics, the First Affiliated Hospital of Anhui Medical University, No. 218 Ji-Xi Road, Hefei 230022, PR China
2. Department of Urology, Anhui Provincial Hospital, Anhui Medical University, No. 17 Lu-Jiang Road, Hefei 230001, PR China
3. Department of Pediatrics, the First Affiliated Hospital of Guangxi Medical University, No. 6 Shuang-Yong Road, Nanning 530021, PR China
Received 2011-11-30; Accepted 2012-2-23; Published 2012-5-5
Abstract
Hypovitaminosis D is highly prevalent in chronic kidney disease (CKD). Recently, vitamin D has sparked widespread interest because of its potential favorable benefits on cardiovascular disease (CVD). Evidence from clinical studies and animal models supports the existence of biphasic cardiovascular effects of vitamin D, in which lower doses suppress CVD and higher doses stimulate CVD. However, the mechanism for the different effects remains unclear. Fibroblast growth factor-23 (FGF-23) is a recently identified member of the FGF family, and thought to be actively involved in renal phosphate and vitamin D homeostasis. More specifically, Vitamin D stimulates FGF-23 secretion and is inhibited by increased FGF-23. Given this background, we hypothesize that FGF-23 may provide a unique tool to explain the biphasic cardiovascular effects of vitamin D in CKD. The data presented in this review support the hypothesis that FGF-23 may be linked with the high cardiovascular risk in CKD through accelerating the onset of vascular calcification, secondary hyperparathyroidism, left ventricular hypertrophy and endothelial dysfunction. Therefore, modulation of FGF-23 may become a potential therapeutic target to lowing cardiovascular risk in CKD. Several clinical interventions, including decreased phosphate intake, phosphate binders, cinacalcet plus concurrent low-dose vitamin D, C-terminal tail of FGF-23 and renal transplantation, have been employed to manipulate FGF-23.
Keywords: Fibroblast growth factor-23, Hypovitaminosis D, Cardiovascular disease, Phosphate, Left ventricular hypertrophy
Introduction
Vitamin D refers to a group of compounds that have antirachitic activity. During the period of its discovery, it was recognized that there were two antirachitic factors with distinct structures, vitamin D3 and vitamin D2. Vitamin D3 can be produced in the vertebrate skin as 7-dehydrocholesterol on exposure to ultraviolet-B from the sun, whereas vitamin D2 comes from plants [1]. Both vitamin D3 and vitamin D2 can be further modified by corresponding enzymes to produce different vitamin D metabolites. After vitamin D enters the body, it circulates bound to vitamin D-binding protein and is rapidly converted into its major circulating form, 25-hydroxyvitamin D [25-(OH)D], by the liver. 25-(OH)D is filtered at the glomerulus and actively reabsorbed into renal tubular cells via megalin and cubulin, where it is converted into the potent hormone 1,25-dihydroxyvitamin D [1,25-(OH)2D] by the enzyme 1α-hydroxylase [2]. 25-(OH)D is the nutritional form of vitamin D. Serum 25-(OH)D is regarded as the best indicator of vitamin D status, because it has a longer biological half-life than 1,25-(OH)2D, and circulates in much higher concentrations. In contrast, 1,25-(OH)2D is the active form of vitamin D. Circulating levels of 1,25-(OH)2D chiefly depend on the ability of renal 1α-hydroxylase to convert 25-(OH)D into 1,25-(OH)2D. This ability is decreased by a reduction in the nephron mass. Therefore, in this review, when vitamin D status was mentioned, the “vitamin D” referred to “25-(OH)D”; when vitamin D therapy was mentioned, the “vitamin D” referred to “1,25-(OH)2D”.
Circulating 1,25-(OH)2D enters the target cells, either in its free form or facilitated by megalin, and binds to the vitamin D receptor (VDR) in the cytoplasm, which then translocates into the nucleus and heterodimerizes with the retinoic X receptor (RXR). The 1,25-(OH)2D-VDR-RXR complex then binds to vitamin D response elements (VDRE) on DNA to increase transcription of vitamin D-regulated genes [3]. The classical physiological function of vitamin D is to maintain the calcium and phosphate homeostasis, accomplished by close coordination with parathyroid hormone (PTH) [4, 5].
Vitamin D status in chronic kidney disease (CKD)
The Kidney Disease Outcome Quality Initiative (K/DOQI) has recently raised concerns of a high prevalence of hypovitaminosis D in patients with CKD [6]. According to serum 25-(OH)D levels, 76% of CKD patients display either vitamin D deficiency [42%; 25-(OH)D≤15ng/ml] or insufficiency [34%; 15ng/ml<25-(OH)D≤30ng/ml] [7]. What's worse, serum 25-(OH)D is below the recommended sufficiency values in >90% of patients if they progress to end-stage renal disease (ESRD) [8]. CKD patients are at particular risk of 25-(OH)D deficiency, including reduced sun exposure, impaired production of the 25-(OH)D precursor molecule and reduced dietary intake [9]. In contrast to 25-(OH)D, circulating levels of 1,25-(OH)2D chiefly depend on the ability of renal 1α-hydroxylase to convert 25-(OH)D into 1,25-(OH)2D. This ability is decreased because of a reduction in the nephron mass [10]. It is therefore necessary to modulate the vitamin D signaling using 1,25-(OH)2D or its analogues to compensate for the compromised vitamin D status which occurs in all stages of CKD, so that the classical functions of 1,25-(OH)2D may be addressed [11].
Biphasic cardiovascular effects of vitamin D in CKD
The increased risk of cardiovascular disease (CVD) in patients with CKD has been well documented [12, 13]. Although it is true that many traditional cardiovascular risk factors are corrected in CKD, the results of intervention may not be as efficacious as those obtained in the general population. Thus, there may also be a unique milieu established in CKD, which leads to excess CVD burden by mechanisms that are as yet not fully understood [14, 15]. Recently, vitamin D has sparked widespread interest because of its potential beneficial effects on CVD [16, 17]. A historical cohort study carried out by Teng et al. showed that incident chronic hemodialysis patients treated with activated injectable vitamin D had a significant survival advantage over a comparable group of chronic hemodialysis patients who did not; cardiovascular-related mortality rates were 7.6 per 100 person-years in the injectable vitamin D group, compared with 14.6 per 100 person-years in the non-vitamin D group [18]. In stratified analyses by vitamin D therapy, after adjustment for age, sex, PTH and albumin, the odds ratio (OR) of all-cause mortality was 2.2 for 25-(OH)D levels <10ng/ml compared with 25-(OH)D levels >30ng/ml; in a similar stratified model of 1,25-(OH)2D, the adjusted OR of all-cause mortality was 3.1 for 1,25-(OH)2D levels of 6-13pg/ml compared with 1,25-(OH)2D levels >13pg/ml [19]. A meta-analysis examined 9 randomized controlled trials and also found an 8% reduction in cardiovascular mortality with supplementation of very modest amounts of vitamin D (~500IU) [20]. More specifically, two other studies compared mortality among hemodialysis patients receiving different types of 1,25-(OH)2D formulations, and found that mortality was lower among patients receiving doxercalciferol (15.4 per 100 person-years) and paricalcitol (15.3 per 100 person-years) versus calcitriol (19.6 per 100 person-years) [21, 22]. However, the study of Tentori et al. raised questions about the previously reported survival advantage associated with vitamin D therapy [23]. In addition, an earlier and smaller study from India also did not show any benefit from having optimal levels of 25-(OH)D in subjects with established CVD; in contrast, the authors reported that very high levels of 25-(OH)D (>89ng/ml) were associated with an increased risk of ischemic heart disease [24]. Interestingly, consistent with these findings in humans, an animal model of CKD showed that the administration of clinically relevant doses of vitamin D reduced arterial calcification [25]; while in another study, rats fed on a diet rich in cholesterol and extremely high in vitamin D (1.8 million units/kg) developed greater arterial calcification [26]. Thus, evidence from clinical studies and animal models supports the existence of biphasic cardiovascular effects of vitamin D, in which lower doses suppress CVD and higher doses stimulate CVD [27]. However, the mechanism for the different effects remains unclear.
Molecular structure and physiological function of fibroblast growth factor-23 (FGF-23)
FGF-23 is a 32-kDa protein with 251 amino acids that is synthesized and secreted by bone cells, mainly osteoblasts. It is composed of an amino-terminal signal peptide (residues 1-24), an “FGF-like sequence” (residues 25-180), and a carboxyl-terminal extended sequence (residues 181-251) [28, 29]. Unlike most other members of the FGF family, since FGF-23 contains a signal peptide and has low affinity for heparin, it can be distributed throughout the body in the blood and act at many distant sites, such as the kidney [30]. FGF-23 signals with highest efficacy through four FGF receptors (FGFR) bound to the transmembrane protein Klotho as a coreceptor. Of the four members of the FGFR family, FGFR1 and FGFR4 are expressed in the distal tubule, FGFR2 is expressed mainly in the macula densa, whereas FGFR3 is expressed both in the proximal tubule and in the distal tubule [31]. FGFR2 is not a likely candidate for mediating FGF-23 effects in the kidney, because it does not bind to FGF-23 [32]. Furthermore, a recent study showed that neither FGFR3 nor FGFR4 is the principal mediator of FGF-23 effects in the kidney [33], suggesting that FGFR1 is the only remaining target for FGF-23 [34]. FGF-23 induces urinary phosphate excretion by suppressing the abundance of the Na/Pi IIa and IIc cotransporters in the brush border of renal proximal tubules [35]. In animal studies, transgenic mice over-expressing human or mouse FGF-23 have severe renal phosphate wasting because of the inhibition of renal Na/Pi cotransporter activity [36, 37], whereas FGF-23 inactivation (FGF-23 null mice and normal mice treated with FGF-23 blocking antibodies) leads to hyperphosphatemia [38, 39]. A pioneer study published in Nature Genetics revealed that missense mutations in FGF-23 gene are responsible for human variants of hypophosphatemic rickets [40]. In addition, FGF-23 also suppresses 1,25-(OH)2D via down-regulation of 1α-hydroxylase and up-regulation of 24-hydroxylase which converts 1,25-(OH)2D into more hydrophilic metabolites with lesser biological activity [41, 42]. On the contrary, the administration of 1,25-(OH)2D increases circulating FGF-23 levels, apparently due to a direct action of vitamin D on FGF-23 via a VDRE located upstream of the FGF-23 promoter, which may be related to future resistance to vitamin D therapy [43, 44].
Metabolism of FGF-23 in CKD
The elevation of serum FGF-23 is a common feature in CKD. This occurs as early as stage 2 CKD, long before any changes in calcium, phosphate, or PTH are apparent [45]. By the time patients reach ESRD, FGF-23 concentrations are often 100- to 1000- fold above the normal range [46], and moreover, circulating FGF-23 in ESRD patients is mostly intact and biologically active [47]. Three possible explanations could account for the event. First, it appears that the massive elevations in FGF-23 levels in CKD reflect both increased secretion into and decreased removal from the circulation. In the report of Bacchetta et al. [48], treatment with corticosteroids could activate osteocytes in pediatric CKD patients, and then significantly stimulate FGF-23 synthesis. On the other hand, Westerberg et al. [49] found that FGF-23 levels and estimated glomerular filtration rate (eGFR) were inversely correlated among individuals with CKD stage 4-5. Second, the other cause of increased levels of FGF-23 may be a compensatory phenomenon to the phosphate retention seen in CKD. Perwad et al. [50] fed normal mice with low-, medium-, or high-phosphate diet for 5 days and measured circulating FGF-23 concentrations using an intact FGF-23 assay. In mice fed with high- (1.65%) phosphate intake, the mean serum FGF-23 concentration was 65±6.5 pg/ml, a value 7-fold higher than that in mice fed with the low- (0.02%) phosphate intake, 9.9±1.0 pg/ml. Ito et al. [51] expressed FGF-23 promoter constructs in transiently transfected K562 cells in media with various concentrations of phosphate and showed an increase in promoter activity with high-phosphate medium. Last but most important, vitamin D therapy in patients with CKD may also contribute to the increased serum levels of FGF-23. In 5/6 nephrectomized rats, intravenous administration of 1,25-(OH)2D, three times a week for 2 weeks, dose-dependently increased serum FGF-23 [52].
FGF-23 may serve as a novel risk marker for the cardiovascular mortality in CKD [53]. The ArMORR study revealed that high FGF-23 levels in patients starting hemodialysis were an independent predictor of 1-year mortality after adjustment for serum phosphate and PTH levels; and in a multivariable adjusted model, subjects whose FGF-23 levels were in the highest quartile had nearly a 600% increase in risk as compared with subjects in the lowest quartile [54]. In addition, in the case of FGF-23, a special characteristic could favor its use as a marker: the fact that FGF-23 concentrations remain stable throughout the day in patients with CKD [55]. Although the mechanistic link between elevated FGF-23 levels and increased cardiovascular mortality in CKD remains to be answered, the data present below focusing on vascular calcification, secondary hyperparathyroidism (SHPT), left ventricular hypertrophy (LVH), and endothelial dysfunction may be supportive of the pathophysiological sequence of vitamin D supplement - FGF-23 excess - cardiovascular susceptibility (Figure 1).
The pathophysiological sequence of vitamin D supplement - FGF-23 excess - cardiovascular susceptibility in CKD.
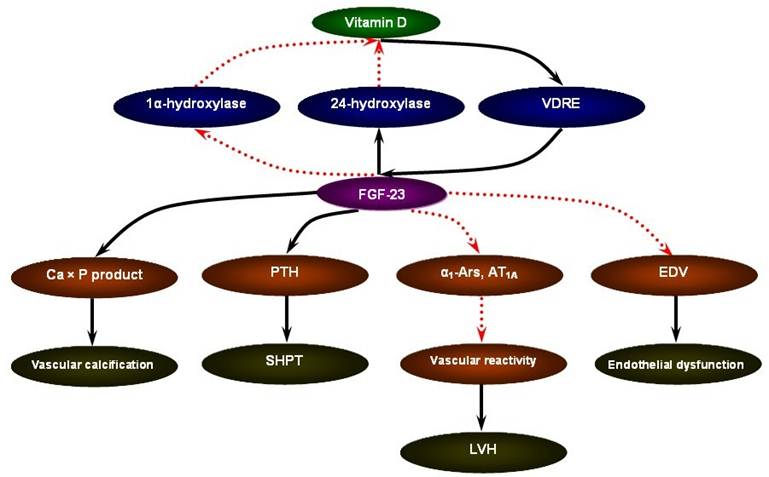
FGF-23 and vascular calcification
Vascular calcification is very prevalent in CKD, especially in ESRD. The mechanisms of vascular calcification are hyperphosphatemia and elevated calcium × phosphate product. Vascular calcification induces stiffening of the vessel wall and reduces vascular compliance, which has been found to be predictive of cardiovascular mortality [56]. FGF-23, a major phosphatonin, decreases serum phosphate concentrations by directly reducing renal reabsorption, and therefore prevents vascular calcification in physiological conditions. FGF-23 null mice on a regular diet develop a mineral profile similar to patients who suffer from ESRD and are treated with high-dosage vitamin D, namely, elevated serum calcium, phosphate and 1,25-(OH)2D levels along with ubiquitous vascular calcification and decreased survival [38, 57]. In the clinical investigation undertaken by Wesseling-Perry et al. [58], irrespective of residual renal function, serum FGF-23 levels correlated with phosphate and calcium × phosphate product. In another study, Gutierrez et al. [59] divided 80 patients into four CKD groups according to the eGFR, and observed that serum levels of FGF-23 increased as kidney function decline, whereas phosphate values did not become abnormal until eGFR fell below 30 ml/min/1.73m2. Thus, it seems that during early stages of CKD, FGF-23 may contribute to maintaining the serum phosphate levels within the normal range. This compensation for decreased nephron mass may be overcome by severe renal failure when overt hyperphosphatemia and vascular calcification develop despite markedly increased FGF-23 levels [60]. There may be a feedback loop existing between serum vitamin D, FGF-23 and mineral disorder. FGF-23 is both the cause of abnormal serum calcium and phosphate levels and their consequence [61].
FGF-23 and SHPT
As kidney function declines, the elevated serum PTH concentrations are frequently the earliest detectable abnormality [62]. Individuals with elevated PTH concentrations are at higher risk of cardiovascular mortality. According to the statistical data reported by Hagström et al. [63], in a community-based cohort of 958 elderly men, a 1-SD increase in serum PTH was associated with a 37% to 38% higher risk for cardiovascular mortality in the crude and multivariable models. Several mechanisms may account for the link between PTH and cardiovascular mortality. First, PTH is directly implicated in atherogenesis through vascular calcification and vascular remodeling [64, 65]. Second, PTH seems also to have detrimental effects on the myocardium through induction of LVH, cardiac calcification and fibrosis [66-68]. Third, SHPT may participate in the pathophysiological mechanism of hyperlipidemia seen in chronic renal failure [69]. Fourth, higher PTH is related with both established cardiovascular risk factors and more recently described risk factors such as inflammation markers, renal dysfunction and cardiac pathology [63, 70-72]. Finally, because PTH is one of the pivotal regulatory hormones in the mineral homeostasis, it is possible that the serum levels of PTH reflect other abnormalities along the same pathway such as vitamin D deficiency, hypercalcemia, hyperphosphatemia and renal failure, which predisposes to a higher risk for cardiovascular mortality [63].
FGF-23 is located upstream of the PTH molecule. A study that measured both FGF23 and PTH in patients with early CKD and healthy controls suggested that FGF23 excess precedes PTH excess, because FGF23 levels were already twice normal and significantly higher in patients with CKD, whereas PTH levels were normal and not significantly different between patients with CKD and healthy participants [73]. Up-regulation of FGF-23 in patients with CKD inhibits renal 1α-hydroxylase and results in early 1,25-(OH)2D deficiency, which may initiate the development of SHPT [74]. Two representative approaches have been employed to identify the significant relationship between FGF-23 and PTH in vivo. Animal models of expression of FGF-23 demonstrate diffuse parathyroid hyperplasia and SHPT [37]; FGF-23 null mice have low PTH concentrations due to very high serum calcium and 1,25-(OH)2D levels [75]. However, the action of FGF-23 on PTH is variable in vitro. Krajisnik et al. [76] treated parathyroid cells with a stabilized form of recombinant FGF-23 [FGF-23(R176Q)] and found that FGF-23(R176Q) exerted an obvious effect on cell apoptosis and reduced the PTH mRNA level in a dose-dependent manner in the concentration rang of 400~2000pg/ml; when the FGF-23(R176Q) concentrations were over 2000pg/ml, they did not detect any significant effect on cell apoptosis, whereas a small but significant increase in cell proliferation was observed. In another study, Ben-Dov et al. showed that FGF-23 suppressed both PTH secretion and PTH gene expression in vitro rat parathyriod cultures through the mitogen-activated protein kinase pathway [77]. The Ben-Dov and Krajisnik studies are the only data that isolate direct effects of FGF23 on the parathyroid, independent of circulating mineral content. Either provide some evidence to support the statement that “FGF23 suppresses PTH”. Conversely, PTH may stimulate FGF-23 secretion by osteoblasts, because the FGF-23 levels of patients with primary hyperparathyroidism are increased, which may be reduced by parathyroidectomy [78]. The mechanisms by which PTH mediates changes in FGF-23 expression remain unclear and may be ascribed to either direct effects on FGF-23 gene expression itself or mediation through other potential regulators of FGF-23 [79]. Therefore, FGF-23 stimulates PTH in vivo or in CKD individuals, whereas suppresses PTH in vitro, and is also increased by excess PTH [80]; FGF-23 is an important predictor of future SHPT and subsequent cardiovascular mortality in patients undergoing CKD, especially when serum phosphate and PTH levels are in the normal range [81].
FGF-23 and LVH
LVH is a prevalent manifestation of CVD and also an independent risk factor for mortality in patients with CKD [82, 83]. Approximately 40% of patients with predialysis CKD and up to 80% of patients initiating hemodialysis manifest LVH [84, 85]. Insufficiency of cardiac diastolic and contractive functions, inappropriate activation of the renin-angiotensin-aldosterone system, alteration of fluid balance and disturbance of collagen are identified as the major determinants of LVH in CKD [86]. Several near recent studies have demonstrated that elevated FGF-23 may be also involved in the LVH onset [47, 87]. In maintenance hemodialysis patients, serum FGF-23 levels were significantly correlated with higher mean left ventricular mass index (LVMI) and lower mean ejection fraction, irrespective of B-type natriuretic peptide and cardiac troponin T [88]. And in transgenic mice overexpressing human FGF-23, interventricular septum thickness diastolic, left ventricular posterior wall thickness diastolic and LVMI relative to body weight were significantly increased, which may be attributed to the impaired vascular reactivity and down-regulation of vasoconstrictor receptors (α1-ARs and AT1A) [89]. The cardiac hypertrophic effects of FGF-23 are mediated by FGFR activation and could be blocked in vitro by the FGFR inhibitor PD173074, but do not require Klotho as coreceptor [90]. Furthermore, matrix metalloproteinases dysmetabolism may also act as a close link between elevated FGF-23 and LVH in patients with CKD [91].
FGF-23 and endothelial dysfunction
CKD is now considered a typical situation of chronic inflammatory state. C-reactive protein (CRP), a nonspecific marker of inflammation, is described as a fundamental biomarker for endothelial dysfunction in patients with CKD [92]. Endothelial dysfunction usually describes binding of monocytes to the endothelial surface, down-regulation of nitric oxide activity, reduced dilatory capacities and an early event of arteriosclerosis [93]. Although some data from clinical observations have suggested that FGF-23 is associated with the elevated levels of CRP in CKD patients [94, 95], there is scant direct evidence supporting the induction of endothelial dysfunction by FGF-23. To the best of our knowledge, pioneer work was conducted by Mirze et al. [96] at the Uppsala University Hospital. His group found that FGF-23 could reduce endothelium-dependent vasodilation both in healthy subjects and in subjects with eGFR<60 ml/min/1.73m2. A more recent report also demonstrated that the high log FGF-23 was a significant independent risk factor of the increased carotid artery intima-media thickness in 128 maintenance hemodialysis patients [97]. However, there is some data suggesting that Klotho, the cofactor of FGF-23, can regulate endothelial function. In the study of Hu et al. [98], despite induction of CKD, Klotho transgenetic mice had preserved levels of Klotho and much less vascular calcification compared with wild-type mice with CKD; inversely, Klotho-haploinsufficient mice with CKD had undetectable levels of Klotho and severe vascular calcification. It is possible that part of the beneficial effects of Klotho on endothelial function in CKD result from improvement of FGF-23 signal transduction [99]. Therefore, further studies in vitro are needed to clarify whether FGF-23 is a marker or a potential initiation for endothelial dysfunction in CKD.
Perspectives
Taken as a whole, the data presented above which come from observational or experimental studies support the hypothesis that FGF-23 may help to explain the biphasic cardiovascular effects of vitamin D in CKD, according to the cross actions between FGF-23 and the multiple metabolic pathways (vascular calcification, SHPT, LVH and endothelial dysfunction). Therefore, modulation of FGF-23 may become a potential therapeutic target for the lowering of cardiovascular risk in CKD.
Several clinical interventions have been employed to manipulate FGF-23. (1) Decreased phosphate intake: Ferrari et al. [100] measured FGF-23 concentrations in 29 healthy young men who were treated sequentially with 5 days of dietary phosphate restriction, followed by an equilibration period, then 5 days of a high-phosphate diet. The mean serum FGF-23 concentration decreased 29.1±6.5% during phosphate restriction, and increased 31.1±9.5% during phosphate supplementation. In addition, dietary phosphate modification can also regulate 1,25-(OH)2D production, mediated in part by changes in circulating FGF-23. Antoniucci et al. studied 13 healthy men during a 4-week dietary phosphate intervention study, and found that serum FGF-23 concentrations decreased significantly from 30.7±8.7 pg/ml during phosphate supplementation to 19.6±7.0 pg/ml during phosphate restriction, accompanied with increased 1,25-(OH)2D levels from 29±10 pg/ml to 40±16 pg/ml [101]. (2) Phosphate binders: A short-term 6-week dose titration study evaluated the effects of two phosphate binders, sevelamer hydrochloride and calcium acetate, on FGF-23 levels in patients with CKD stages 3 to 4. Sevelamer-treated patients presented a significant reduction in FGF-23 (107 pg/ml at baseline versus 54 pg/ml at the 6th week), whereas this was not observed in calcium-treated patients [102]. Conformably, Gonzalez-Parra et al. showed that serum FGF-23 concentrations in 18 patients with CKD stages 3 were significantly decreased after 4 weeks treatment with lanthanum carbonate [103]. Even more excitingly, in a 1-year prospective observational study of incident hemodialysis patients, treatment with phosphate binders was associated with a significant survival advantage compared with no treatment (13.6 versus 23.5 deaths per 100 person-years) [104]. (3) Cinacalcet and concurrent low-dose vitamin D: Wetmore et al. [105] used samples from ACHIEVE trial to perform a per-protocol analysis of the effects of treatment on FGF-23, and they found that the treatment regimen using Cinacalcet plus fixed low-dose calcitriol analogs could result in a relative decrease in FGF-23 levels compared with an approach using escalating doses of calcitriol analogs alone, after a 27-week intervention period (-9.7% versus 4.1%). (4) C-terminal tail of FGF-23: Goetz et al. [106] injected the isolated 72-residue-long C-terminal tail of FGF-23 into healthy rats and demonstrated that it could block the pathogenic actions of intact FGF-23, which may hold promise of providing the first causative pharmacotherapy. C-terminal tail of FGF-23, generating an endogenous inhibitor of intact FGF-23, removes the binding site for the binary FGFR-Klotho complex. (5) Renal transplantation: Economidou et al. [107] performed a prospective study to investigate FGF-23 levels in 18 patients with ESRD before and after renal transplantation. Compared with pretransplantation, FGF-23 levels decreased by 89% at the end of 3 months posttransplantation, and moreover, all patients had normal FGF-23 levels (<50 pg/ml) at 12 months posttransplantation. The excessive and rapid reduction may be due to increased urinary excretion [108].
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81000306) and the Post-Doctoral Foundation of Anhui Medical University (No. 2009KJ02).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Beale MG, Chan JC, Oldham SB. et al. Vitamin D: the discovery of its metabolites and their therapeutic applications. Pediatrics. 1976;57:729-41
2. Stamp TC. Vitamin D metabolism. Recent advances. Arch Dis Child. 1973;48:2-7
3. Haussler MR, Haussler CA, Jurutka PW. et al. The vitamin D hormone and its nuclear receptor: molecular actions and disease states. J Endocrinol. 1997;154:S57-73
4. Breidenbach A, Schlumbohm C, Harmeyer J. Peculiarities of vitamin D and of the calcium and phosphate homeostatic system in horses. Vet Res. 1998;29:173-86
5. Amizuka N, Kwan MY, Goltzman D. et al. Vitamin D3 differentially regulates parathyroid hormone/parathyroid hormone-related peptide receptor expression in bone and cartilage. J Clin Invest. 1999;103:373-81
6. National Kidney Foundation. K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis. 2003;42:S1-201
7. Barreto DV, Barreto FC, Liabeuf S. et al. Vitamin D affects survival independently of vascular calcification in chronic kidney disease. Clin J Am Soc Nephrol. 2009;4:1128-35
8. London GM, Guérin AP, Verbeke FH. et al. Mineral metabolism and arterial functions in end-stage renal disease: potential role of 25-hydroxyvitamin D deficiency. J Am Soc Nephrol. 2007;18:613-20
9. Querfeld U, Mak RH. Vitamin D deficiency and toxicity in chronic kidney disease: in search of the therapeutic window. Pediatr Nephrol. 2010;25:2413-30
10. Williams S, Malatesta K, Norris K. Vitamin D and chronic kidney disease. Ethn Dis. 2009;19:S5-S8 -S11
11. Hu P, Hu B, Wang J. et al. Modulation of vitamin D signaling is a potential therapeutic target to lower cardiovascular risk in chronic kidney disease. Med Sci Monit. 2011;17:HY14-20
12. Oh J, Wunsch R, Turzer M. et al. Advanced coronary and carotid arteriopathy in young adults with childhood-onset chronic renal failure. Circulation. 2002;106:100-5
13. Mitsnefes MM. Cardiovascular complications of pediatric chronic kidney disease. Pediatr Nephrol. 2008;23:27-39
14. Rakhit DJ, Marwick TH, Armstrong KA. et al. Effect of aggressive risk factor modification on cardiac events and myocardial ischaemia in patients with chronic kidney disease. Heart. 2006;92:1402-8
15. Kaisar MO, Isbel NM, Johnson DW. Recent clinical trials of pharmacologic cardiovascular interventions in patients with chronic kidney disease. Rev Recent Clin Trials. 2008;3:79-88
16. Reddy Vanga S, Good M, Howard PA. et al. Role of vitamin D in cardiovascular health. Am J Cardiol. 2010;106:798-805
17. Santoro D, Gitto L, Ferraro A. et al. Vitamin D status and mortality risk in patients with chronic kidney disease. Ren Fail. 2011;33:184-91
18. Teng M, Wolf M, Ofsthun MN. et al. Activated Injectable Vitamin D and Hemodialysis Survival: A Historical Cohort Study. J Am Soc Nephrol. 2005;16:1115-25
19. Wolf M, Shah A, Gutierrez O. et al. Vitamin D levels and early mortality among incident hemodialysis patients. Kidney Int. 2007;72:1004-13
20. Judd SE, Tangpricha V. Vitamin D deficiency and risk for cardiovascular disease. Am J Med Sci. 2009;338:40-4
21. Teng M, Wolf M, Lowrie E. et al. Survival of Patients Undergoing Hemodialysis with Paricalcitol or Calcitriol Therapy. N Engl J Med. 2003;349:446-56
22. Tentori F, Hunt WC, Stidley CA. et al. Mortality risk among hemodialysis patients receiving different vitamin D analogs. Kidney Int. 2006;70:1858-65
23. Tentori F, Albert JM, Young EW. et al. The survival advantage for haemodialysis patients taking vitamin D is questioned: findings from the Dialysis Outcomes and Practice Patterns Study. Nephrol Dial Transplant. 2009;24:963-72
24. Rajasree S, Rajpal K, Kartha CC. et al. Serum 25-hydroxyvitamin D3 levels are elevated in South Indian patients with ischemic heart disease. Eur J Epidemiol. 2001;17:567-71
25. Mathew S, Lund RJ, Chaudhary LR. et al. Vitamin D receptor activators can protect against vascular calcification. J Am Soc Nephrol. 2008;19:1509-19
26. Kunitomo M, Kinoshita K, Bandô Y. Experimental atherosclerosis in rats fed a vitamin D, cholesterol-rich diet. J Pharmacobiodyn. 1981;4:718-23
27. Reis JP, Michos ED, von Mühlen D. et al. Differences in vitamin D status as a possible contributor to the racial disparity in peripheral arterial disease. Am J Clin Nutr. 2008;88:1469-77
28. Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventro-lateral thalamic nucleus of the brain. Biochem Biophy Res Comm. 2000;277:494-8
29. Nitta K. Relationship between Fibroblast Growth Factor-23 and Mineral Metabolism in Chronic Kidney Disease. Int J Nephrol. 2010;2010:167984
30. Brownstein CA, Zhang J, Stillman A. et al. Increased bone volume and correction of HYP mouse hypophosphatemia in the Klotho/HYP mouse. Endocrinology. 2010;151:492-501
31. Hughes SE. Differential expression of the fibroblast growth factor receptor (FGFR) multigene family in normal human adult tissues. J Histochem Cytochem. 1997;45:1005-19
32. Kurosu H, Ogawa Y, Miyoshi M. et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281:6120-3
33. Liu S, Vierthaler L, Tang W. et al. FGFR3 and FGFR4 Do not Mediate Renal Effects of FGF23. J Am Soc Nephrol. 2008;19:2342-50
34. Komaba H, Goto S, Fujii H. et al. Depressed expression of Klotho and FGF receptor 1 in hyperplastic parathyroid glands from uremic patients. Kidney Int. 2010;77:232-8
35. Shaikh A, Berndt T, Kumar R. Regulation of phosphate homeostasis by the phosphatonins and other novel mediators. Pediatr Nephrol. 2008;23:1203-10
36. Saito H, Kusano K, Kinosaki M. et al. Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1alpha, 25-dihydroxyvitamin D3 production. J Biol Chem. 2003;278:2206-11
37. Bai X, Miao D, Li J, Goltzman D, Karaplis AC. Transgenic mice overexpressing human fibroblast growth factor 23 (R176Q) delineate a putative role for parathyroid hormone in renal phosphate wasting disorders. Endocrinology. 2004;145:5269-79
38. Stubbs JR, Liu S, Tang W. et al. Role of hyperphosphatemia and 1,25-dihydroxyvitamin D in vascular calcification and mortality in fibroblastic growth factor 23 null mice. J Am Soc Nephrol. 2007;18:2116-24
39. Aono Y, Yamazaki Y, Yasutake J. et al. Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. J Bone Miner Res. 2009;24:1879-88
40. ADHR Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26:345-8
41. Perwad F, Zhang MY, Tenenhouse HS. et al. Fibroblast growth factor 23 impairs phosphorus and vitamin D metabolism in vivo and suppresses 25-hydroxyvitamin D-1alpha-hydroxylase expression in vitro. Am J Physiol Renal Physiol. 2007;293:F1577-83
42. Jüppner H, Wolf M, Salusky IB. FGF-23: More than a regulator of renal phosphate handling? J Bone Miner Res. 2010;25:2091-7
43. Wesseling-Perry K. FGF-23 in bone biology. Pediatr Nephrol. 2010;25:603-8
44. Levine BS, Kleeman CR, Felsenfeld AJ. The journey from vitamin D-resistant rickets to the regulation of renal phosphate transport. Clin J Am Soc Nephrol. 2009;4:1866-77
45. Isakova T, Wahl P, Vargas GS. et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79:1370-8
46. Gutiérrez OM, Januzzi JL, Isakova T. et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation. 2009;119:2545-52
47. Shimada T, Urakawa I, Isakova T. et al. Circulating fibroblast growth factor 23 in patients with end-stage renal disease treated by peritoneal dialysis is intact and biologically active. J Clin Endocrinol Metab. 2010;95:578-85
48. Bacchetta J, Dubourg L, Harambat J. et al. The influence of glomerular filtration rate and age on fibroblast growth factor 23 serum levels in pediatric chronic kidney disease. J Clin Endocrinol Metab. 2010;95:1741-8
49. Westerberg PA, Linde T, Wikström B. et al. Regulation of fibroblast growth factor-23 in chronic kidney disease. Nephrol Dial Transplant. 2007;22:3202-7
50. Perwad F, Azam N, Zhang MY. et al. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology. 2005;146:5358-64
51. Ito M, Sakai Y, Furumoto M. et al. Vitamin D and phosphate regulate fibroblast growth factor-23 in K-562 cells. Am J Physiol Endocrinol Metab. 2005;288:E1101-9
52. Saito H, Maeda A, Ohtomo S. et al. Circulating FGF-23 is regulated by 1alpha, 25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem. 2005;280:2543-9
53. Stubbs JR, Egwuonwu S. Is fibroblast growth factor 23 a harbinger of mortality in CKD? Pediatr Nephrol. 2011 [Epub ahead of print]
54. Gutiérrez OM, Mannstadt M, Isakova T. et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359:584-92
55. Oliveira RB, Moysés RM. FGF-23: state of the art. J Bras Nefrol. 2010;32:323-31
56. Tamei N, Ogawa T, Ishida H. et al. Serum fibroblast growth factor-23 levels and progression of aortic arch calcification in non-diabetic patients on chronic hemodialysis. J Atheroscler Thromb. 2011;18:217-23
57. Razzaque MS, St-Arnaud R, Taguchi T. et al. FGF-23, vitamin D and calcification: the unholy triad. Nephrol Dial Transplant. 2005;20:2032-5
58. Wesseling-Perry K, Pereira RC, Wang H. et al. Relationship between plasma fibroblast growth factor-23 concentration and bone mineralization in children with renal failure on peritoneal dialysis. J Clin Endocrinol Metab. 2009;94:511-7
59. Gutierrez O, Isakova T, Rhee E. et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol. 2005;16:2205-15
60. Komaba H, Fukagawa M. FGF23: a key player in mineral and bone disorder in CKD. Nefrologia. 2009;29:392-6
61. Fliser D, Kollerits B, Neyer U. et al. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) Study. J Am Soc Nephrol. 2007;18:2600-8
62. Ix JH, Shlipak MG, Wassel CL. et al. Fibroblast growth factor-23 and early decrements in kidney function: the Heart and Soul Study. Nephrol Dial Transplant. 2010;25:993-7
63. Hagström E, Hellman P, Larsson TE. et al. Plasma parathyroid hormone and the risk of cardiovascular mortality in the community. Circulation. 2009;119:2765-71
64. Coen G, Mantella D, Sardella D. et al. Asymmetric dimethylarginine, vascular calcifications and parathyroid hormone serum levels in hemodialysis patients. J Nephrol. 2009;22:616-22
65. Song GJ, Fiaschi-Taesch N, Bisello A. Endogenous parathyroid hormone-related protein regulates the expression of PTH type 1 receptor and proliferation of vascular smooth muscle cells. Mol Endocrinol. 2009;23:1681-90
66. Cha H, Jeong HJ, Jang SP. et al. Parathyroid hormone accelerates decompensation following left ventricular hypertrophy. Exp Mol Med. 2010;42:61-8
67. Di Leo C, Gallieni M, Bestetti A. et al. Cardiac and pulmonary calcification in a hemodialysis patient: partial regression 4 years after parathyroidectomy. Clin Nephrol. 2003;59:59-63
68. Amann K, Ritz E, Wiest G. et al. A role of parathyroid hormone for the activation of cardiac fibroblasts in uremia. J Am Soc Nephrol. 1994;4:1814-9
69. Nishizawa Y, Miki T, Okui Y. et al. Deranged metabolism of lipids in patients with chronic renal failure: possible role of secondary hyperparathyroidism. Jpn J Med. 1986;25:40-5
70. Ogard CG, Engelmann MD, Kistorp C. et al. Increased plasma N-terminal pro-B-type natriuretic peptide and markers of inflammation related to atherosclerosis in patients with primary hyperparathyroidism. Clin Endocrinol (Oxf). 2005;63:493-8
71. De Boer IH, Gorodetskaya I, Young B. et al. The severity of secondary hyperparathyroidism in chronic renal insufficiency is GFR-dependent, race-dependent, and associated with cardiovascular disease. J Am Soc Nephrol. 2002;13:2762-9
72. Walker MD, Fleischer JB, Di Tullio MR. et al. Cardiac structure and diastolic function in mild primary hyperparathyroidism. J Clin Endocrinol Metab. 2010;95:2172-9
73. Zisman AL, Wolf M. Recent advances in the rapidly evolving field of fibroblast growth factor 23 in chronic kidney disease. Curr Opin Nephrol Hypertens. 2010;19:335-42
74. Zoccali C. FGF-23 in dialysis patients: ready for prime time? Nephrol Dial Transplant. 2009;24:1078-81
75. Shimada T, Kakitani M, Yamazaki Y. et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113:561-8
76. Krajisnik T, Björklund P, Marsell R. et al. Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol. 2007;195:125-31
77. Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V. et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117:4003-8
78. Yamashita H, Yamashita T, Miyamoto M. et al. Fibroblast growth factor (FGF)-23 in patients with primary hyperparathyroidism. Eur J Endocrinol. 2004;151:55-60
79. Wesseling-Perry K, Salusky I. Redefining the pathogenesis of CKD-MBD: the critical role of FGF-23. Bol Pediatr. 2010;50:11-6
80. Imel EA, Econs MJ. Fibroblast growth factor 23: roles in health and disease. J Am Soc Nephrol. 2005;16:2565-75
81. Razzaque MS. Does FGF23 toxicity influence the outcome of chronic kidney disease? Nephrol Dial Transplant. 2009;24:4-7
82. Butler KG. Hemoglobin levels, cardiovascular disease, and left ventricular hypertrophy in patients with chronic kidney disease. Case study of the anemic patient. Nephrol Nurs J. 2002;29:189-92
83. Paoletti E, Bellino D, Gallina AM. et al. Is left ventricular hypertrophy a powerful predictor of progression to dialysis in chronic kidney disease? Nephrol Dial Transplant. 2011;26:670-7
84. Levin A, Singer J, Thompson CR. et al. Prevalent left ventricular hypertrophy in the predialysis population: identifying opportunities for intervention. Am J Kidney Dis. 1996;27:347-54
85. Middleton RJ, Parfrey PS, Foley RN. Left ventricular hypertrophy in the renal patient. J Am Soc Nephrol. 2001;12:1079-84
86. Cerasola G, Nardi E, Palermo A. et al. Epidemiology and pathophysiology of left ventricular abnormalities in chronic kidney disease: a review. J Nephrol. 2011;24:1-10
87. Nakai K, Komaba H, Fukagawa M. New insights into the role of fibroblast growth factor 23 in chronic kidney disease. J Nephrol. 2010;23:619-25
88. Negishi K, Kobayashi M, Ochiai I. et al. Association between fibroblast growth factor 23 and left ventricular hypertrophy in maintenance hemodialysis patients. Comparison with B-type natriuretic peptide and cardiac troponin T. Circ J. 2010;74:2734-40
89. Liu P, Bai X, Wang H. et al. Hypophosphatemia-mediated hypotension in transgenic mice overexpressing human FGF-23. Am J Physiol Heart Circ Physiol. 2009;297:H1514-20
90. Faul C, Amaral AP, Oskouei B. et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121:4393-408
91. Peiskerová M, Kalousová M, Kratochvílová M. et al. Fibroblast growth factor 23 and matrix-metalloproteinases in patients with chronic kidney disease: are they associated with cardiovascular disease? Kidney Blood Press Res. 2009;32:276-83
92. Tripepi G, Mallamaci F, Zoccali C. Inflammation markers, adhesion molecules, and all-cause and cardiovascular mortality in patients with ESRD: searching for the best risk marker by multivariate modeling. J Am Soc Nephrol. 2005;16:S83-8
93. Stenvinkel P. Endothelial dysfunction and inflammation-is there a link? Nephrol Dial Transplant. 2001;16:1968-71
94. Kojima F, Uchida K, Ogawa T. et al. Plasma levels of fibroblast growth factor-23 and mineral metabolism in diabetic and non-diabetic patients on chronic hemodialysis. Int Urol Nephrol. 2008;40:1067-74
95. Manghat P, Fraser WD, Wierzbicki AS. et al. Fibroblast growth factor-23 is associated with C-reactive protein, serum phosphate and bone mineral density in chronic kidney disease. Osteoporos Int. 2010;21:1853-61
96. Mirza MA, Larsson A, Lind L. et al. Circulating fibroblast growth factor-23 is associated with vascular dysfunction in the community. Atherosclerosis. 2009;205:385-90
97. Balci M, Kirkpantur A, Gulbay M. et al. Plasma fibroblast growth factor-23 levels are independently associated with carotid artery atherosclerosis in maintenance hemodialysis patients. Hemodial Int. 2010;14:425-32
98. Hu MC, Shi M, Zhang J. et al. Klotho Deficiency Causes Vascular Calcification in Chronic Kidney Disease. J Am Soc Nephrol. 2011;22:124-36
99. Donate-Correa J, Mora-Fernández C, Martínez-Sanz R. et al. Expression of FGF23/KLOTHO system in human vascular tissue. Int J Cardiol. 2011 [Epub ahead of print]
100. Ferrari SL, Bonjour JP, Rizzoli R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab. 2005;90:1519-24
101. Antoniucci DM, Yamashita T, Portale AA. Dietary phosphorus regulates serum fibroblast growth factor-23 concentrations in healthy men. J Clin Endocrinol Metab. 2006;91:3144-9
102. Oliveira RB, Cancela AL, Graciolli FG. et al. Early control of PTH and FGF23 in normophosphatemic CKD patients: a new target in CKD-MBD therapy? Clin J Am Soc Nephrol. 2010;5:286-91
103. Gonzalez-Parra M, Gonzalez-Casaus ML, Galán A. et al. Lanthanum carbonate reduces FGF23 in chronic kidney disease Stage 3 patients. Nephrol Dial Transplant. 2011;26:2567-71
104. Isakova T, Gutiérrez OM, Chang Y. et al. Phosphorus binders and survival on hemodialysis. J Am Soc Nephrol. 2009;20:388-96
105. Wetmore JB, Liu S, Krebill R. et al. Effects of cinacalcet and concurrent low-dose vitamin D on FGF23 levels in ESRD. Clin J Am Soc Nephrol. 2010;5:110-6
106. Goetz R, Nakada Y, Hu MC. et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci USA. 2010;107:407-12
107. Economidou D, Dovas S, Papagianni A. et al. FGF-23 Levels before and after Renal Transplantation. J Transplant. 2009;2009:379082
108. Evenepoel P, Meijers BK, de Jonge H. et al. Recovery of hyperphosphatoninism and renal phosphorus wasting one year after successful renal transplantation. Clin J Am Soc Nephrol. 2008;3:1829-36
Author contact
![]() Corresponding author: Tel.: +86 551 2922058. E-mail: hupeng28com.cn (P. Hu).
Corresponding author: Tel.: +86 551 2922058. E-mail: hupeng28com.cn (P. Hu).