Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
The role of TGF-β signaling...
Therapeutic Targeting of...
Pre-clinical Data
Clinical Data
Developing Concepts and Future...
Conclusions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(7):964-978. doi:10.7150/ijbs.4564 This issue Cite
Review
Complexities of TGF-β Targeted Cancer Therapy
Erin C. Connolly1, Julia Freimuth1, Rosemary J. Akhurst1,2 ![]()
1. UCSF Helen Diller Family Comprehensive Cancer Center, University of California at San Francisco, California 94143-0512, USA
2. Department of Anatomy, University of California at San Francisco, California 94143-0512, USA
Received 2012-5-7; Accepted 2012-6-23; Published 2012-7-12
Abstract
Many advanced tumors produce excessive amounts of Transforming Growth Factor-β (TGF-β) which, in normal epithelial cells, is a potent growth inhibitor. However, in oncogenically activated cells, the homeostatic action of TGF-β is often diverted along alternative pathways. Hence, TGF-β signaling elicits protective or tumor suppressive effects during the early growth-sensitive stages of tumorigenesis. However, later in tumor development when carcinoma cells become refractory to TGF-β-mediated growth inhibition, the tumor cell responds by stimulating pathways with tumor progressing effects. At late stages of malignancy, tumor progression is driven by TGF-β overload. The tumor microenvironment is a target of TGF-β action that stimulates tumor progression via pro-tumorigenic effects on vascular, immune, and fibroblastic cells. Bone is one of the richest sources of TGF-β in the body and a common site for dissemination of breast cancer metastases. Osteoclastic degradation of bone matrix, which accompanies establishment and growth of metastases, triggers further release of bone-derived TGF-β. This leads to a vicious positive feedback of tumor progression, driven by ever increasing levels of TGF-β released from both the tumor and bone matrix. It is for this reason, that pharmaceutical companies have developed therapeutic agents that block TGF-β signaling. Nonetheless, the choice of drug design and dosing strategy can affect the efficacy of TGF-β therapeutics. This review will describe pre-clinical and clinical data of four major classes of TGF-β inhibitor, namely i) ligand traps, ii) antisense oligonucleotides, iii) receptor kinase inhibitors and iv) peptide aptamers. Long term dosing strategies with TGF-β inhibitors may be ill-advised, since this class of drug has potentially highly pleiotropic activity, and development of drug resistance might potentiate tumor progression. Current paradigms for the use of TGF-β inhibitors in oncology have therefore moved towards the use of combinatorial therapies and short term dosing, with considerable promise for the clinic.
Keywords: Transforming growth factor-β (TGF-β)
Introduction
Targeting a tumor promoting agent for neutralization seems like a clear-cut strategy for cancer therapy. But what if the tumor promoter of interest can be tumor suppressive in a different context? And what if the molecule or signaling pathway of interest has a broad impact on multiple biological programs? How might this influence therapeutic strategies, or alter the outcome of targeting this agent? This review seeks to stimulate discussion of these questions through exploring the Transforming Growth Factor-β (TGF-β) signaling pathway as a target in oncology.
TGF-β Structure and Signaling
In 1978 DeLarco and Todaro described the partial purification of Sarcoma Growth Factors (SGFs) and their ability to induce anchorage-independent growth in normal rat kidney cells [1-2]. Two years later, Roberts et al. [3] and Moses et al. [4] independently purified TGF-β as one component of SGF. These findings initiated both, the description of the TGF-β ligands and the birth of the TGF-β signaling field [4-6]. TGF-β is now known to be the most potent growth inhibitor for normal epithelial, hematopoietic and immune cells, and plays an important function in normal tissue homeostasis [7]. TGF-β ligands are members of the TGF-β superfamily, a family which is comprised of more than 30 closely related proteins including bone morphogenetic proteins (BMPs), activins, inhibins and nodal [8]. In humans three isoforms of TGF-β (TGF-β1, TGF-β2 and TGF-β3) have been described [7]. These isoforms share 75% amino acid sequence homology and have demonstrated comparable signaling activities in vitro while expression patterns of the three isoforms differ between cell and tissue types [9], and knockout mouse studies have demonstrated distinct roles for the different isoforms in vivo [10]. TGF-β ligands are secreted from the cell as homodimers in their latent precursor form, which are activated at the responding cell surface by proteolytical cleavage of the latency-associated peptide (LAP). In its latent form TGF-β cannot bind to its receptor, thus processing of the propeptide into its active state is of regulatory importance for TGF-β bioavailability. Despite the great shared homology between TGF-β ligands, LAP isoforms (LAPβ1, β2, β3) share only 34-38% amino acid homology suggesting a mechanism for differential TGF-β regulation. For example, direct interaction of αvβ6 integrin with the RGD-integrin binding site in LAPs of latent TGF-β1 and TGF-β3 can efficiently activate the signaling cascade. In contrast, the LAP isoform of TGF-β2 is unique in lacking this RGD-binding sequence and therefore cannot be activated by αvβ6 integrin [11-12]. The mature TGF-β2 homodimer is also unique in having a much lower binding affinity for the TGF-β type II receptor, due to inter-isoform divergence at amino-acids Lys25, Ile92, and Lys94 of the mature bioactive TGF-β2 peptide [13-14]. TGF-β2 is therefore dependent on β-glycan for high affinity binding to the signaling receptor complex and, unlike TGF-β1 and TGF-β3, shows weak activity on endothelial and hematopoietic cells that do not express β-glycan [13-14].
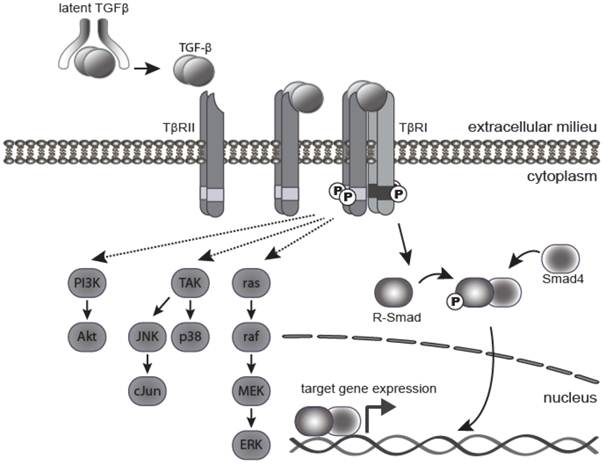
Active TGF-β1 and 3 bind with high affinity and selectivity to the transmembrane TGF-β type II receptor (TβRII). This in turn recruits and activates the TGF-β type I receptor (TβR1 or ALK5) (Figure 1). Activated TβR1 then initiates canonical Smad signaling by phosphorylation of receptor-associated Smads (R-Smads), Smad2, and Smad3. The activated R-Smads form a stable heterohexameric complex with Smad4, the common mediator Smad, and translocate into the nucleus. In association with other DNA binding transcription factors, the Smad complex attains high affinity binding to Smad-binding elements within the promoter region of TGF-β target genes, culminating in TGF-β dependent transcription [15]. TGF-β can also signal through mechanisms independent of Smad activation, including the PI3 kinase, MAPK, TRAF6-TAK1 and RhoA-Rock pathways [16] (Figure 1). The canonical Smad pathway is central to the growth inhibitory action of TGF-β, however, the relative contribution of Smad and non-Smad pathways to other TGF-β induced processes, such as EMT and apoptosis, is still the subject of continued investigation.
TGF-β signaling pathway. TGF-β ligand is secreted as a latent precursor protein, bound to LAP. Activation of TGF-β involves cleavage of LAP from the ligand, which then binds to the type II receptor, and drives hetero-tetramerization with the type I receptor. The canonical signaling pathway involves phosphorylation of R-Smads (mainly Smad2 and Smad3) by activated TβRI. Phosphorylated R-Smads form a complex with the Co-Smad (Smad4), which translocates into the nucleus to bind gene promoters and activate expression of target genes. There are several non-canonical (non-Smad) signaling pathways, whereby TGF-β signals through the TGF-β receptors to activate TGF-β activated kinase 1 (Tak1), Ras and PI3K as well as other pathways.
The role of TGF-β signaling in tumorigenesis and progression
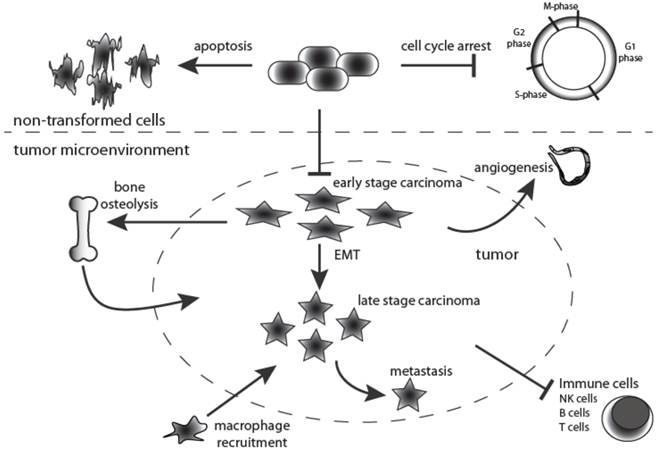
One of the key functions of TGF-β signaling is to maintain epithelial, endothelial and hematopoietic cell homeostasis (Figure 2). However, in pathological situations its homeostatic action is hijacked and diverted along several alternative routes, particularly during cancer progression when loss of tumor suppressors and mutation of oncogenes disrupt the intracellular signaling networks of the tumor cell. The current consensus is that TGF-β signaling has a dual role in cancer. TGF-β signaling elicits a preventative or tumor suppressing effect during the earlier stages of tumorigenesis, when the epithelial cells retain exquisite growth sensitivity to this ligand. Later in tumor development, when carcinoma cells become refractory to TGF-β-mediated growth inhibition and acquire oncogenic mutations, the intracellular signaling circuitry of the cells is altered leading to tumor progressing effects, acting via an array of cellular and molecular mechanisms [17] (Figure 2).
Dual roles for TGF-β signaling during tumorigenesis. TGF-β is a multipotent cytokine that is involved in many cellular processes. Moreover, its action is context-dependent. In the normal untransformed cell it functions as an inducer of apoptosis while at the same time controlling cell differentiation and proliferation. Moreover, TGF-β1 is fundamentally implicated in many aspects of tumorigenesis by directly acting on the tumor cell as well as influencing the tumor microenvironment. During early stages of tumorigenesis it inhibits proliferation of transformed cells but at later stages it supports tumor growth, and enhances tumor invasion and metastasis, macrophage recruitment, tumor angiogenesis and systemic and local tumor immunosuppression. Black arrows indicate TGF-β action.
On the whole, the most commonly mutated TGF-β pathway genes in cancer are TGFBR2, TGFBR1, SMAD4 and SMAD2 [18]. Mutations are invariably loss of function, and tend to be restricted to cancers of the GI tract, such as colon, pancreas, and gastric cancer. They have been observed particularly in cancers that have acquired microsatellite instability (MIS). Concordantly, TGFBR2 is a mutational hotspot for MIS inactivation due to possession of a 10 base-pair poly-adenine repeat within its coding sequence [19]. In breast and skin cancer, however, TGF-β pathway mutations are uncommon. A study of 34 matched primary and recurrent breast tumors demonstrated that, despite no detection of TGFBR2 mutations in primary tumors, 12% of recurrent breast tumors contained receptor activity-attenuating point mutations. These findings suggest that, in the minority of breast tumors that do mutate TGFBR2, this is a late event [20]. Likewise, mutations in TGFBR1 are relatively rare in breast or skin cancer [18]. Loss of heterozygosity (LOH) on chromosome 18q, that harbors SMAD4, is seen in 30% of breast tumors, but specific SMAD4 mutations within this large region of LOH are only seen in 12% of tumors [21]. On the other hand, LOH at either SMAD2 and/or SMAD4, which are closely linked on human chromosome 18q, was reported in the majority of 17 human skin squamous cell carcinoma (SCC) specimens examined. However, in this study it is not clear which gene(s) were driving the large regions of LOH, since mutational studies were not undertaken. Nevertheless, the authors reported down-regulation of Smad proteins in many human skin SCC tumors [22]. Whether this was due to mutation, epigenetic or transcriptional down-regulation of the genes remains to be revealed. In conclusion, it appears that rewiring rather than mutation of the TGF-β signaling pathway drives malignant transformation of skin and breast tumor cells.
Mouse models of breast and skin cancer have been used by numerous investigators to demonstrate the biphasic role of TGF-β during tumorigenesis. The ideal therapeutic design would be suppression of the oncogenically-acquired TGF-β tumor promoting activity while reactivating the cell autonomous, tumor suppressive arm of the TGF-β signaling pathway. However, there does not appear to be a definitive “switch” from tumor suppressor to promoter, but rather a multitude of genetic, epigenetic and cellular events involving both the tumor cell and the tumor microenvironment. The lack of a “switch” from tumor suppressor to promoter may present a challenge on a case by case basis, in determining dosing strategies required to achieve strong efficacy with minimal adverse effects.
TGF-β as Tumor Suppressor
The tumor suppressive role of TGF-β signaling has been manifested in many mouse models in which TGF-β1 was over-expressed or under-expressed, demonstrating the importance of growth suppression at early stages of tumorigenesis [23-27]. TGF-β signaling is capable of opposing mitogenic stimuli, mainly by inhibiting cell cycle progression through G1-arrest, but also by inducing apoptosis (Figure 2). The anti-proliferation effects of TGF-β are mediated by suppression of c-Myc via mobilization of cyclin-dependent kinase inhibitors, p15, p21 and p27. Interestingly however, disruption of TGF-β signaling in the absence of a proliferative signal or oncogenic mutation does not induce cell proliferation [28]. In the mouse skin model, TGF-β1 over-expression acts in a tumor suppressive manner [23, 29] and Tgfb1 genetic haplo-insufficiency results in enhanced papilloma numbers [24]. While these studies plainly demonstrate the tumor suppressive action of TGF-β, its mechanism(s) may be multifold. In addition, to the clear cytostatic effects of TGF-β [30-32], in some epithelial cell types this ligand can also induce apoptosis [33-36] as well as senescence [37-40]. In particular, Boulanger and Smith [41-43] postulated that TGF-β acted as a tumor suppressor by inducing senescence of the mammary stem cell population since this ligand was able to diminish the self-renewing capability of pluripotent epithelial cells of the mammary gland.
Using genetic knockout studies, TGF-β signaling has also been shown to be a “guardian of the genome”. In keratinocytes, gene knockout of Tgfb1 or expression of a dominant negative version of the type II receptor, ΔTβRII, resulted in enhanced genomic instability and subsequently, increased rates of aneuploidy and chromosome breaks preceding accelerated malignant transformation [44]. Barcellos-Hoff's group provided mechanistic evidence for a more central role of TGF-β signaling in maintenance of genomic stability. They demonstrated in vitro and in vivo that the DNA damage response, specifically phosphorylation of ATM and downstream targets p53, Chk2, and Rad17, is impaired by genetic loss of Tgfb1 or pharmacological inhibition of TβRI [45-46]. The above studies indicate that loss of TGF-β signaling in early tumorigenesis provides the tumor with a growth advantage, and an environment conducive to accumulation of further mutations by down-regulating the DNA repair pathway. It is for these reasons, that inhibition of the TGF-β signaling pathway has not been considered by some as an appropriate global therapeutic strategy for oncology.
Tumor Promotion by TGF-β
In addition to the view that TGF-β is predominantly tumor suppressing, this signaling pathway clearly also has a major role in tumor progression (Figure 2). The current consensus is that TGF-β signaling stimulates tumor progression through three broad biological effects: 1) cell autonomous induction of epithelial to mesenchymal transition (EMT) [47]; 2) dampening of immune surveillance, both cell autonomously and non-cell autonomously [48]; and 3) indirect facilitation of tumor cell proliferation via its effects on stromal fibroblasts, angiogenesis and ECM, that in turn modulate the tumor cell [28, 49-50]. Once the tumor cell has undergone certain genetic and/or epigenetic changes that attenuate the growth suppressive pathway of TGF-β, targeted over expression of TGF-β1 can drive malignant progression and metastasis. This has been seen in both, the mouse mammary and the skin tumor models [25, 29, 51-52] as well as melanoma, prostate cancer and other types of tumor [49], and is consistent with the fact that many advanced human and murine tumors secrete this ligand in abundance [53-58]. Even once the growth inhibitory pathway is attenuated, both breast carcinoma cells and skin SCC cells can still respond to TGF-β in other ways, such as altered transcriptional programs that result in enhanced tumor cell migration, invasion, extravasation and cell survival [17, 59], as well as by changes in the profile of cytokines that the tumor cell secretes. These in turn contribute to recruitment and polarization of macrophages and neutrophils [60], as well as tumor cell evasion from host cell immune surveillance.
Epithelial to Mesenchymal Transition in Migration and Invasion
The term epithelial to mesenchymal transition (EMT) describes a multi-step event during which cells lose numerous epithelial characteristics and gain the properties typical for mesenchymal cells. Transitions in cell phenotype from epithelial to mesenchymal (EMT) or mesenchymal to epithelial (MET), play a crucial role during embryonic development and tumorigenesis, and require complex changes in gene expression, cell architecture and migratory and invasive behavior. Studies on human and mouse tumors suggest that the same molecular processes that drive developmental EMT are reactivated in the tumor cell to drive tumor progression towards invasive metastatic carcinomas [61].
One of the essential molecules for formation and maintenance of the epithelial phenotype is the adhesion molecule E-cadherin (encoded by Cdh1) which is typically located at cell-cell adhesion junctions. Loss of E-cadherin is consistently observed during EMT and is currently regarded as a hallmark of EMT [62-63]. At the same time, up regulation of Snail, Slug, Vimentin, and Fibronectin leads to acquisition of motility and invasive properties, and allows the cells to migrate through the extracellular matrix and form metastases at distant sites [64]. The TGF-β/Smad pathway is sufficient for a complete phenotypic switch in the transcriptional program from epithelial to a mesenchymal cell type [29, 65-68]. Of note, the extent of cellular and molecular changes that occur along the pathway towards EMT depends on both the cell type and the number of acquired oncogenic mutations. Some epithelial cells undergo only a limited amount of change towards EMT. Nevertheless, even small alterations in migration and cellular plasticity can impact invasion and metastasis significantly. In certain model systems, epithelial cells can undergo a complete loss of expression of all epithelial molecular markers accompanied by acquisition of a completely fibroblastoid or even myofibroblastoid phenotype. This is specifically true in the mouse skin model of chemically-induced carcinoma, where there can be frequent appearance of fibroblastic spindle cell carcinoma (SpCC) that are ultimately derived from keratinocytes of squamous cell carcinoma (SqCC) that have undergone EMT. In this system, the spindle phenotype is driven by TGF-β, but dependent on synergy with activation of the oncogenic H-ras signaling pathway [69-70]. Such an overt EMT of the entire tumor does not generally occur during human tumorigenesis. When EMT does occur it often does so transiently and reversibly yet is still induced by TGF-β/Smad signaling [71]. This variable extent of EMT in different systems, and the contribution of EMT towards metastasis, has resulted in some confusion in the literature as to how to define EMT and how central this event is to the spread of cancer [68]. In our view, this phenomenon is an important driver of tumor dissemination, and can be both TGF-β dependent and independent. HGF, acting through the Met receptor, is a major player in inducing EMT independently of TGF-β. Indeed, studies from the Moses lab [72-73] have shown that genetic inhibition of TGF-β signaling within tumor stromal cells can potentiate invasion and metastasis, specifically through elevation in HGF/Met signaling to the adjacent tumor cell.
It should be noted that EMT is sufficient but not necessary for invasion or metastasis because these two processes can each occur without EMT, often supported by co-migratory bone marrow-derived cells of the tumor stroma [74]. It has therefore been hypothesized that EMT is irrelevant to cancer progression, in part because many tumor metastases tend to be epithelial rather than mesenchymal. However, EMT is known to be plastic and reversible [61, 71], until or unless the mesenchymal phenotype becomes fixed by subsequent epigenetic changes and/or further genetic mutations. EMT might occur transiently to promote cancer cell intravasation into the blood or lymph systems, but the phenotype of the tumor at the secondary metastatic site is determined by the stromal compartments of that site, rather than the innate properties of the tumor cell. Indeed, during cancer spread there must be selection for cells that not only move and survive in the vascular and/or lymphatic systems, but that can re-establish a colony at a secondary site. This sequence of events has been modeled by the metastatic skin carcinoma cell line, E4. This SqCC carcinoma line reversibly transforms from fully epithelial to fully mesenchymal in culture, dependent on the addition of TGF-β [70, 75-76]. When injected subcutaneously into a mouse, it grows as a spindle tumor that depends on TGF-β for its spindle phenotype. In contrast, if E4 cells are injected intra-peritoneally to colonize the peritoneal cavity, they form squamous colonies on the mesothelial lining of the abdomen [70]. The plasticity and reversibility of EMT in response to changing local TGF-β levels is therefore clearly demonstrated in vitro and in vivo.
Epithelial to Mesenchymal Transition in Driving the Stem Cell Phenotype
Regardless of its role in migration and invasion, TGF-β induced EMT might be even more attractive as a druggable target because TGF-β induced EMT is thought to drive cells towards a more “stem cell-like” phenotype. Mesenchymal Stem Cells (MSCs) were first reported in the hematopoietic system, but have more recently been described in many solid tumors, such as breast, colon and brain [77]. It has been reported that induction of EMT either by TGF-β1 or its downstream targets, Snail or Twist, promoted the expression of cell surface markers associated with cancer stem cells (CSCs) in immortalized human mammary epithelial cells (HMECs) [77]. Furthermore, TGF-β can polarize CSCs into a multi-potential cell. Battula et al. [78] demonstrated that HMECs stably expressing TGF-β1, Snail, or Twist, exhibited a cell surface marker profile very similar to that of MSCs. Along with expression of these stem cell surface markers, TGF-β -induced HMECs showed a remarkable similarity of 70% in gene expression profile to bone marrow-derived MSCs. Indeed, these cells were more similar to MSCs than to other mammary tumor cell types.
Interestingly, it has been demonstrated that MSCs preferentially home to wounds and to tumors [78-81] , a property shared by EMT-induced HMECs (EMT-HMECs). In vitro, EMT-HMECs invaded towards breast cancer cells (MDA-MB-231 cells) at similar rates to that of bone marrow-derived MSCs, and in vivo they were able to home to wounded tissue in a similar fashion to MSCs [78]. This TGF-β induced stem cell-like phenotype may therefore be critical for tumor progression and metastasis because of its effects on tumor cell dissemination and homing, as well as colony-initiating activity. It was therefore postulated that inhibiting TGF-β may reduce the “stem cell-like” compartment of the tumor. This is also important as cancer stem cells are attributed with having enhanced chemotherapeutic drug resistance [82-83].
Dampening of Immune Surveillance and Pro-Tumorigenic Polarization of Myeloid Cells
Lastly, TGF-β can suppress or modulate the immune response. Broadly speaking, many of the TGF-β signaling effects on both adaptive and innate immune cells of the tumor microenvironment result from the ability of this cytokine to polarize innate immune cells towards an alternative differentiation status. Macrophages and neutrophils of the innate immune system are attracted towards TGF-β in the tumor, and driven towards a “type 2” phenotype by this ligand [84-85]. The type 2 macrophage or neutrophil is thought to be a relatively immature state of the cell which is consistent with the role of TGF-β in maintenance of primitive stem-like phenotypes. Regardless of whether the type 2 phenotype represents arrested or alternative differentiation, the outcome is the same, namely a cell that delivers pro-tumorigenic cytokines to the tumor milieu [17]. TGF-β blunts the normal anti-tumor functions of type 1 differentiated T-cells, macrophages and neutrophils, and stimulates the release of pro-tumorigenic cytokines (including IL-11 and yet more TGF-β), from type 2 immune cells [48]. Thus TGF-β signaling within the tumor microenvironment suppresses the fully differentiated anti-tumor “cytotoxic” arm of the immune system. Genetic mouse models of T cell-specific loss of TGF-β signaling (CD4 promoter driven ΔTβRII) showed enhanced tumor eradication due to increased tumor-specific cytotoxic T lymphocyte (CTL) response compared to wild type littermates [86]. In line with these results, inhibition of TGF-β signaling by monoclonal antibodies also led to increased cytotoxic activity of CTLs [87].
Additionally, it has recently been observed that IL-17 expressing, and thus tumor promoting, TH cells (TH17) are elevated within the tumor-infiltrating lymphocyte population (TILs) of melanoma and breast cancers [88]. These effects may be tumor type-specific since other studies have shown a decrease of TH17 cells in ovarian cancer and non-Hodgkin's lymphoma [89-90]. Appearance of tumor-associated TH17 cells is linked to the influence of TGF-β on the fate of CD4+ precursors. These normally differentiate into tumor suppressing Tregs, but in the presence of TGF-β and IL-6 are diverted to differentiate along the TH17 pathway. The transcription factors RORγt and STAT3 are known to be critical for the development of TH17 cells, and TGF-β induced Smad2 binds and synergizes with RORγt to drive the TH17 arm of CD4+ cell differentiation [91].
One mechanism through which TH17 cells may promote tumorigenesis and/or progression is by promoting angiogenesis. IL-17 is a well-established angiogenic cytokine that stimulates migration and cord formation of endothelial cells in vitro and of blood vessel formation in vivo [92]. These observations suggest that the true efficacy of TGF-β inhibitors may be through their ability to reprogram the tumor microenvironment.
Therapeutic Targeting of TGF-β signaling
As a result of the wide variety of effects of TGF-β on tumorigenesis, blockade of TGF-β and its signaling pathway provides multiple therapeutic opportunities (Figure 3). There are many TGF-β signaling antagonist agents under development at both the pre-clinical and clinical stages. The major classes of TGF-β inhibitors addressed in this review include ligand traps [1], antisense oligonucleotides (ASO) [5], small molecule receptor kinase inhibitors [4], and peptide aptamers [6]. Ligand traps serve as a sink for the excess TGF-β produced by tumor cells and fibroblasts during cancer progression, which increases with aggressiveness and tumor stage [28, 34, 93-94]. Ligand traps include anti-ligand neutralizing antibodies and soluble decoy receptor proteins that incorporate the ectodomains from either TβRII or TβRIII/betaglycan protein. Neutralizing antibodies have been raised against individual ligands or may be designed to block all three isomers. ASOs can also be used to reduce the bioavailability of active TGF-β ligands in the local tumor microenvironment by blocking TGF-β synthesis. ASOs are single-stranded polynucleotide molecules, 13-25 nucleotide in length, that hybridize to complementary RNA, inhibiting mRNA function, and preventing protein synthesis through accelerated mRNA degradation by RNase H [95].
Inhibition of TGF-β signaling pathway. The TGF-β signaling pathway is often elevated in human tumors, and thus a clinical target. This has led to the development of a range of anti-TGF-β-signaling drugs for cancer therapy. The four major classes of TGF-β inhibitors include ligand traps (e.g. 1D11 or Fresolimumab), antisense oligonucleotides (ASO) like Trabedersen, small molecule receptor kinase inhibitors such as LY2109761 or LY2157299, and peptide aptamers (e.g. Trx-SARA).
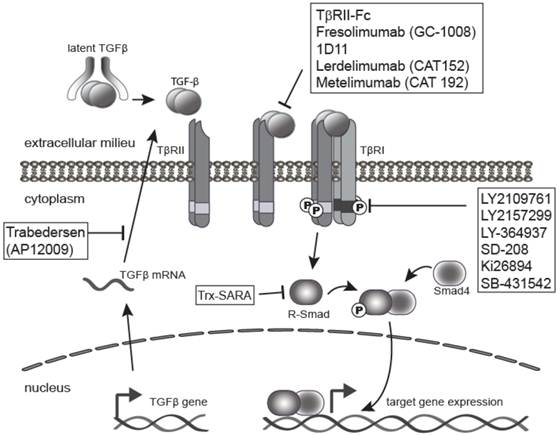
Another therapeutic strategy is to block TβRI activity through the use of small molecule receptor kinase inhibitors that act via ATP-competitive inhibition of the kinase catalytic activity of the receptor. Lastly, targeting intracellular TGF-β signaling molecules, such as Smads, is possible with the use of peptide aptamers, although this is the least explored therapeutic strategy. Aptamers are small peptide molecules containing a target-binding and a scaffolding domain that stabilize and interfere with the function of the target. Aptamers may therefore be designed specifically against Smad2 versus Smad3, and against multimeric transcriptional complexes containing Smads and other transcription factors, transcriptional co-activators, or co-repressors. This approach therefore lends itself to design of more specific targets downstream of the receptor, and thus has the potential for targeting specific subsets of TGF-β responses. This review will describe current pre-clinical and clinical data related to these four major sub-classes of TGF-β antagonists.
Pre-clinical Data
Ligand traps
Pre-clinically, breast cancers that metastasize to the bone have been a focus of anti-TGF-β antibody studies. The bone is the principal reservoir of TGF-β in the body, where it plays an osteogenic function in regulation of both bone mass and bone matrix properties. In cancer, the normal remodeling balance between bone resorption and formation is disrupted. In the case of osteolytic bone metastasis, there is an increase in osteoclastic bone resorption, resulting in excessive secretion of active TGF-β into the bone microenvironment, which in turn triggers a positive feedback loop of TGF-β tumor promoting activity [96]. Anti-ligand antibody therapy has the potential to stop this vicious cycle, by neutralizing the excess pool of TGF-β. Genzyme Corporation (now owned by Sanofi), developed a pan-neutralizing anti-mouse TGF-β monoclonal antibody, 1D11, which binds all three TGF-β isoforms and reduces their biological activity. In the 4T1 immune competent mouse model of breast cancer metastasis to the lung, treatment with 1D11 demonstrated suppression of metastasis. While inhibition of angiogenesis was reported in the primary tumor, the main mechanism of action appeared to be a robust enhancement of the CD8+ T-cell mediated antitumor immune response [97]. Tumor-derived TGF-β polarizes CD8+ lymphocytes to promote tumorigenesis by inhibiting tumor cell apoptosis and supporting tumor vascularization through the induction of IL-17. Nam et al. [87] demonstrated that 1D11 treatment reduced IL-17 levels in the tumor microenvironment and enhanced tumor apoptosis in the 4T1 mouse breast cancer model.
On top of the efficacy of 1D11 in inhibiting tumor burden it has also been shown that treatment with 1D11 can rescue bone loss due to osteolytic bone metastasis in breast cancer models. In vivo treatment with 1D11 reduced osteolytic lesions in the MDA-MB-231 cardiac injection model. One proposed mechanism of TGF-β-supported metastasis is through TGF-β induced expression of Gli2 (GLI family zinc finger 2), a hedgehog signaling molecule. Gli2 regulates the expression of parathyroid hormone related protein (PTHrP) a major osteolytic factor [96]. In vitro, treatment of MDA-MB-231 cells with 1D11, showed reduced expression of both Gli2 and PTHrP, and treatment in vivo significantly lowered the number of osteoclasts per millimeter in mouse long bones bearing tumor metastases, as determined by TRAP staining in drug-dosed versus vehicle control mice. In the same mouse model it was further reported that anti-TGF-β treatment increased bone volume and improved bone architecture [98].
An alternative approach to avert TGF-β signaling is the employment of recombinant Fc-fusion proteins containing the soluble ectodomain of either TβRII (TβRII-Fc) or the type III receptor, betaglycan. The in vivo expression of TβRII-Fc has shown to reduce the incidence of breast tumor metastasis in transgenic mice [99]. Administration of TβRII-Fc in the MMTV-PMT transgenic mouse model also demonstrated an increase in apoptosis in primary tumors, as well as a reduction in metastasis [93]. Additionally, treatment of MDA-MB-231 human breast cancer cells with the soluble betaglycan ectodomain in a xenograft mouse model, also demonstrated a block to both angiogenesis and metastasis [100]. More recently, expression of the soluble type II receptor TβRII-Fc has been coupled to an oncolytic adenovirus (Ad.sTβRII-Fc) and infection of MDA-MB-231 cells with Ad.sTβRII-Fc in vitro, inhibited TGF-β signaling. When administered directly into subcutaneous MDA-MB-231 tumors in nude mice, this resulted in reduction of tumor growth [101]. Furthermore, Hu et al. [102] evaluated the systemic administration of Ad.sTβRII-Fc on breast cancer bone metastases in a nude mouse model. Their study demonstrated that intravenous delivery of Ad.sTβRII-Fc resulted in viral replication and expression of TβRII-Fc in skeletal tumors as well as a signification reduction of primary tumor growth and osteolytic bone destruction.
Antisense oligonucleotides (ASOs)
The reduction of TGF-β synthesis by ASOs is another therapeutic strategy to reduce excess TGF-β levels within the tumor microenvironment. ASOs are designed to hybridize to their complementary RNA sequence and accelerate mRNA degradation. However, ASOs have some limitations which need to be taken into account when using them. These limitations include unpredictable RNA binding affinity, possible non-specific/off target effects and the challenge of delivery of relatively large molecules into the target cell. In the TGF-β1-driven metastasis mouse model of PyMT-induced mammary cancer, TGF-β1 ASOs partially reduced tumor metastasis. A partial rather than full reduction in metastasis was attributed to further autocrine TGF-β production, synthesized in cancer and/or stroma cells [94]. These results highlight a possible limitation of the ASO strategy.
Among the TGF-β ligands, TGF-β2 has a unique role in both glioblastoma and pancreatic cancer, since its expression correlates with poor clinical outcome. In fact, it has been reported that TGF-β2, but not TGF-β1, induces a pronounced immunosuppressive phenotype in these tumor types through the activation of Foxp3 [103]. Therefore, ASOs to TGF-β2 have been investigated as an approach for treatment of pancreatic cancer. AP12009 (Trabedersen) was developed by Antisense Pharma as an ASO specifically targeting human TGF-β2 RNA, and previously shown to inhibit TGF-β2 expression, decrease cellular proliferation and migration of glioma cells in vitro [104]. This drug produced promising results for treatment of glioblastoma in the clinic (see below). In an orthotopic mouse model of human metastatic pancreatic cancer, using the L3.6pl human pancreatic cancer cell line implanted into the pancreases of BALB/Cnu/nu mice, treatment with AP 12009 demonstrated a significant reduction in tumor growth, vascularization, and metastasis [105].
Small molecule receptor kinase inhibitors
In spite of the fact that ligand traps and ASOs limit the bioavailability of the active TGF-β ligands, they fail to directly block receptor signaling. Small molecule inhibitors of the TGF-β receptor kinases have an advantage here, although reduced drug specificity of kinase inhibitors compared to ASOs or monoclonoal antibodies may be a challenge. Current preclinical data suggest that the majority of small molecule inhibitors of TβRI/ALK5 also inhibit the related activin and nodal receptors, ACVR1B/ALK4 and ACVR1C/ALK7, but with reduced affinity [106-108]. The targeting of receptor kinases by small molecules has been an abundant area of experimental drug development in the last few years precisely because of ease of drug production and the practicality of drug delivery by the oral route.
GlaxoSmithKline has developed a small molecule inhibitor of TβRI, SB-431542, which has now been widely used in basic research studies. For example, in vitro it has been shown to block TGF-β induced transcription of fibronectin and collagen in renal epithelial carcinoma cells, as well as inhibit the proliferation of glioma and osteosarcoma cells [109]. However, this drug is pharmacokinetically unstable, a major hurdle to in vivo studies. Ki26894, also a TβRI/ALK5 kinase inhibitor, has been shown to block TGF-β signaling, invasion, and motility of the bone metastatic breast cancer cell line, MDA-MB-231-D. Ki26894 treatment of nude mice one day prior to intra-cardiac inoculation of MDA-MB-231-D cells resulted in decreased metastasis and prolonged mouse survival [110]. The use of TβRI/ALK5 inhibitor LY364937, in a MDA-MB-435-F-L orthotopic xenograft model into nude mice, also demonstrated reduction in the formation of early bone and lung metastases after treatment [111]. Similar findings with the TβRI inhibitor SD-208 (Scios Inc.), showed that tumor cell pre-treatment with drug before tumor inoculation into a mouse model of human melanoma, prevented the development of osteolytic bone metastases compared with vehicle. Additionally, mice with established bone metastases showed a significant reduction in size of osteolytic lesions after four weeks of SD-208 treatment compared to vehicle-treated mice [112].
TGF-β secretion by tumors suppresses the antitumor immune response. Dendritic cells (DCs) play an important role in this response through antigen presentation and activation of cytotoxic T lymphocytes (CTLs). In this context, Tanaka et al. [113] reported a microenvironment-mediated anti-tumor effect of SB-431542 treatment in vitro through induction of DC maturation. Using an orthotopic model of colon carcinoma it was observed that intra-peritoneal administration of SB-431542 significantly induced cancer-specific CTL activities. Furthermore, treatment with SB-431542 or another TβRI inhibitor, Ki26894 in an in vitro model of Multiple Myeloma (MMy) also demonstrated that TGF-β signaling affected tumor stromal cell differentiation. Blockage of TGF-β signaling with either of these inhibitors released stromal cells from MMy-induced differentiation arrest, resulting in terminal differentiation of osteoblasts (OBs) from mesenchymal stem cells. The OBs were then in turn able to inhibit MMy cell growth in the bone marrow and prevented bone destruction. These results suggest that TGF-β suppression of OB differentiation not only accelerates bone loss but also creates a tumor niche to enhance tumor growth and survival [114].
The objective of TGF-β inhibition is to target its tumor promoting properties, both cell autonomous and microenvironmental, while avoiding inhibition of its tumor suppression arm. This goal begs the following question: How long should TGF-β signaling be suppressed and what are the long term effects of this suppression? LY2109761, a dual inhibitor of TβRI/II has shown promising effects on inhibiting the formation of metastases in several short term mouse tumor models, including breast [115], colon [116], and pancreatic [117] cancer. However, in a long term drug dosing study in a mouse model of chemically-induced skin carcinogenesis, a new phenomenon for this drug class has emerged. We demonstrated that despite the ability of LY2109761 to inhibit EMT in vitro, and despite its short-term effects in suppressing components of SpSC EMT in vivo, sustained pharmacologic inhibition of TGF-β signaling led to biochemical resistance of tumor cells to the drug. This led to undesirable and oppositional effects in driving EMT, with the potential for promoting further carcinoma progression. The tumors acquired constitutively elevated levels of P-Smad2, despite the presence of the drug, that drove expression of genes characteristic of invasion, inflammation, and ”stemness” [76].
It is in fact a dirty little secret of chemotherapy that most cancers do acquire resistance to cytotoxic drugs. Clinically, many small molecule inhibitors such as EGFR (erlotinib), ABL/PDGFR/KIT (imatinib), and VEGFR/RAF/PDGFR (sorafenib), have produced impressive initial clinical responses, including disease remission in a subset of patients. However, this outcome is habitually followed by eventual disease progression [118-119]. This inconvenient fact emphasizes the need to understand both the acute and chronic effects of signaling pathway suppression. In the case of Ly2109761 it would appear that the tumor cells acquired biochemical resistance to a non-cytotoxic drug. Clearly, for this to happen there must be some selective advantage to the tumor in driving resistance. Elucidating the mechanisms of Ly2109761 drug-resistance, whether it is acquired or due to outgrowth of a pre-existing cell population, should lead to an understanding of alternative pathways that might drive the cancer cell. Various hypotheses exist based on studies with inhibitors of Her2 and Raf kinases in various tumor types. These include negative feedback loops, the re-wiring of the signaling pathway via alternative downstream effectors, and mutational activation of the drug target, namely the kinase receptor [119-122]. The paradoxical activation of the therapeutic target suggests that long term suppression of a signaling pathway may not be efficacious when used as monotherapy.
Peptide Aptamers
Another possible strategy to block TGF-β signaling for cancer is to interfere with the TGF-β signaling molecules, the Smads, that act downstream of the type I receptor. This may be achieved by using peptide aptamers. Peptide aptamers, as the name suggests, are small peptides that can bind specifically to certain proteins. They have two domains, a target-binding domain and a scaffolding domain that stabilizes the resultant molecular complex. A few peptide aptamers have been designed that bind to Smad2 and Smad3, consequently disrupting their interactions with Smad4. The Trx-SARA aptamer is an example. Treatment with Trx-SARA has been reported to reduce the levels of Smad2/3 in complex with Smad4 after TGF-β stimulation. Furthermore, Trx-SARA treatment has been shown to inhibit TGF-β-induced EMT in NMuMG murine mammary epithelial cells in vitro [123].
Clinical Data
Ligand traps
Fully humanized pan-TGF-β monoclonal neutralizing antibodies were developed by Genzyme for use in patients, including Lerdelimumab [124-125], Metelimumab [126] and GC-1008 (Fresolimumab) [127]. Here we will focus on publically available results from the GC-1008 trials. Genzyme sponsored a two part clinical trial of GC-1008 in patients with advanced renal cell carcinoma (RCC) and malignant melanoma (MM). Preliminary data from Part 1 of this study was presented at the 2008 ASCO Annual Meeting [127]. Part 1 was a multi-center Phase I/II safety and efficiency trial of GC-1008 in a cohort of patients, mainly with advanced MM (n=22) but also including a single patient with RCC (n=1), all of whom had at least one prior failed therapy [127]. The patients were treated by intravenous administration of GC-1008 at one of six dosages (0.1, 0.3, 1, 3, 10 or 15 mg/kg). If there were no dose limiting toxicities (DLTs) within the first 28 days of first dosing, three further doses were administered at two week intervals. No DLTs were observed and thus 15 mg/kg was determined to be the highest safe dose tested. Standard Response Evaluation Criteria in Solid Tumors (RECIST) was used to determine clinical response of the tumors. Patients achieving stable disease (SD) or partial responses (PR) were offered extended treatment with the drug for four further doses at two weeks intervals. As reported in 2008, five patients had achieved SD or better and therefore received extended treatment. Furthermore, of those five responders, three patients had mixed responses, including shrinkage of metastases in liver and at other sites. One MM patient achieved a PR with a greater than 75% reduction in the target lesion. The most frequently reported drug-related side effects were skin rashes/lesions including eruptive non-malignant keratoacanthomas (KA) and squamous cell carcinoma (SCC) on sun-damaged skin, as well as gingival bleeding and fatigue.
The objective of the second part of the trial was to determine the frequency of GC-1008 induced adverse skin events in patients with MM. Patients in this part of the study received intravenous dosing of GC-1008 at 10 mg/kg or 15 mg/kg on study days 0, 28, 42 and 56 [128]. Biopsies of non-melanoma skin lesions from MM patients who had received multiple doses of the antibody in Part 1 of the trial were also screened by histopathology for KA versus SCC. KA and well-differentiated SCC are difficult to distinguish from each other, but the current interpretation of the data is that GC-1008-associated skin lesions were predominantly non-malignant KA which often spontaneously resolved or improved after discontinuation of the drug. The severity of skin lesion development appeared to be associated with the higher dose and longer duration of GC-1008 exposure in these oncology trials, since neither KA nor SCC were observed in single dose Phase I trials for idiopathic pulmonary fibrosis or focal segmental glomerulo-sclerosis [128].
Antisense oligonucleotides (ASOs)
AP12009 (Trabedersen) is an ASO that specifically inhibits TGF-β2 expression. It was initially tested in a Phase I/II study in patients with high-grade glioma where AP 12009 treatment showed a significant survival benefit over standard chemotherapy [104]. In more recent studies, the Antisense Pharma group who developed this drug moved into the treatment of advanced pancreatic carcinoma (PanCa, stage III/IV, n=23), malignant melanoma (MM, stage III/IV, n=5), and colorectal carcinoma (stage III/IV, n=5), and undertook a dose escalation trial. Patients received i.v. Trabedersen monotherapy as a second to fourth line therapy, with escalating doses in one of two treatment schedules (schedule 1: 7 days on, 7 days off; schedule 2: 4 days on, 10 days off; both up to 10 cycles). AP12009 was well-tolerated, with the only adverse event being transient thrombocytopenia. Phase II selected a dose of 140 mg/m2/d to treat PanCa and MM according to schedule 2 dosing. Median overall survival of the PanCa patients (n=9) had not yet been reached as of December 2010 (12.9 months in Dec. 2010). One of the nine PanCa patients had a complete response of liver metastasis while other promising efficacy data included 1 metastatic DTIC-(5-(3,3-dimethyl-1-triazeno)-imidazole-4-carboxamide) resistant melanoma patient who was still alive 19.7 months after the start of treatment (status Dec 2010). Additionally, three other patients with stage IV melanoma, were treated with Trabedersen as the third or fourth line of therapy, and survived for 11.4, 13.8, and 18.6 further months [129]. Earlier clinical trials of Trabedersen in glioblastoma suggested that the effect of this drug might be through alterations in host immunity, since intra-tumoral injection of Trabedersen into a patient with multiple brains tumors not only led to regression of the target tumor, but to reduction of tumors in the contralateral brain hemisphere [130].
Receptor kinase inhibitors
LY2157299 is a small molecule inhibitor which is selective for the kinase domain of the type 1 TGF-β receptor. LY2157299 is currently in a Phase I escalation study in patients with metastatic malignancies. The goals of this study are to determine the safety and pharmacokinetics of LY2157299. 28 patients with Grade IV glioma have so far been treated by 2008 [131]. In a previous First Human Dose (FHD) study, patients with advanced/metastatic malignancies were divided into cohorts of 3 patients each and treated with 40 mg or 80 mg LY2157299 daily. These doses were well tolerated and pharmacokinetic profiles were consistent with the pre-clinical based pharmacokinetic/pharmacodynamic modeling [131]. To determine a potential maximum tolerated dose, patients were treated in a new FHD study at 160, 240 and 300 mg/day, 14 days on 14 days off, with 6 - 12 patients at each dosage level. Standard evaluation criteria, cardiac safety and tumor responses were assessed. Two drug-related dose limiting toxicities included pulmonary embolism and thrombocytopenia in the second cycle of drug dosing, but no medically significant cardiotoxicities were observed. At the lowest dose of 160 mg/day, three patients were treated for >20 cycles. Currently the study is reporting 3 confirmed PR at 160 mg/day and 1 unconfirmed PR at 300 mg/day. In some patients, down regulation of P-Smad2 levels, as a biomarker, in peripheral blood mononuclear cells indicated that drug target inhibition was effective at the 160 mg/day dose level, and at this dose an anti-tumor response was seen in at least 3 patients with durable responses beyond 1 year. The current conclusions of the study are that the 14 days on/ 14 days off treatment is safe at all dose levels and a maximum tolerated dose was not observed. Given the overall safety profile and likely anti-tumor effect, LY2157299 is being investigated in Phase II studies [132].
Developing Concepts and Future Prospects
While clearly TGF-β signaling inhibition results in a significant reduction in metastasis in mouse models, clinically the effects have been less robust then hoped for. Additionally, it has been more difficult to demonstrate inhibition of primary tumors. These facts suggest that combinatorial therapy may increase the efficacy of TGF-β inhibitors in a clinical setting. Ionizing radiotherapy is known to induce TGF-β in both the tumor and tumor microenvironment. This increase in TGF-β results in an enhanced DNA damage response [46]. Therefore, treatment with a TGF-β inhibitor would sensitize the tumor to the radiation. In the subcutanecous 4T1 breast cancer mouse model, animals treated with the pan TGF-β neutralizing antibody 1D11 24 hours prior to radiotherapy showed marked reduction in primary tumor growth compared to the single agent treatment group [133]. Additionally, the TβRI/II kinase inhibitor LY2109761 in combination with temozolomine (clinical standard) and radiotherapy in a glioblastoma model delayed tumor growth compared to controls [134]. The TβRI kinase inhibitor LY2157299 in combination with temozolomine and radiotherapy is in a Phase I trial in glioma patients (clinical trial ID NCT01220271). Additionally, a clinical trial evaluating the effects of the neutralizing TGF-β antibody, GC-1008, in combination with radiotherapy in metastatic breast cancer is currently recruiting participants (clinical trial ID NCT01401062).
Furthermore, it has been previously shown through knockdown of the TGF-β signaling pathway that loss of TGF-β signaling can enhance the therapeutic efficacy of various cytotoxic agents such as rapamycin [135] and doxorubicin [136]. In the 4T1 mouse model of triple negative breast cancer the combination of ixabepilone (a microtubule stabilizer), capecitabine (a pyrimidine analogue) and the TGF-β pan neutralizing antibody 1D11 demonstrated reduction in primary tumor growth and metastasis [137]. The combination of ixabepilone and capecitabine has shown some effectiveness in breast cancer patients which have failed anthracyclin and taxane therapy [138]. A clinical trial evaluating the effects the TβRI kinase inhibitor LY2157299 in combination with gemcitabine (nucleoside analog) in metastatic pancreatic cancer is currently recruiting participants (clinical trial ID NCT01373164).
Conclusions
TGF-β has a predominant role in a variety of cancer types during progression and metastasis. Increased quantities of TGF-β in the tumor and tumor microenvironment promote tumorigenesis by inducing EMT, re-programming of immune surveillance, and/or indirect facilitation of tumor cell proliferation, thereby making it a very druggable target. To date, there are three major therapeutic designs targeting TGF-β in clinical trials: TGF-β antibodies, antisense oligonucleotides, and receptor kinase inhibitors. Each of these strategies has different pharmacokinetic/pharmacodynamic properties and mechanisms of action. These differences have distinct limitation in respect of delivery, specificity and toxicity. However, all strategies are faced with the fact that TGF-β signaling is involved in many normal physiological functions. As highlighted by the LY2109761 DMBA/TPA mouse study long term suppression of this pathway may lead to harmful off-target effects. With this concern in mind, TGF-β inhibitors may be a therapeutic benefit within a combinatorial therapy setting for oncology. This may be especially true for multiple myeloma and breast cancer, where attenuation of TGF-β signaling may not only reduce metastatic spread but also help maintain bone mass in patients with osteolytic metastases.
Acknowledgements
Dedicated to Jasmine Ahluwahlia PhD. Research in the authors' lab was funded by NIH grants R01-CA116019, R01-GM060514, P50-CA58207, R21-CA164772 to R.J.A., and a gift from the Bouque Estate to R.J.A.. E.C.C. was funded by an institutional research service award from the National Cancer Institute (T32 CA108462).
Competing Interests
The authors have declared that no competing interest exists.
References
1. de Larco JE, Todaro GJ. Growth factors from murine sarcoma virus-transformed cells. Proc Natl Acad Sci U S A. 1978;75:4001-5
2. Todaro GJ, De Larco JE. Growth factors produced by sarcoma virus-transformed cells. Cancer Res. 1978;38:4147-54
3. Roberts AB, Lamb LC, Newton DL, Sporn MB, De Larco JE, Todaro GJ. Transforming growth factors: isolation of polypeptides from virally and chemically transformed cells by acid/ethanol extraction. Proc Natl Acad Sci U S A. 1980;77:3494-8
4. Moses HL, Branum EL, Proper JA, Robinson RA. Transforming growth factor production by chemically transformed cells. Cancer Res. 1981;41:2842-8
5. Roberts AB, Anzano MA, Lamb LC, Smith JM, Sporn MB. New class of transforming growth factors potentiated by epidermal growth factor: isolation from non-neoplastic tissues. Proc Natl Acad Sci U S A. 1981;78:5339-43
6. Seyedin SM, Thompson AY, Bentz H, Rosen DM, McPherson JM, Conti A. et al. Cartilage-inducing factor-A. Apparent identity to transforming growth factor-beta. J Biol Chem. 1986;261:5693-5
7. Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169-78
8. Kingsley DM. The TGF-beta superfamily: new members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 1994;8:133-46
9. Wu MY, Hill CS. Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev Cell. 2009;16:329-43
10. Kulkarni AB, Thyagarajan T, Letterio JJ. Function of cytokines within the TGF-beta superfamily as determined from transgenic and gene knockout studies in mice. Curr Mol Med. 2002;2:303-27
11. Annes JP, Rifkin DB, Munger JS. The integrin alphaVbeta6 binds and activates latent TGFbeta3. FEBS Lett. 2002;511:65-8
12. Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J. et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319-28
13. De Crescenzo G, Hinck C, Shu Z, Zúñiga J, Yang J, Tang Y. et al. Three key residues underlie the differential affinity of the TGFbeta isoforms for the TGFbeta type II receptor. J Mol Biol. 2006;359(2):508
14. Qian SW, Burmester JK, Tsang ML, Weatherbee JA, Hinck AP, Ohlsen DJ. et al. Binding affinity of transforming growth factor-beta for its type II receptor is determined by the C-terminal region of the molecule. J Biol Chem. 1996;271:30656-62
15. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685-700
16. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577-84
17. Connolly EC, Akhurst RJ. The Complexities of TGF-beta Action During Mammary and Squamous Cell Carcinogenesis. Curr Pharm Biotechnol. 2011;12(12):2138-49
18. Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41-58
19. Parsons R, Myeroff LL, Liu B, Willson JK, Markowitz SD, Kinzler KW. et al. Microsatellite instability and mutations of the transforming growth factor beta type II receptor gene in colorectal cancer. Cancer Res. 1995;55:5548-50
20. Lucke CD, Philpott A, Metcalfe JC, Thompson AM, Hughes-Davies L, Kemp PR. et al. Inhibiting mutations in the transforming growth factor beta type 2 receptor in recurrent human breast cancer. Cancer Res. 2001;61:482-5
21. Jakob J, Nagase S, Gazdar A, Chien M, Morozova I, Russo JJ. et al. Two somatic biallelic lesions within and near SMAD4 in a human breast cancer cell line. Genes Chromosomes Cancer. 2005;42:372-83
22. Hoot KE, Lighthall J, Han G, Lu SL, Li A, Ju W. et al. Keratinocyte-specific Smad2 ablation results in increased epithelial-mesenchymal transition during skin cancer formation and progression. J Clin Invest. 2008;118:2722-32
23. Cui W, Fowlis DJ, Bryson S, Duffie E, Ireland H, Balmain A. et al. TGFbeta1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell. 1996;86:531-42
24. Mao JH, Saunier EF, de Koning JP, McKinnon MM, Higgins MN, Nicklas K. et al. Genetic variants of Tgfb1 act as context-dependent modifiers of mouse skin tumor susceptibility. Proc Natl Acad Sci U S A. 2006;103:8125-30
25. Muraoka RS, Koh Y, Roebuck LR, Sanders ME, Brantley-Sieders D, Gorska AE. et al. Increased malignancy of Neu-induced mammary tumors overexpressing active transforming growth factor beta1. Mol Cell Biol. 2003;23:8691-703
26. Forrester E, Chytil A, Bierie B, Aakre M, Gorska AE, Sharif-Afshar AR. et al. Effect of conditional knockout of the type II TGF-beta receptor gene in mammary epithelia on mammary gland development and polyomavirus middle T antigen induced tumor formation and metastasis. Cancer Res. 2005;65:2296-302
27. Pierce DF Jr, Gorska AE, Chytil A, Meise KS, Page DL, Coffey RJ Jr. et al. Mammary tumor suppression by transforming growth factor beta 1 transgene expression. Proc Natl Acad Sci U S A. 1995;92:4254-8
28. Massague J. TGFbeta in Cancer. Cell. 2008;134:215-30
29. Han G, Lu SL, Li AG, He W, Corless CL, Kulesz-Martin M. et al. Distinct mechanisms of TGF-beta1-mediated epithelial-to-mesenchymal transition and metastasis during skin carcinogenesis. J Clin Invest. 2005;115:1714-23
30. Chen CR, Kang Y, Siegel PM, Massague J. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell. 2002;110:19-32
31. Gomis RR, Alarcon C, Nadal C, Van Poznak C, Massague J. C/EBPbeta at the core of the TGFbeta cytostatic response and its evasion in metastatic breast cancer cells. Cancer Cell. 2006;10:203-14
32. Seoane J, Pouponnot C, Staller P, Schader M, Eilers M, Massague J. TGFbeta influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat Cell Biol. 2001;3:400-8
33. Pardali K, Moustakas A. Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta. 2007;1775:21-62
34. Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807-21
35. Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N. et al. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol. 2008;10:1199-207
36. Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol Cell. 2008;31:918-24
37. Senturk S, Mumcuoglu M, Gursoy-Yuzugullu O, Cingoz B, Akcali KC, Ozturk M. Transforming growth factor-beta induces senescence in hepatocellular carcinoma cells and inhibits tumor growth. Hepatology. 2010;52:966-74
38. Shukla A, Ho Y, Liu X, Ryscavage A, Glick AB. Cripto-1 alters keratinocyte differentiation via blockade of transforming growth factor-beta1 signaling: role in skin carcinogenesis. Mol Cancer Res. 2008;6:509-16
39. Tremain R, Marko M, Kinnimulki V, Ueno H, Bottinger E, Glick A. Defects in TGF-beta signaling overcome senescence of mouse keratinocytes expressing v-Ha-ras. Oncogene. 2000;19:1698-709
40. Vijayachandra K, Lee J, Glick AB. Smad3 regulates senescence and malignant conversion in a mouse multistage skin carcinogenesis model. Cancer Res. 2003;63:3447-52
41. Boulanger CA, Smith GH. Reducing mammary cancer risk through premature stem cell senescence. Oncogene. 2001;20:2264-72
42. Boulanger CA, Wagner KU, Smith GH. Parity-induced mouse mammary epithelial cells are pluripotent, self-renewing and sensitive to TGF-beta1 expression. Oncogene. 2005;24:552-60
43. Wagner KU, Boulanger CA, Henry MD, Sgagias M, Hennighausen L, Smith GH. An adjunct mammary epithelial cell population in parous females: its role in functional adaptation and tissue renewal. Development. 2002;129:1377-86
44. Glick AB, Weinberg WC, Wu IH, Quan W, Yuspa SH. Transforming growth factor beta 1 suppresses genomic instability independent of a G1 arrest, p53, and Rb. Cancer Res. 1996;56:3645-50
45. Ewan KB, Henshall-Powell RL, Ravani SA, Pajares MJ, Arteaga C, Warters R. et al. Transforming growth factor-beta1 mediates cellular response to DNA damage in situ. Cancer Res. 2002;62:5627-31
46. Kirshner J, Jobling MF, Pajares MJ, Ravani SA, Glick AB, Lavin MJ. et al. Inhibition of transforming growth factor-beta1 signaling attenuates ataxia telangiectasia mutated activity in response to genotoxic stress. Cancer Res. 2006;66:10861-9
47. Derynck R, Akhurst RJ. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nat Cell Biol. 2007;9:1000-4
48. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010;10:554-67
49. Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117-29
50. Wakefield LM, Roberts AB. TGF-beta signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev. 2002;12:22-9
51. Lu SL, Reh D, Li AG, Woods J, Corless CL, Kulesz-Martin M. et al. Overexpression of transforming growth factor beta1 in head and neck epithelia results in inflammation, angiogenesis, and epithelial hyperproliferation. Cancer Res. 2004;64:4405-10
52. Weeks BH, He W, Olson KL, Wang XJ. Inducible expression of transforming growth factor beta1 in papillomas causes rapid metastasis. Cancer Res. 2001;61:7435-43
53. Derynck R, Goeddel DV, Ullrich A, Gutterman JU, Williams RD, Bringman TS. et al. Synthesis of messenger RNAs for transforming growth factors alpha and beta and the epidermal growth factor receptor by human tumors. Cancer Res. 1987;47:707-12
54. Ivanovic V, Todorovic-Rakovic N, Demajo M, Neskovic-Konstantinovic Z, Subota V, Ivanisevic-Milovanovic O. et al. Elevated plasma levels of transforming growth factor-beta 1 (TGF-beta 1) in patients with advanced breast cancer: association with disease progression. Eur J Cancer. 2003;39:454-61
55. Naef M, Ishiwata T, Friess H, Buchler MW, Gold LI, Korc M. Differential localization of transforming growth factor-beta isoforms in human gastric mucosa and overexpression in gastric carcinoma. Int J Cancer. 1997;71:131-7
56. Nakamura M, Katano M, Kuwahara A, Fujimoto K, Miyazaki K, Morisaki T. et al. Transforming growth factor beta1 (TGF-beta1) is a preoperative prognostic indicator in advanced gastric carcinoma. Br J Cancer. 1998;78:1373-8
57. Oft M, Akhurst RJ, Balmain A. Metastasis is driven by sequential elevation of H-ras and Smad2 levels. Nat Cell Biol. 2002;4:487-94
58. Saito H, Tsujitani S, Oka S, Kondo A, Ikeguchi M, Maeta M. et al. An elevated serum level of transforming growth factor-beta 1 (TGF-beta 1) significantly correlated with lymph node metastasis and poor prognosis in patients with gastric carcinoma. Anticancer Res. 2000;20:4489-93
59. Padua D, Massague J. Roles of TGFbeta in metastasis. Cell Res. 2009;19:89-102
60. Bierie B, Chung CH, Parker JS, Stover DG, Cheng N, Chytil A. et al. Abrogation of TGF-beta signaling enhances chemokine production and correlates with prognosis in human breast cancer. J Clin Invest. 2009;119:1571-82
61. Lindley LE, Briegel KJ. Molecular characterization of TGFbeta-induced epithelial-mesenchymal transition in normal finite lifespan human mammary epithelial cells. Biochem Biophys Res Commun. 2010;399:659-64
62. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871-90
63. Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28:15-33
64. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265-73
65. Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. 1994;127:2021-36
66. Piek E, Moustakas A, Kurisaki A, Heldin CH, ten Dijke P. TGF-(beta) type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J Cell Sci. 1999;112( Pt 24):4557-68
67. Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. Embo J. 2004;23:1155-65
68. Akhurst RJ. TGFb signaling in epithelial/mesenchymal transition and invasion/metastasis. In: (ed.) Derynck R, Miyazono K. TGFbeta. US: CSHL. 2006
69. Oft M, Peli J, Rudaz C, Schwarz H, Beug H, Reichmann E. TGF-beta1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 1996;10:2462-77
70. Portella G, Cumming SA, Liddell J, Cui W, Ireland H, Akhurst RJ. et al. Transforming growth factor beta is essential for spindle cell conversion of mouse skin carcinoma in vivo: implications for tumor invasion. Cell Growth Differ. 1998;9:393-404
71. Giampieri S, Pinner S, Sahai E. Intravital imaging illuminates transforming growth factor beta signaling switches during metastasis. Cancer Res. 2010;70:3435-9
72. Cheng N, Chytil A, Shyr Y, Joly A, Moses HL. Transforming growth factor-beta signaling-deficient fibroblasts enhance hepatocyte growth factor signaling in mammary carcinoma cells to promote scattering and invasion. Mol Cancer Res. 2008;6:1521-33
73. Bhowmick NA, Chytil A, Plieth D, Govska AE, Dumont N, Shappell S. et al. TGFbeta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelial. Science. 2004;303:848-51
74. Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M. et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23-35
75. Lacher MD, Tiirikainen MI, Saunier EF, Christian C, Anders M, Oft M. et al. Transforming growth factor-beta receptor inhibition enhances adenoviral infectability of carcinoma cells via up-regulation of Coxsackie and Adenovirus Receptor in conjunction with reversal of epithelial-mesenchymal transition. Cancer Res. 2006;66:1648-57
76. Connolly EC, Saunier EF, Quigley D, Luu MT, de Sapio A, Hann B. et al. Outgrowth of drug-resistant carcinomas expressing markers of tumor aggression after long term TβRI/II kinase inhibition with LY2109761. Cancer Research. 2011;71:1-11
77. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704-15
78. Battula VL, Evans KW, Hollier BG, Shi Y, Marini FC, Ayyanan A. et al. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells. 2010;28:1435-45
79. Bittira B, Shum-Tim D, Al-Khaldi A, Chiu RC. Mobilization and homing of bone marrow stromal cells in myocardial infarction. Eur J Cardiothorac Surg. 2003;24:393-8
80. Herdrich BJ, Lind RC, Liechty KW. Multipotent adult progenitor cells: their role in wound healing and the treatment of dermal wounds. Cytotherapy. 2008;10:543-50
81. Spees JL, Whitney MJ, Sullivan DE, Lasky JA, Laboy M, Ylostalo J. et al. Bone marrow progenitor cells contribute to repair and remodeling of the lung and heart in a rat model of progressive pulmonary hypertension. FASEB J. 2008;22:1226-36
82. Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10:415-24
83. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741-51
84. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L. et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN. Cancer Cell. 2009;16:183-94
85. Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22:231-7
86. Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med. 2001;7:1118-22
87. Nam JS, Terabe M, Kang MJ, Chae H, Voong N, Yang YA. et al. Transforming growth factor beta subverts the immune system into directly promoting tumor growth through interleukin-17. Cancer Res. 2008;68:3915-23
88. Su X, Ye J, Hsueh EC, Zhang Y, Hoft DF, Peng G. Tumor microenvironments direct the recruitment and expansion of human Th17 cells. J Immunol. 2010;184:1630-41
89. Horlock C, Stott B, Dyson PJ, Morishita M, Coombes RC, Savage P. et al. The effects of trastuzumab on the CD4+CD25+FoxP3+ and CD4+IL17A+ T-cell axis in patients with breast cancer. Br J Cancer. 2009;100:1061-7
90. Yang ZZ, Novak AJ, Ziesmer SC, Witzig TE, Ansell SM. Malignant B cells skew the balance of regulatory T cells and TH17 cells in B-cell non-Hodgkin's lymphoma. Cancer Res. 2009;69:5522-30
91. Martinez GJ, Zhang Z, Reynolds JM, Tanaka S, Chung Y, Liu T. et al. Smad2 positively regulates the generation of Th17 cells. J Biol Chem. 2010;285:29039-43
92. Takahashi H, Numasaki M, Lotze MT, Sasaki H. Interleukin-17 enhances bFGF-, HGF- and VEGF-induced growth of vascular endothelial cells. Immunol Lett. 2005;98:189-93
93. Muraoka RS, Dumont N, Ritter CA, Dugger TC, Brantley DM, Chen J. et al. Blockade of TGF-beta inhibits mammary tumor cell viability, migration, and metastases. J Clin Invest. 2002;109:1551-9
94. Muraoka-Cook RS, Kurokawa H, Koh Y, Forbes JT, Roebuck LR, Barcellos-Hoff MH. et al. Conditional overexpression of active transforming growth factor beta1 in vivo accelerates metastases of transgenic mammary tumors. Cancer Res. 2004;64:9002-11
95. Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259-93
96. Drabsch Y, ten Dijke P. TGF-beta signaling in breast cancer cell invasion and bone metastasis. J Mammary Gland Biol Neoplasia. 2011;16:97-108
97. Nam JS, Terabe M, Mamura M, Kang MJ, Chae H, Stuelten C. et al. An anti-transforming growth factor beta antibody suppresses metastasis via cooperative effects on multiple cell compartments. Cancer Res. 2008;68:3835-43
98. Biswas S, Nyman JS, Alvarez J, Chakrabarti A, Ayres A, Sterling J. et al. Anti-transforming growth factor ss antibody treatment rescues bone loss and prevents breast cancer metastasis to bone. PLoS One. 2011;6:e27090
99. Yang YA, Dukhanina O, Tang B, Mamura M, Letterio JJ, MacGregor J. et al. Lifetime exposure to a soluble TGF-beta antagonist protects mice against metastasis without adverse side effects. J Clin Invest. 2002;109:1607-15
100. Bandyopadhyay A, Lopez-Casillas F, Malik SN, Montiel JL, Mendoza V, Yang J. et al. Antitumor activity of a recombinant soluble betaglycan in human breast cancer xenograft. Cancer Res. 2002;62:4690-5
101. Seth P, Wang ZG, Pister A, Zafar MB, Kim S, Guise T. et al. Development of oncolytic adenovirus armed with a fusion of soluble transforming growth factor-beta receptor II and human immunoglobulin Fc for breast cancer therapy. Hum Gene Ther. 2006;17:1152-60
102. Hu Z, Zhang Z, Guise T, Seth P. Systemic delivery of an oncolytic adenovirus expressing soluble transforming growth factor-beta receptor II-Fc fusion protein can inhibit breast cancer bone metastasis in a mouse model. Hum Gene Ther. 2010;21:1623-9
103. Hinz S, Pagerols-Raluy L, Oberg HH, Ammerpohl O, Grussel S, Sipos B. et al. Foxp3 expression in pancreatic carcinoma cells as a novel mechanism of immune evasion in cancer. Cancer Res. 2007;67:8344-50
104. Hau P, Jachimczak P, Schlingensiepen R, Schulmeyer F, Jauch T, Steinbrecher A. et al. Inhibition of TGF-beta2 with AP 12009 in recurrent malignant gliomas: from preclinical to phase I/II studies. Oligonucleotides. 2007;17:201-12
105. Schlingensiepen KH, Jaschinski F, Lang SA, Moser C, Geissler EK, Schlitt HJ. et al. Transforming growth factor-beta 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci. 2011;102:1193-200
106. Fu K, Corbley MJ, Sun L, Friedman JE, Shan F, Papadatos JL. et al. SM16, an orally active TGF-beta type I receptor inhibitor prevents myofibroblast induction and vascular fibrosis in the rat carotid injury model. Arterioscler Thromb Vasc Biol. 2008;28:665-71
107. Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD. et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65-74
108. Tojo M, Hamashima Y, Hanyu A, Kajimoto T, Saitoh M, Miyazono K. et al. The ALK-5 inhibitor A-83-01 inhibits Smad signaling and epithelial-to-mesenchymal transition by transforming growth factor-beta. Cancer Sci. 2005;96:791-800
109. Laping NJ, Grygielko E, Mathur A, Butter S, Bomberger J, Tweed C. et al. Inhibition of transforming growth factor (TGF)-beta1-induced extracellular matrix with a novel inhibitor of the TGF-beta type I receptor kinase activity: SB-431542. Mol Pharmacol. 2002;62:58-64
110. Ehata S, Hanyu A, Fujime M, Katsuno Y, Fukunaga E, Goto K. et al. Ki26894, a novel transforming growth factor-beta type I receptor kinase inhibitor, inhibits in vitro invasion and in vivo bone metastasis of a human breast cancer cell line. Cancer Sci. 2007;98:127-33
111. Bandyopadhyay A, Agyin JK, Wang L, Tang Y, Lei X, Story BM. et al. Inhibition of pulmonary and skeletal metastasis by a transforming growth factor-beta type I receptor kinase inhibitor. Cancer Res. 2006;66:6714-21
112. Mohammad KS, Javelaud D, Fournier PG, Niewolna M, McKenna CR, Peng XH. et al. TGF-beta-RI kinase inhibitor SD-208 reduces the development and progression of melanoma bone metastases. Cancer Res. 2011;71:175-84
113. Tanaka H, Shinto O, Yashiro M, Yamazoe S, Iwauchi T, Muguruma K. et al. Transforming growth factor beta signaling inhibitor, SB-431542, induces maturation of dendritic cells and enhances anti-tumor activity. Oncol Rep. 2010;24:1637-43
114. Takeuchi K, Abe M, Hiasa M, Oda A, Amou H, Kido S. et al. Tgf-Beta inhibition restores terminal osteoblast differentiation to suppress myeloma growth. PLoS One. 2010;5:e9870
115. Korpal M, Yan J, Lu X, Xu S, Lerit DA, Kang Y. Imaging transforming growth factor-beta signaling dynamics and therapeutic response in breast cancer bone metastasis. Nat Med. 2009;15:960-6
116. Zhang B, Halder SK, Zhang S, Datta PK. Targeting transforming growth factor-beta signaling in liver metastasis of colon cancer. Cancer Lett. 2009;277:114-20
117. Melisi D, Ishiyama S, Sclabas GM, Fleming JB, Xia Q, Tortora G. et al. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol Cancer Ther. 2008;7:829-40
118. Engelman JA, Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Current opinion in genetics & development. 2008;18:73-9
119. Corcoran RB, Dias-Santagata D, Bergethon K, Iafrate AJ, Settleman J, Engelman JA. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Science signaling. 2010;3:ra84
120. Di Fiore F, Sesboue R, Michel P, Sabourin JC, Frebourg T. Molecular determinants of anti-EGFR sensitivity and resistance in metastatic colorectal cancer. Br J Cancer. 2010;103:1765-72
121. Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol. 2010;28:1254-61
122. Sierra JR, Cepero V, Giordano S. Molecular mechanisms of acquired resistance to tyrosine kinase targeted therapy. Molecular cancer. 2010;9:75
123. Zhao BM, Hoffmann FM. Inhibition of transforming growth factor-beta1-induced signaling and epithelial-to-mesenchymal transition by the Smad-binding peptide aptamer Trx-SARA. Mol Biol Cell. 2006;17:3819-31
124. Mead AL, Wong TT, Cordeiro MF, Anderson IK, Khaw PT. Evaluation of anti-TGF-beta2 antibody as a new postoperative anti-scarring agent in glaucoma surgery. Invest Ophthalmol Vis Sci. 2003;44:3394-401
125. Thompson JE, Vaughan TJ, Williams AJ, Wilton J, Johnson KS, Bacon L. et al. A fully human antibody neutralising biologically active human TGFbeta2 for use in therapy. J Immunol Methods. 1999;227:17-29
126. Cordeiro MF, Gay JA, Khaw PT. Human anti-transforming growth factor-beta2 antibody: a new glaucoma anti-scarring agent. Invest Ophthalmol Vis Sci. 1999;40:2225-34
127. Morris JC, Shapiro GI, Tan AR, Lawrence DP, Olencki TE, Dezube BJ. et al. Phase I/II study of GC1008: A human anti-transforming growth factor-beta (TGFβ) monoclonal antibody (MAb) in patients with advanced malignant melanoma (MM) or renal cell carcinoma (RCC). ASCO Annual Meeting. 2008
128. Lonning S, Mannick J, McPherson JM. Antibody Targeting of TGF-beta in Cancer Patients. Curr Pharm Biotechnol. 2011;12:2176-89
129. Bogdahn U, Hau P, Olyushin V, Mahapatra AK, Parfenov V, Venkataramana NK. et al. Targeted therapy with AP 12009 in recurrent or refractory glioblastoma patients: Results of a phase Iib study; ASCO Annual Meeting. J Clin Oncol. 2008;26(suppl):abstr2018
130. Bogdahn U, Hau P, Stockhammer G, Venkataramana NK, Mahapatra AK, Suri A. et al. Targeted therapy for high-grade glioma with the TGF-beta2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro Oncol. 2011;13:132-42
131. Calvo-Aller E, Baselga J, Glatt S, Cleverly A, Lahn M, Arteaga CL. et al. First human dose escalation study in patients with metastatic malignancies to determine safety and pharmacokinetics of LY2157299, a small molecule inhibitor of the transforming growth factor-beta receptor I kinase; ASCO Annual Meeting. J Clin Oncol. 2008;26(suppl):abstr14554
132. Rodon Ahnert J, Baselga J, Calvo E, Seoane J, Brana I, Sicart E. et al. First human dose (FHD) study of the oral transforming growth factor-beta receptor I kinase inhibitor LY2157299 in patients with treatment-refractory malignant glioma. J Clin Oncol. 2011;29(suppl):abstr3011
133. Bouquet S, Pilones K. Inhibition of transforming growth factor-beta increase the therapeutic benefit of radiotherapy in murine mammary tune. AACR. 2010
134. Zhang M, Kleber S, Rohrich M, Timke C, Han N, Tuettenberg J. et al. Blockade of TGF-beta signaling by the TGFbetaR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 2011;71:7155-67
135. Gadir N, Jackson DN, Lee E, Foster DA. Defective TGF-beta signaling sensitizes human cancer cells to rapamycin. Oncogene. 2008;27:1055-62
136. Filyak Y, Filyak O, Stoika R. Transforming growth factor beta-1 enhances cytotoxic effect of doxorubicin in human lung adenocarcinoma cells of A549 line. Cell Biol Int. 2007;31:851-5
137. Harper J, Green T. Neutralization of TGF-beta enhance the efficacy of chemotherapeutics in preclinical nodel of triple negative breast cancer. AACR. 2010
138. Hortobagyi GN, Gomez HL, Li RK, Chung HC, Fein LE, Chan VF. et al. Analysis of overall survival from a phase III study of ixabepilone plus capecitabine versus capecitabine in patients with MBC resistant to anthracyclines and taxanes. Breast Cancer Res Treat. 2010;122:409-18
Author contact
![]() Corresponding author: Rosemary J. Akhurst, UCSF Helen Diller Family Cancer Research Building, San Francisco, CA 94158-9001, USA, 415-502-3179; email: RAkhurstucsf.edu
Corresponding author: Rosemary J. Akhurst, UCSF Helen Diller Family Cancer Research Building, San Francisco, CA 94158-9001, USA, 415-502-3179; email: RAkhurstucsf.edu