Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
INTRODUCTION
RESULT AND DISCUSSION
MATERIALS AND METHODS
SUPPLEMENTARY MATERIAL
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(8):1142-1155. doi:10.7150/ijbs.4588 This issue Cite
Research Paper
Tissue-Specific Transcriptome Profiling of Plutella Xylostella Third Instar Larval Midgut
Wen Xie1*, Yanyuan Lei1*, Wei Fu1, Zhongxia Yang1, Xun Zhu1, Zhaojiang Guo1, Qingjun Wu1, Shaoli Wang1, Baoyun Xu1, Xuguo Zhou2 ![]() , Youjun Zhang1
, Youjun Zhang1 ![]()
1. Department of Plant Protection, Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences, Beijing, 100081, P. R. China.
2. Department of Entomology, University of Kentucky, Lexington, KY 40546-0091, USA.
* These authors contributed equally.
Received 2012-5-13; Accepted 2012-8-11; Published 2012-9-14
Abstract
The larval midgut of diamondback moth, Plutella xylostella, is a dynamic tissue that interfaces with a diverse array of physiological and toxicological processes, including nutrient digestion and allocation, xenobiotic detoxification, innate and adaptive immune response, and pathogen defense. Despite its enormous agricultural importance, the genomic resources for P. xylostella are surprisingly scarce. In this study, a Bt resistant P. xylostella strain was subjected to the in-depth transcriptome analysis to identify genes and gene networks putatively involved in various physiological and toxicological processes in the P. xylostella larval midgut.
Using Illumina deep sequencing, we obtained roughly 40 million reads containing approximately 3.6 gigabases of sequence data. De novo assembly generated 63,312 ESTs with an average read length of 416bp, and approximately half of the P. xylostella sequences (45.4%, 28,768) showed similarity to the non-redundant database in GenBank with a cut-off E-value below 10-5. Among them, 11,092 unigenes were assigned to one or multiple GO terms and 16,732 unigenes were assigned to 226 specific pathways. In-depth analysis indentified genes putatively involved in insecticide resistance, nutrient digestion, and innate immune defense. Besides conventional detoxification enzymes and insecticide targets, novel genes, including 28 chymotrypsins and 53 ABC transporters, have been uncovered in the P. xylostella larval midgut transcriptome; which are potentially linked to the Bt toxicity and resistance. Furthermore, an unexpectedly high number of ESTs, including 46 serpins and 7 lysozymes, were predicted to be involved in the immune defense.
As the first tissue-specific transcriptome analysis of P. xylostella, this study sheds light on the molecular understanding of insecticide resistance, especially Bt resistance in an agriculturally important insect pest, and lays the foundation for future functional genomics research. In addition, current sequencing effort greatly enriched the existing P. xylostella EST database, and makes RNAseq a viable option in the future genomic analysis.
Keywords: Illumina sequencing, expressed sequence tag, Plutella xylostella, midgut, insecticide resistance
INTRODUCTION
The diamondback moth, Plutella xylostella (Lepidoptera: Plutellidae), is one of the most devastating insect pests in more than 100 countries around the world; affecting cruciferous plants, especially Brassica oleracea crops including cabbage, brussels sprout, broccoli, cauliflower, and turnip [1]. Estimated global damage and control costs for this insect pest exceed 1billion USD annually. This troublesome pest has been especially problematic in many parts of China since the 1970s, where the only successful form of control has been the use of insecticides. However, Plutella xylostella has developed a robust resistance to many chemical and biological pesticides, including organophosphates, pyrethroids, agricultural antibiotics, and Bacillus thuringiensis Berliner (Bt) toxins, one of the most successful microbial insecticides used worldwide for suppressing pest populations, especially lepidopterans [2].
Plutella xylostella has been studied extensively as a model system for insect physiology and insecticide resistance, including cuticle function [3], chemosensory proteins [4], hormonal regulation [5], insect immunity and defense [6, 7, 8], insect-plant interaction [9], and the mechanistic study of insecticide resistance [10, 11], especially against Bt toxins [12]. The target site for Bt toxins is believed to be the midgut, a dynamic tissue which plays a vital role in metabolism, digestion, and detoxification. In Lepidoptera, previous studies have focused on the role of proteases, lipases and carbohydrases in digestion, and carboxylesterases, glutathione-s-transferases and cytochrome P450s in xenobiotic metabolism in the midgut [13, 14, 15]. With the advent of genomics and its “omics” tools, current research looked more closely at the physiological and toxicological changes at a global level instead of focusing on individual genes in the midgut. Meunier et al studied the transcriptional responses of spruce budworm, Choristoneura fumiferana, larval midgut when challenged with a Cry1Ab Bt toxin at a sublethal concentration [16]. Eum et al investigated the immune-inducible genes in P. xylostella using ESTs and cDNA microarray [7]. Etebari et al documented the host-parasitoid interactions in P. xylostella larvae using an Illumina-based transcriptome profiling technique [8]. Most recently, He et al preformed the most comprehensive transcriptome analysis covering several developmental stages and different susceptible levels of P. xylostella to chlorpyrifos and fipronil, respectively [17]. For non-model organisms, without a fully sequenced genome, a robust EST database is essential for any downstream “omics”-based analyses, especially for the RNAseq. The time- and tissue-specific nature of transcriptome sequencing offers an unparalleled opportunity to investigate the temporal and spatial changes of gene expressions related to a specific biological question. Although a whole body transcriptome is currently available [8, 17], tissue-specific gene expression profiles in P. xylostella are lacking.
In this study, we used the second generation Illumina sequencing platform to provide a comprehensive view of the genes expressed in the larval midgut of a Bt resistant P. xylostella. We generated over three billion bases of high-quality DNA sequences and investigated the potential roles of these predicted proteins involved in various physiological and toxicological processes in P. xylostella larval midgut. In our effort to analyze the midgut transcriptome of P. xylostella, we focused on genes potentially involved in insecticide resistance, digestion, immune and defensive response, and peritrophic membrane integrity. This transcriptome sequencing effort has dramatically increased the number of known genes for this insect model and provides an invaluable resource for the subsequent RNAseq analysis as well as for P. xylostella genome annotation.
RESULT AND DISCUSSION
Sequencing summary
To obtain an overview of the transcriptional profile of the midgut of the diamond back moth, Plutella xylostella (Lepidoptera: Plutellidae), a cDNA sample was prepared and sequenced using the Illumina sequencing platform. After cleaning and quality checks to remove the low quality reads, we obtained 39 million reads with an average length of 90bp from one plate of sequencing. To facilitate sequence assembly, these raw reads were assembly and resulted in 213,674 contigs with Trinity [18] (Table 1). The average size of a contig was 189bp and further assembled into 63,312 unigenes with an average size of 416bp, including 3,333 unigenes (5.26%) which are over 1,000bp in length (Table 1; Figure 1). The N50 of all contigs and unigenes are 262bp and 499bp, respectively. The size distribution of these contigs and unigenes are shown in Figure 1. The resultant parameters are comparable to a recent whole body transcriptomic sequencing efforts to inventory genes differentially expressed among developmental stages and between insecticide resistant and susceptible P. xylostella [Table 1]. To examine the quality of newly assembled P. xylostella midgut transcriptome, we selected 5 unigenes randomly for the RT-PCR validation. The resultant PCR products were visualized on 1% agarose gel first and then cleaned for the direct sequencing. The identity of these PCR products (4/5) was confirmed by the conventional Sanger sequencing.
Sequencing summary in Plutella xylostella larval midgut transcriptome
| Sequencing Summary | Midgut specific transcriptome | Whole body transcriptomea |
|---|---|---|
| Total number of reads | 39,764,230 | 27,514,263-29,793,272 |
| Total base pairs (bp) | 3,578,780,700 | 4,127,139,450-4,468,990,800 |
| Average read length (bp) | 90 | 75b |
| Total number of contig | 213,674 | 223,409-313,859 |
| Mean length of contigs (bp) | 189 | 153-161 |
| N50d of contigs | 262 | 152-168 |
| Total number of unigenes | 63,312 | 171,262c |
| Mean length of unigenes | 416 | 436-468 |
| N50d of unigenes | 499 | 470-521 |
| Sequences with E-value < 10-5 | 28,768 (45.4%) | 38,255 (22.3%) |
| Reference | this study | [17] |
aWhole body transcriptome included 6 libraries covering 4 developmental stages and 2 resistant 3rd instar larvae.
bPaired-end sequencing (75bp in each single-ended).
c171,262 non-redundant sequences from clustered results of all six libraries range from 54,869 to 73,194 unigenes.
dN50 size of contigs or unigenes was calculated by sorting all the sequences by their respective lengths, and then adding the length from longest to shortest until the summed length exceeded 50% of the total length of all sequences.
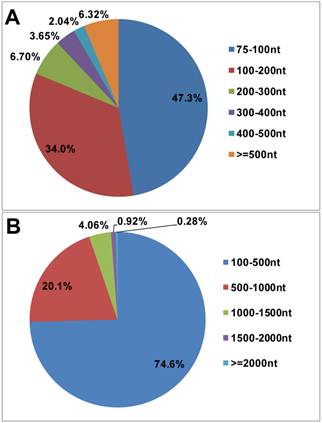
Length distribution of assembled sequences in P. xylostella larval midgut transcriptome. The average length of contig (A) and unigene (B) in P. xylostella larval midgut transcriptome were 84 and 416bp, respectively.
Functional annotation
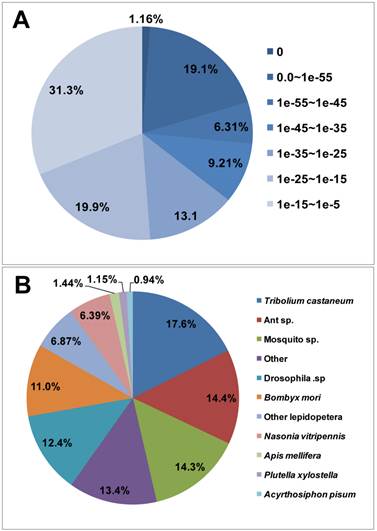
For annotation, the unigenes were first searched using BLASTx against the non-redundant (nr) NCBI protein database with a cut-off E-value of 10-5. Using this approach, 28,768 genes (45.4% of all unigenes sequences) returned above the cut-off BLAST hits (Additional file 1: Table S1). Without a fully sequenced P. xylostella genome, more than half (54.6%) of the 63,312 assembled sequences could not be matched to known genes. The E-value distribution of the top hits in the nr database showed that 26.5% of the mapped sequences exhibit strong homology (smaller than 1.0E-45), whereas 73.5% of the homolog sequences have an E-value ranged between 1.0E-5 to 1.0E-45 (Figure 2A). For species distribution, 17.6% of the unigenes sequences have top matches (first hit) trained with sequences from the red flour beetle (Tribolium castaneum), followed by the ants (14.4%), mosquitoes (14.3%), fruitflies (12.4%), silkworm (Bombyx mori) (11.0%), and other Lepidoptera species (6.87%) (Figure 2B). There are 335 unigene sequences (1.15%) with the highest homology to genes from P. xylostella and the majority of these hits match to cytochrome P450 and trypsin-like serine proteinase (data not shown).
E-value and species distribution of the top BLASTX hits. The BLASTX search was carried out against the NCBI (National Center for Biotechnology Information) nr database. The search results were summarized based on the distribution of their E-value (A) and taxonomic status (B), respectively.
Functional classification and pathway analysis
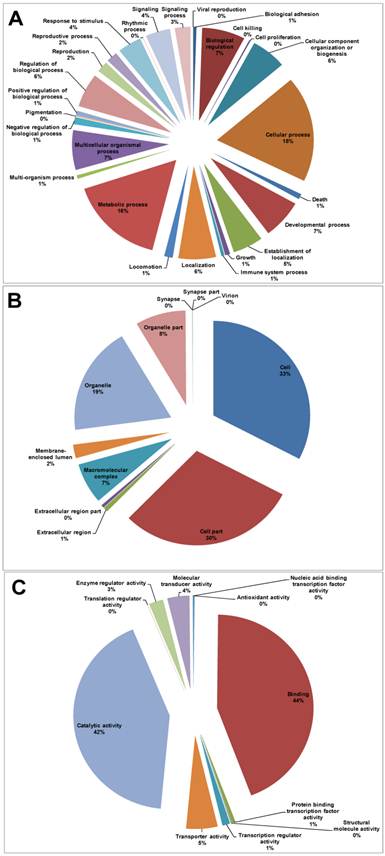
GO assignments were used to classify the functions of the predicted P. xylostella midgut genes. Based on sequence homology, 11,092 unigene sequences can be categorized into 49 functional groups (Additional file 1: Table S3, Figure 3). In each of the three main categories (biological process, cellular component and molecular function) of the GO classification, “Cell”, “Cellular process”, “Cell part”, “Binding” and “Metabolic process” are the dominant terms. In contrast, few genes fall into terms of “Cell killing” and “Translation regulator activity”, “Synapse part” and “Virion” (Figure 3).
Distribution of Gene Ontology (GO) terms in P. xylostella larval midgut transcriptome. (A) Biological Process, (B) Cellular Component, (C) Molecular Function.
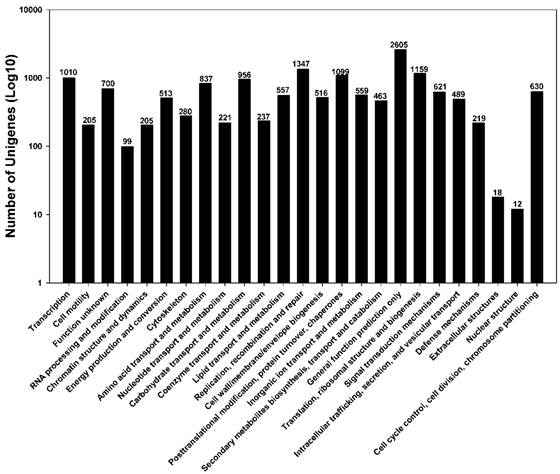
To further evaluate the completeness of our transcriptome library and the effectiveness of our annotation process, we searched the annotated sequences for genes involved in the Cluster of Orthologous Groups (COG, http://www.ncbi.nlm.nih.gov/COG/) classifications. In total, out of 28,768 nr hits, 15,557 unigene sequences (54.1%) have a COG classification (Figure 4). Among the 25 COG categories, “General function prediction only cluster” (2605, 16.7%) represents the largest group, followed by “Replication, recombination and repair” (1347, 8.66%), and “Translation, ribosomal structure and biogenesis” (1159, 7.45%). “Nuclear structure” (12, 0.077%), “Extracellular structures” (18, 0.12%), and “RNA processing and modification” (99, 0.64%) are the least represented categories (Figure 4).
Distribution of Clusters of Orthologous Groups (COG) in P. xylostella larval midgut transcriptome. Among 28,768 nr hits, 8,583 sequences have a COG classification among the 25 categories.
To identify the biological pathways that are active in P. xylostella midgut, 28,768 annotated sequences were mapped to the reference canonical pathways in Kyoto Encyclopedia of Genes and Genomes (KEGG) [19]. In total, 16,732 sequences were assigned to 226 KEGG pathways. The pathways with most representation by the unique sequences are Metabolic pathways (3226 members), Spliceosome (705 members), and Purine metabolism (628 members) (Additional File 1: Table S4). These annotations provide a valuable resource to study specific physiological processes, functions, and pathways involved in the P. xylostella midgut.
Putative SNPs and SSRs
There were a total of 4,953 putative single nucleotide polymorphisms (SNPs) wherein 2,013 were transversions and 2940 were transitions, respectively (Table 2, Additional file 1: Table S5). Additionally, 2,351 simple sequence repeats (SSRs or microsatellites) were identified, of which 60% were dinucleotide repeats, followed by 37.5% trinucleotide and 2.1% tetranucleotide repeats (Table 3, Additional file 1: Table S6). Molecular markers, SNPs and SSRs, identified in this study lay a foundation for the better understanding of the adaptation and ecology of P. xylostella [20]. The identity of predicted molecular markers, however, needs to be validated in future research to exclude false positives and sequencing errors.
Putative SNPs in Plutella xylostella larval midgut transcriptome
| SNP type | Number of Occurrence |
|---|---|
| Transition | 2,940 |
| A-G | 1,448 |
| C-T | 1,492 |
| Transversion | 2,013 |
| A-C | 441 |
| A-T | 564 |
| C-G | 565 |
| G-T | 443 |
| Total | 4,953 |
Microsatellite loci predicted in Plutella xylostella larval midgut transcriptome
| No. of repeats | Di- nucleotide repeats | Tri- nucleotide repeats | Tetra- nucleotide repeats | Penta- nucleotide repeats | Hexa- nucleotide repeats |
|---|---|---|---|---|---|
| 5 | 1096 | 624 | 38 | 2 | - |
| 6 | 196 | 186 | 11 | - | - |
| 7 | 51 | 59 | - | - | - |
| 8 | 29 | 12 | - | - | - |
| 9 | 16 | - | - | - | - |
| 10 | 8 | - | - | - | - |
| 11 | 14 | - | - | - | - |
| 12 | 7 | - | - | - | - |
| 15 | - | 1 | - | - | - |
| 16 | - | - | - | - | 1 |
| Subtotal | 1417 | 882 | 49 | 2 | 1 |
Putative transcription factors
Of the 63,312 unigene sequences, 31,978 unigenes sequences with an open reading frame longer than 150 bp were used for the transcription factor (TF) prediction. After the HMMER search with 75 TF families from the Drosophila Transcription Factor Database (http://www.flytf.org/), 1107 unigenes were predicted as putative TF (Additional file 1: Table S7). Among them, the pfam family for “Zinc finger, C2H2 type” represents the largest TF family (407, 36.8%), followed by “Ras family” (77, 6.96%), and “Zinc-finger associated domain (zf-AD)” (67, 6.05%). (Table 4, Additional file 1: Table S7).
Top transcription factor (TF) families in Plutella xylostella larval midgut transcriptomea
| TF related pfam ID | Pfam domain description | Number of occurrence in midgut |
|---|---|---|
| PF00096 | Zinc finger, C2H2 type | 407 |
| PF00071 | Ras family | 77 |
| PF07776 | Zinc-finger associated domain (zf-AD) | 67 |
| PF00651 | BTB/POZ domain | 47 |
| PF00412 | LIM domain | 30 |
| PF00046 | Homeobox domain | 29 |
| PF00010 | Helix-loop-helix DNA-binding domain | 26 |
| PF00628 | PHD-finger | 26 |
| PF00856 | SET domain | 26 |
| PF05485 | THAP domain | 24 |
| PF00505 | HMG (high mobility group) box | 23 |
| PF00439 | Bromodomain | 21 |
aDrosophila transcription factor database was used as a reference for the TF domain family search in this study.
Genes putatively involved in insecticide resistance
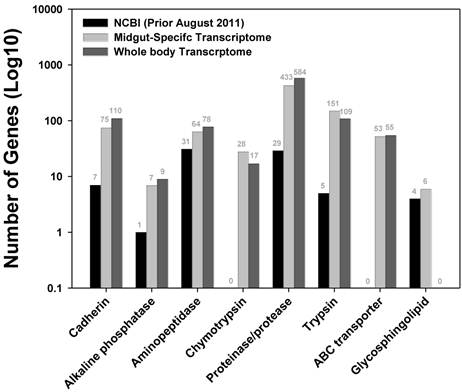
Plutella xylostella has developed resistance to various synthetic and biological pesticides, and has been an important model system to study the molecular mechanisms underlying the development of insecticide resistance. Sequences encoding enzymes potentially involved in xenobiotic detoxification and the targets of the major classes of synthetic insecticides were extracted and compared with sequences from the NCBI protein database. Genes potentially associated with the Bt toxicity and resistance in the P. xylostella larval midgut are listed in Figure 5, including alkaline cadherin, phosphatase, aminopeptidase, chymotrypsin, proteinase/protease, trypsin, ABC transporter, and glycosphingolipid. Among them, chymotrypsin was identified from the P. xylostella for the first time. In total, we obtained 28 chymotrypsin and 53 ABC transporter related sequences. After removing redundant sequences, we identified 18 different chymotrypsin sequences (Additional file 2: Table S8) and 35 ABC transporter sequences (Additional file 2: Table S9). The number of genes of interest obtained in this larval midgut transcriptome is comparable to a whole body sequencing effort (Figure 5), reflecting the tissue (spatial)-specific nature of transcriptome sequencing approach. Recently, an ABCC2 gene in Heliothis virescens was genetically linked to the Cry1Ac resistance. A loss-of-function mutation in ABCC2 led to the loss of Cry1Ac binding to membrane vesicles, suggesting ABC transporters may play a key role in the mode of action of Bt toxins [21]. Moreover, Baxter et al (2011) cloned an ABCC2 gene in P. xylostella, and genetically mapped it onto a locus controlling the Bt Cry1Ac resistance [12].
Genes putatively associated with Bt toxicity and resistance in P. xylostella larval midgut transcriptome. Black bar represents the existing P. xylostella sequence available in the NCBI protein database (as of August 2011, red bar denotes the number of sequences obtained in a tissue-specific transcriptome (this study), and green bar includes the number of sequences acquired through a whole body transcriptome [17].
ESTs potentially involved in the insecticide metabolic resistance are summarized in Table 5, including conventional detoxification enzymes such as cytochrome P450 monooxygenase, carboxylesterase and glutathione S-transferase; and putative insecticide targets, including neuropeptide receptor, glutamate receptor and ryanodine receptor. Based on the closest BLAST hits in the NCBI nr database, transcripts encoding putative P450s were assigned to appropriate CYP clades and families (Table 6). Specifically, among 156 P450 unigenes annotated in the NCBI nr database, 74 contained CYP family information and 41 GST unigenes annotated in the NCBI nr database, 23 contained GST class information. The remaining 74 P450 unigenes were subdivided into 4 clades and 13 families, including 3 families of CYP304, CYP305 and CYP306 in CYP2 clade, 6 families of CYP6, CYP321, CYP337, CYP347, CYP354 and CYP366 in CYP3 clade, 1 CYP4 family in CYP4 clade, and 3 families of CYP301, CYP314 and CYP333 in mitochondrial CYP clade (Table 6). The majority of annotated P450s belonged to the CYP3 clade (41/74), and followed by CYP4 (13/74), mitochondrial (13/74), and CPY2 (7/74). At the family level, CYP6 (26) and CYP4 (13) are the most abundant P450 families. Four genes (CYP6B8, CYP6B9, CYP6B28 and CYP6B27) have been identified from Helicoverpa zea in the CYP6 family, which are associated with xenobiotics resistance [22, 23].
Genes of interest in Plutella xylostella larval midgut transcriptome
| Genes | NCBIa | Midgut specificb | Whole bodyc |
|---|---|---|---|
| Metabolic insecticide resistance and insecticide targets | |||
| Cytochrome P450 monooxygenase | 17 | 156 | 235 |
| Carboxylesterase | 9 | 28 | 40 |
| Glutathione S-transferase | 7 | 41 | 40 |
| Acetylcholinesterase | 4 | 1 | 5 |
| Nicotinic acetylcholine Receptor | 25 | 1 | 22 |
| GABA receptor | 6 | 2 | 7 |
| Neuropeptide receptor | 1 | 43 | 31 |
| Glutamate receptor | 0 | 10 | 48 |
| G-protein coupled receptor | 0 | 14 | 8 |
| Ryanodine receptor | 0 | 22 | 60 |
| Lipophorin receptor | 1 | 8 | 6 |
| Sodium channel | 14 | 6 | 13 |
| Chloride channel | 5 | 19 | 21 |
| NADH dehydrogenase | 0 | 31 | 44 |
| NADH oxidoreductase | 0 | 14 | 27 |
| Catalase | 0 | 20 | 21 |
| Peroxidase | 0 | 21 | 36 |
| Superoxide dismutase | 3 | 8 | 11 |
| Nutrient digestion | |||
| Trehalase | 0 | 11 | 11 |
| Serine proteinase all types | 5 | 45 | 26 |
| Cysteine proteinase all types | 0 | 9 | 11 |
| Carboxypeptidase all types | 0 | 71 | 109 |
| Dipeptidyl-peptidase | 0 | 19 | 20 |
| α-amylase | 0 | 15 | 10 |
| glucosidase | 0 | 62 | 77 |
| Lipase | 16 | 149 | 153 |
| Innate immune defense | |||
| Toll-like receptor (TLR) | 0 | 5 | 4 |
| β-1,3-glucan | 0 | 13 | 38 |
| β-1,3-glucanase | 1 | 8 | 5 |
| Peptidoglycan recognition protein | 2 | 10 | 12 |
| Defensin | 0 | 1 | 0 |
| Lysozyme | 0 | 7 | 7 |
| Serine protease inhibitor (Serpin) | 6 | 46 | 78 |
| Transferrin | 1 | 14 | 19 |
| Peritrophic membrane biosynthesis, metabolization and remodelling | |||
| Chitin synthase | 2 | 14 | 24 |
| Chitinase | 3 | 38 | 39 |
| Chitin deacetylase | 0 | 12 | 9 |
| Peritrophin-like, Mucin-like | 51 | 1 | 46 |
aNumber of Plutella xylostella sequences available at the NCBI protein database (as of August 2011).
bNumber of sequences obtained in a tissue-specific transcriptome (this study, in shade).
bNumber of sequences obtained in a whole body transcriptome [17].
Different CYPs P450 clans, families, and GSTs classes in midgut transcriptome of Plutella xylostella
| Detoxification enzymes | #Occurrence | Family members with corresponding number |
|---|---|---|
| CYPs P450 | ||
| Cyp2 clade (3 families) | ||
| CYP304 | 04 | CYPCCCIVA1(1) , CYPCCCIVF2(3) |
| CYP305 | 01 | CYPCCCVB1(1) |
| CYP306 | 02 | CYPCCCVIA1(2) |
| Cyp3 clade (6 families) | ||
| CYP6 | 26 | CYPVIAB13(5), CYPVIAB5(4), CYPVIAE27(1), CYPVIAE32(5), CYPVIAE9(1), CYPVIAN5(5), CYPVIBK1(1), CYPVIBQ4(1), CYPVIK1(3) |
| CYP321 | 06 | CYPCCCXXIA1(3), CYPCCCXXIB1(3) |
| CYP337 | 02 | CYPCCCXXXVIIA1(1), CYPCCCXXXVIIB1(1) |
| CYP347 | 01 | CYPCCCXLVIIA1(1) |
| CYP354 | 04 | CYPCCCLIVA5(4) |
| CYP366 | 02 | CYPCCCLXVIA1(2) |
| Cyp4 clade (1 family) | ||
| CYP4 | 13 | CYPIV(1), CYPIVAB2(1), CYPIVCG1(1), CYPIVG11(1), CYPIVG4(1), CYPIVG47(2), CYPIVM1(2), CYPIVM2(1), CYPIVM5(1), CYPIVM7(1), CYPIVV2(1) |
| Mitochondrial CYP clade (3 family) | ||
| CYP301 | 02 | CYPCCCIA1(1), CYPCCCIB1(1) |
| CYP314 | 05 | CYPCCCXIVA1(5) |
| CYP333 | 06 | CYPCCCXXXIIIA3(2), CYPCCCXXXIIIB10(1), CYPCCCXXXIIIB11(3) |
| GSTs classes | ||
| Delta | 2 | Delta(1), Delta3(1) |
| Epsilon | 3 | Epsilon2(1), Epsilon4(1), Epsilon6(1) |
| Omega | 10 | Omega1(7),Omega2(1), Omega3(2) |
| Theta | 4 | Theta(3), Theta1(1) |
| Zeta | 4 | Zeta(2), Zeta1(2) |
Meanwhile, the remaining 23 GST unigenes were subdivided into 5 classes, among which the Omega class was the main group (10/23) which is identified in some Lepidopteran species, for example, Spodoptera litura [24], Bombyx mori [25], but absent in Trialeurodes vaporariorum[26], Acyrthosiphon pisum and Myzus persicae[27]. Furthermore, glutamate receptors are synaptic receptors located primarily on the membranes of neuronal cells and mediate neuronal communication at synapses throughout vertebrate and invertebrate nervous systems [28]. Ryanodine receptors (RyRs) are the major cellular mediator of calcium-induced calcium release in animal cells. Diamide insecticides control insects by the activation of RyRs which leads to uncontrolled calcium release in muscle [29]. In this study, unigenens encoding the glutamate receptor (10) and ryanodine receptor (22), respectively, were identified. After consolidating the redundant sequences, we assembled 8 different glutamate receptor sequences (Additional file 1: Table S2) and 9 ryanodine receptor sequences (Additional file 2: Table S10).
Putative digestive enzymes
The lepidopteran midgut plays key roles in the nutrient digestion and allocation. Digestive enzymes identified from this sequencing effort include trehalase, carboxypeptidase, dipeptidyl-peptidase, α-amylase, glucosidase and lipase (Table 5), chymotrypsin, proteinase/protease, aminopeptidase, and trypsin (Figure 5). Trehalase plays a pivotal role in various physiological processes, including flight metabolism [30], chitin synthesis [31], and cold tolerance [32] through the hydrolysis of trehalose, a principal hemolymph sugar in insects which is an indispensable substrate for energy production and macromolecular biosynthesis [33]. Trehalase was divided into the soluble (Tre-1) and the membrane-bound (Tre-2) trehalases [34]. In total, 11 trehalose related sequences were obtained. After consolidating the redundant sequences, we identified 8 different trehalase sequences containing Tre-1 and Tre-2 (Additional file 2: Table S11).
The most abundant digestive enzymes in this study are carboxypeptidase and aminopeptidase, a group of specialized exopeptidases that break down dietary proteins into amino acids and small peptides. Current studies of insect digestive exopeptidases have been focused primarily on aminopeptidases due to the fact that midgut aminopeptidases may serve as the receptors for Bt endotoxins [35-38]. In this study, a wealth of aminopeptidases and carboxypeptidases were uncovered, and these finding will undoubtedly facilitate the future research on P. xylostella midgut exopeptidases.
Candidate genes involved in the immune and defense response
Although the insect midgut has traditionally been viewed as a tissue primarily involved in digestion and detoxification, several studies have also placed increasing emphasis on the immune responsiveness of this tissue [39-41]. Some immune responses induced by the presence of microbes in food can be transmitted from insect parent to their offspring [41, 42]. Recent studies have identified a significant number of immune- and metabolic-related genes in the midgut. Transcriptomic analysis on parasitized versus non-parasitized P. xylostella larvae led to the identification of DsIV, a gene expressed by ichnoviruses, a symbiotic polydnaviruses associated with ichneumonid parasitoids, Diadegma semiclausum [8]. DsIV plays a major role in host immune suppression and developmental regulation. In our P. xylostella larval midgut EST database, the most abundant immune response genes are putative serine proteinase inhibitors or serpins. After consolidating the original 64 unigenes, we identified 21 different serpin sequences (Additional file 2: Table S12). Among them, 5 correspond to the existing P. xylostella serpins (3 serpin-1, 1 serpin-2, and 1 serpin-3) [43], and the remaining 16 unigenes are novel serpins (Additional file 2: Table S12).
Among the 6 different PGRPs identified in this study (Additional file 2: Table S13), two match the known P. xylostella PGRPs, and the other four are new genes (PGRP-LC, PGRP-SC2, PGRP-D and PGRP-B). Based on the InterProScan analysis, these new PGRPs all possess a conserved amidase-like domain (IPR002502), a typical active site for PGRPs. Further analysis shows that P. xylostella PGRP-B is very similar to PGRP-B from the Eri silkworm, Samia cynthia ricini [44], and PGRP-LB from the fruit fly, Drosophila melanogaster [45]. Both of these PGRPs are constitutively expressed in the larval midgut. PGRP-LC in D. melanogaster serves as a signal-transducing innate immune receptor [46, 47]. In P. xylostella, the functional characterization of PGRP-LC, -SC2, -D and -B homologues identified in this study could therefore be a logical first step to understand how peptidoglycan fragments are recognized by the lepidopteran immune system.
The primary structure of defensin-like-1 protein genes (Unigene51045_mk) from P. xylostella midgut was compared with other lepidopteran defensins in Additional file 2: Figure S1. The amino acid alignments show a highly conserved six cysteine residues in these defensin-like proteins (Additional file 2: Figure S1). As part of the innate immune defense against pathogens, a group of 5 ESTs putatively encoding lysozymes were indentified in the P. xylostella larval midgut (Additional file 2: Table S14). The deduced amino acid sequence alignments show the alpha-lactalbumin motif (lysozyme C signature) in the P. xylostella lysozyme-like protein 1 (LLP1, Unigene58181_mk) (Additional file 2: Figure S2). However, the P. xylostella LLP1 lacks the catalytic residues Glu and Asp (marked with * in Additional file 2: figure S2), which are conserved in classical lysozymes and necessary for their enzymatic activity [48]. In tasar silkworm, Antheraea mylitta, the lack of this critical residue is thought to account for the loss of muramidase activity [48], and the similar enzymatic consequences would therefore be hypothesized for the P. xylostella LLP1 homologue.
Genes putatively involved in the peritrophic membrane biosynthesis, metabolization and remodeling
The midgut is involved in the biosynthesis, degradation, and remodeling of the peritrophic matrix (PM), a permeable protein shield protecting the midgut epithelium and a likely target site for Bt toxins [49]. Fourteen chitin synthase related sequences were obtained; in which 8 unigenes correspond to the chitin synthase 1(Table 5). P. xylostella chitin synthase 1 were found to express in all developmental stages [50]. After consolidating the redundant sequences, 4 different chitin synthase sequences (Unigene23722_mk, Unigene60458_mk, Unigene4562_mk, and Unigene12371_mk) were identified (Additional file 2: Table S15). Sequence comparisons of predicted amino acid sequence of the longest P. xylostella chitin synthase (Unigene4562_mk) with other lepidopteran chitin synthase are shown in Additional file 2: Figure S3. In addition, 9 putative chitin deacetylase sequences containing chitin deacetylase 1, 2, 4, 5a and 5b were consolidated from the initial pool of 21 unigenes by removing the redundant sequences (Additional file 2: Table S16).
Housekeeping genes
Normalization of target gene expression levels is a critical step in the qRT-PCR analysis, a molecular biology tool used extensively for the downstream functional characterization of the annotated candidate genes [51]. The use of reference genes as internal controls is the most common method for normalizing the qRT-PCR data [52]. In this study, referring to a list of human reference genes [53], 28,768 annotated unigenes were subjected to the housekeeping genes search. A total of 1808 unigenes grouped into 119 clusters were annotated as the putative reference genes (Additional file 2: Table S17). Among them, the most well represented housekeeping genes in the P. xylostella larval midgut are myosin (142, 7.85%), follows by ATPase (113, 6.25%) and proteasome (89, 4.92%). (Table 7, Additional file 2: Table S17). To standardize the qRT-PCR analysis in P. xylostella, a parallel study is currently underway to select the appropriate reference genes from these newly annotated housekeeping genes.
Top 20 housekeeping genes in Plutella xylostella larval midgut transcriptome
| Gene | Number of occurrence |
|---|---|
| myosin | 142 |
| ATPase | 113 |
| proteasome | 89 |
| splicing factor | 84 |
| longation factor | 70 |
| lectin | 54 |
| dynein | 50 |
| eukaryotic translation initiation factor 3 | 49 |
| tubulin | 40 |
| protein kinase C | 38 |
| collagen | 38 |
| chitinase | 38 |
| clathrin | 37 |
| glycoprotein | 36 |
| ubiquitin specific protease (USP) | 31 |
| NADH dehydrogenase (ubiquinone) | 31 |
| ATP synthase | 29 |
| coatomer protein complex | 27 |
| sorting nexin (SNX) | 26 |
| actin | 24 |
MATERIALS AND METHODS
Plutella xylostella colony maintenance
The PXR colony has been maintained since 2005 and subjected to the laboratory selection with Cry1Ac at the Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences.. Plutella xylostella colonies were provisioned with cabbage seedlings (Brassica oleracea L., cv Jingfeng 1), and kept at 26°C with a 12:12 (L: D) photoperiod.
RNA isolation and cDNA library construction for transcriptome analysis
Fresh midgut tissue was dissected from 3rd instar P. xylostella. Midgut total RNA was extracted with Trizol reagent (Invitrogen) according to the manufacturer's instructions. The quality and integrity of resultant total RNA was checked on 1% agarose gel first, and further examined with a 2100 Bioanalyzer (Agilent Technologies) using a minimum RIN (RNA Integrated Number) value of 8. Poly (A)-containing RNA was then separated from total RNA using the Dynabeads® mRNA purification kit (Invitrogen), and the quality was checked on a denaturing gel. The cDNA library for transcriptome sequencing was prepared using the ScriptSeq™ mRNA-Seq Library Preparation Kit (Illumina, San Diego, CA) following manufacturer's recommendations.
Sequence analysis
The cDNA library was sequenced on the Illumina sequencing platform (GAII). The insert size of the library is approximately 200 bp and both ends of the cDNA were sequenced. Image deconvolution and quality value calculations were performed using the Illumina GA pipeline 1.3. The raw reads were cleaned by removing adaptor sequences, empty reads and low quality sequences (reads with unknown sequences 'N'). The reads obtained were assembled using Trinity [18]. The result unigenes were used for the blast search and annotation against an NCBI nr database using an E-value cut-off of 10-5. Functional annotation by gene ontology terms (GO; http://www.geneontology.org) was analyzed by Blast2go software. The COG and KEGG pathway annotation were performed using Blastall software against Cluster of Orthologous Groups database and Kyoto Encyclopedia of Genes and Genomes database, respectively. The data sets are available at the NCBI Short Read Archive (SRA) with the accession number: SRX101299.
Identification of putative molecular markers
SNPs were predicted using the Short Oligonucleotide Alignment Program 2 (SOAP2) software package [SOAPsnp, 54]). Briefly, all high quality reads were used for mapping with arbitrary criteria of at least 5 reads supporting the consensus or variant. The identification and localization of microsatellites were accomplished using a PERL5 script (MIcroSAtellite MISA) [55]. The script can identify both perfect and compound microsatellites, which are interrupted by certain number of bases.
Identification and classification of putative transcription factors
ESTScan [56] was used to detect coding sequences in assembled EST sequences. Sequences that resulted in peptides longer than 50 amino acids were used to predict TFs. TF prediction was carried out according to He et al [57] with some modifications. The HMM profiles of 75 TF families were downloaded from the Drosophila Transcription Factor Database (http://www.flytf.org/) and PFAM [58]. An HMMER search with a threshold E-value of 0.01 was carried out and unique genes with significant hits to the HMM profiles were annotated as TFs.
SUPPLEMENTARY MATERIAL
Additional File 1Table S1-S7.
Figure S1-S3 and Table S8-S17.
Acknowledgements
Authors are grateful to the two anonymous reviewers for their critical comments and suggestions. Special thanks go to Dr. John Obrycki (University of Kentucky) for his comments on an earlier draft. The current study was supported by the National Natural Youthful Science Foundation of China (Grant No. 30900948), the Special Fund for Agro-scientific Research in the Public Interest (201103021), the National Science and technology support task (2012BAD19B06), the National Basic Research and Development Program of China (2009CB119004), and the Key Laboratory of Biology and Genetic Improvement of Horticultural Crops, Ministry of Agriculture, P. R. China.
Author Contributions
Conceived and designed the experiments: WX YYL XGZ YJZ. Performed the experiments: WX YYL. Analyzed the data: WX XGZ. Contributed reagents/materials/analysis tools: WX YYL XGZ YJZ. Wrote the paper: WX WF ZXY QJW SLW BYX XGZ YJZ.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Talekar NS. Biology, ecology, and management of the diamondback moth. Annu Rev Entomol. 1993;38:275-301
2. Tabashnik BE, Van Rensburg JBJ, Carriere Y. Field-evolved insect resistance to Bt crops: Definition, theory, and data. J Econ Entomol. 2009;102:2011-2025
3. Altre JA, Vandenberg JD, Cantone FA. Pathogenicity of Paecilomyces fumosoroseus isolates to diamondback moth, Plutella xylostella: correlation with spore size, germination speed, and attachment to cuticle. J Invertebr Pathol. 1999;73:332-8
4. Liu X, Luo Q, Zhong G. et al. Molecular characterization and expression pattern of four chemosensory proteins from diamondback moth, Plutella xylostella (Lepidoptera: Plutellidae). J Biochem. 2010;148:189-200
5. Lee DW, Shrestha S, Kim AY. et al. RNA interference of pheromone biosynthesis-activating neuropeptide receptor suppresses mating behavior by inhibiting sex pheromone production in Plutella xylostella (L.). Insect Biochem Molec. 2011;41:236-43
6. Huang F, Shi M, Yang YY. et al. Changes in hemocytes of Plutella xylostella after parasitism by Diadegma semiclausum. Arch Insect Biochem. 2009;70:177-87
7. Eum JH, Seo YR, Yoe SM. et al. Analysis of the immune-inducible genes of Plutella xylostella using expressed sequence tags and cDNA microarray. Dev Comp Immunol. 2007;31:1107-20
8. Etebari K, Palfreyman RW, Schlipalius D. et al. Deep sequencing-based transcriptome analysis of Plutella xylostella larvae parasitized by Diadegma semiclausum. BMC Genomics. 2011;12:446
9. Yang L, Fang Z, Dicke M. et al. The diamondback moth, Plutella xylostella, specifically inactivates Mustard Trypsin Inhibitor 2 (MTI2) to overcome host plant defence. Insect Biochem Molec. 2009;39:55-61
10. Baxter SW, Chen M, Dawson A. et al. Mis-spliced transcripts of nicotinic acetylcholine receptor α6 are associated with field evolved spinosad resistance in Plutella xylostella (L.). PLoS Genetic. 2010;6:e1000802
11. Bautista MA, Miyata T, Miura K. et al. RNA interference-mediated knockdown of a cytochrome P450, CYP6BG1, from the diamondback moth, Plutella xylostella, reduces larval resistance to permethrin. Insect Biochem Molec. 2009;39:38-46
12. Baxter SW, Badenes-Pérez FR, Morrison A. et al. Parallel evolution of Bt toxin resistance in lepidoptera. Genetics. 2011 doi: 10.1534/genetics.111.130971
13. Simpson RM, Newcomb RD, Gatehouse HS. et al. Expressed sequence tags from the midgut of Epiphyas postvittana (Walker) (Lepidoptera: Tortricidae). Insect Mol Biol. 2007;16:675-690
14. Coates BS, Sumerford DV, Hellmich RL. et al. Mining an Ostrinia nubilalis midgut expressed sequence tag (EST) library for candidate genes and single nucleotide polymorphisms (SNPs). Insect Mol Biol. 2008;17:607-620
15. Pauchet Y, Wilkinson P, Vogel H. et al. Pyrosequencing the Manduca sexta larval midgut transcriptome: messages for digestion, detoxification and defence. Insect Mol Biol. 2010;19:61-75
16. Meunier L, Prefontaine G, Van Munster M. et al. Transcriptional response of Choristoneura fumiferana to sublethal exposure of Cry1Ab protoxin from Bacillus thuringiensis. Insect Mol Biol. 2006;15:475-483
17. He W, You M, Vasseur L. et al. Developmental and insecticide-resistant insights from the de novo assembled transcriptome of the diamondback moth, Plutella xylostella. Genomics. 2012;99:169-177
18. Grabherr MG, Haas BJ, Yassour M. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644-52
19. Kanehisa M, Goto S, Kawashima S. et al. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:277-280
20. Behura SK. Molecular marker systems in insects: current trends and future avenues. Mol Ecol. 2006;15:3087-3113
21. Gahan LJ, Pauchet Y, Vogel H. et al. An ABC transporter mutation is correlated with insect resistance to Bacillus thuringiensis Cry1Ac toxin. PLoS Genetic. 2010;6:e1001248
22. Li X, Schuler MA, Berenbaum MR. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annu Rev Entomol. 2007;52:231-253
23. Hopkins BW, Longnecker MT, Pietrantonio PV. Transcriptional overexpression of CYP6B8/CYP6B28 and CYP6B9 is a mechanism associated with cypermethrin survivorship in field-collected Helicoverpa zea (Lepidoptera: Noctuidae) moths. Pest Manag Sci. 2011;67:21-5
24. Huang Y, Xu Z, Lin X. et al. Structure and expression of glutathione S-transferase genes from the midgut of the Common cutworm, Spodoptera litura (Noctuidae) and their response to xenobiotic compounds and bacteria. J Insect Physiol. 2011;57:1033-44
25. Yamamoto K, Teshiba S, Shigeoka Y. et al. Characterization of an omega-class glutathione S-transferase in the stress response of the silkmoth. Insect Mol Biol. 2011;20:379-86
26. Karatolos N, Pauchet Y, Wilkinson P. et al. Pyrosequencing the transcriptome of the greenhouse whitefly, Trialeurodes vaporariorum reveals multiple transcripts encoding insecticide targets and detoxifying enzymes. BMC Genomics. 2011;12:56
27. Ramsey JS, Rider DS, Walsh TK. et al. Comparative analysis of detoxification enzymes in Acyrthosiphon pisum and Myzus persicae. Insect Mol Biol. 2010;19:155-164
28. Benton R, Vannice KS, Gomez-Diaz C. et al. Variant ionotropic glutamate receptors as chemosensory receptors in Drosophila. Cell. 2009;136:149-162
29. Lahm GP, Cordova D, Barry JD. New and selective ryanodine receptor activators for insect control. Bioorgan Med Chem. 2009;17:4127-33
30. Clegg JS, Evans DR. Blood trehalose and flight metabolism in the blowfly. Science. 1961;134:54-55
31. Tatun N, Singtripop T, Sakurai S. Dual control of midgut trehalase activity by 20-hydroxyecdysone and an inhibitory factor in the bamboo borer Omhisa fuscidentalis Hampson. J Insect Physiol. 2008;54:351-357
32. Tatun N, Singtripop T, Tungjitwitayakul J. et al. Regulation of soluble and membrane-bound trehalase activity and expression of the enzyme in the larval midgut of the bamboo borer Omphisa fuscidentalis. Insect Biochem Molec. 2008;38:788-795
33. Friedman S. Trehalose regulation, one aspect of metabolic homeostasis. Annu Rev Entomol. 1978;23:389-407
34. Becker A, Schloder P, Steele JE. et al. The regulation of trehalose metabolism in insects. Experientia. 1996;52:433-439
35. Sangadala S, Walters FS, English LH. et al. A mixture of Manduca sexta aminopeptidase and phosphatase enhances Bacillus thuringiensis insecticidal Cry1Ac toxin binding and 86Rb+-K+ efflux in vitro. J Biol Chem. 1994;269:10088-10092
36. Knight PJK, Crickmore N, Ellar DJ. The receptor for Bacillus thuringiensis Cry1a(C) delta-endotoxin in the brush border membrane of the lepidopteran Manduca sexta is aminopeptidase N. Mol Microbiol. 1994;11:429-436
37. Gill M, Ellar D. Transgenic Drosophila reveals a functional in vivo receptor for the Bacillus thuringiensis toxin Cry1Ac1. Insect Mol Biol. 2002;11:619-625
38. Rajagopal R, Sivakumar S, Agrawal N. et al. Silencing of midgut aminopeptidase N of Spodoptera litura by double-stranded RNA establishes its role as Bacillus thuringiensis toxin receptor. J Biol Chem. 2002;277:46849-46851
39. Zaidman-Remy A, Herve M, Poidevin M. et al. The Drosophila amidase PGRP-LB modulates the immune response to bacterial infection. Immunity. 2006;24:463-473
40. Freitak D, Wheat CW, Heckel DG. et al. Immune system responses and fitness costs associated with consumption of bacteria in larvae of Trichoplusia ni. BMC Biology. 2007;5:56
41. Freitak D, Heckel DG, Vogel H. Bacterial feeding induces changes in immune-related gene expression and has trans-generational impacts in the cabbage looper (Trichoplusia ni). Front Zool. 2009;6:7
42. Freitak D, Heckel DG, Vogel H. Dietary dependent trans-generational immune priming in an insect herbivore. Proc Biol Sci. 2009;276:2617-2624
43. Song KH, Jung MK, Eum JH. et al. Proteomic analysis of parasitized Plutella xylostella larvae plasma. J Insect Physiol. 2008;54:1270-80
44. Hashimoto K, Mega K, Matsumoto Y. et al. Three peptidoglycan recognition protein (PGRP) genes encoding potential amidase from eri-silkworm, Samia cynthia ricini. Comp Biochem Physiol B Biochem Mol Biol. 2007;148:322-328
45. Zaidman-Remy A, Herve M, Poidevin M. et al. The Drosophila amidase PGRP-LB modulates the immune response to bacterial infection. Immunity. 2006;24:463-473
46. Maillet F, Bischoff V, Vignal C. et al. The Drosophila peptidoglycan recognition protein PGRP-LF blocks PGRP-LC and IMD/JNK pathway activation. Cell Host Microbe. 2008;3:293-303
47. Choe KM, Lee H, Anderson KV. Drosophila peptidoglycan recognition protein LC (PGRP-LC) acts as a signal-transducing innate immune receptor. P Natl Acad Sci USA. 2005;102:1122-6
48. Jain D, Nair DT, Swaminathan GJ. et al. Structure of the induced antibacterial protein from tasar silkworm, Antheraea mylitta. Implications to molecular evolution. J Biol Chem. 2001;276:41377-41382
49. Hegedus D, Erlandson M, Gillott C. et al. New insights into peritrophic matrix synthesis, architecture, and function. Annu Rev Entomol. 2009;54:285-302
50. Ashfaq M, Sonoda S, Tsumuki H. Development and tissue-specific expression CHS1 from Plutella xylostella and its response to chlorfluazuron. Pestic Biochem Phys. 2007;89:20-30
51. Vandesompele J, De Preter K, Pattyn F. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034
52. Thellin O, Zorzi W, Lakaye B. et al. Housekeeping genes as internal standards: use and limits. J Biotechnol. 1999;75:291-295
53. Eisenberg E, Levanon E Y. Human housekeeping genes are compact. Trends in Genetics. 2003;19:362-5
54. Li R, Yu C, Li Y. et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966-1967
55. Thiel T, Michalek W, Varshney RK. et al. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet. 2003;106:411-422
56. Iseli C, Jongeneel CV, Bucher P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol. 1999:138-148
57. He QL, Cui SJ, Gu JL. et al. Analysis of floral transcription factors from Lycoris longituba. Genomics. 2010;96:119-27
58. Finn RD, Tate J, Mistry J. et al. The Pfam protein families database. Nucleic Acids Res. 2008;36:281-288
Author contact
![]() Corresponding author: Dr. Youjun Zhang, Department of Entomology, Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences, 12 Zhongguancun Nandajie, Haidian District, Beijing 100081, China. Phone: 86-10-82109518; Fax: 86-10-82109518; Email: zhangyjcaas.net.cn. Dr. Xuguo "Joe" Zhou, Department of Entomology, University of Kentucky, S-225 Agricultural Science Center North, Lexington, KY 40546-0091. Phone: 859-257-3125; Fax: 859-323-1120; Email: xuguozhouedu
Corresponding author: Dr. Youjun Zhang, Department of Entomology, Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences, 12 Zhongguancun Nandajie, Haidian District, Beijing 100081, China. Phone: 86-10-82109518; Fax: 86-10-82109518; Email: zhangyjcaas.net.cn. Dr. Xuguo "Joe" Zhou, Department of Entomology, University of Kentucky, S-225 Agricultural Science Center North, Lexington, KY 40546-0091. Phone: 859-257-3125; Fax: 859-323-1120; Email: xuguozhouedu