Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(8):1178-1187. doi:10.7150/ijbs.4458 This issue Cite
Research Paper
DLX4 Upregulates TWIST and Enhances Tumor Migration, Invasion and Metastasis
Lianmei Zhang1*, Manman Yang1*, Lin Gan1, Tao He1, Xiuli Xiao1, M. David Stewart2, Xiaoyan Liu1, Luquan Yang1, Tiandan Zhang1, Yongxiang Zhao3 ![]() , Junjiang Fu1
, Junjiang Fu1 ![]()
1. Key Laboratory of Epigenetics and Oncology, The Research Center for Preclinical Medicine, Luzhou Medical College, Luzhou, China 646000
2. Department of Biology & Biochemistry, University of Houston, Houston, TX, USA 77204
3. Biological Targeting Diagnosis and Therapy Research Center, Guangxi Medical University, Nanning, China 530021
* Co-first authors
Received 2012-4-11; Accepted 2012-9-25; Published 2012-10-13
Abstract
The distal-less homeobox gene 4 (DLX4) is a member of the DLX family of homeobox genes. Although absent from most normal adult tissues, DLX4 is widely expressed in leukemia, lung, breast, ovarian and prostate cancers. However the molecular targets, mechanisms and pathways that mediate the role of DLX4 in tumor metastasis are poorly understood. In this study, we found that DLX4 induces cancer cells to undergo epithelial to mesenchymal transition (EMT) through TWIST. Overexpression of DLX4 increased expression of TWIST expression in cancer cell lines, resulting in increased migratory and invasive capacity. Likewise, knocking down expression of DLX4 decreased TWIST expression and the migration ability of cancer cell lines. DLX4 bound to regulatory regions of the TWIST gene. Both western blotting and immunohistochemistry staining showed that the expression of DLX4 and TWIST are correlated in most of breast tumors. Taken together, these data from both cell models and tumor tissues demonstrate that DLX4 not only upregulates TWIST expression but also induces EMT and tumor metastasis. Altogether, we propose a new pathway in which DLX4 drives expression of TWIST to promote EMT, cancer migration, invasion and metastasis.
Keywords: DLX4, TWIST, E-Cadherin, breast cancer, metastasis
Introduction
TWIST, also known as TWIST1, is a class II member of the highly conserved family of basic helix-loop-helix (bHLH) transcription factors. TWIST is overexpressed in breast, gastric, hepatocellular, prostate and bladder cancers. Its upregulation correlates with low E-cadherin expression, high cancer aggressiveness and poor patient survival rate (1-3). Overexpression of TWIST leads to antiapoptosis, drug resistance, angiogenesis, epithelial to mesenchymal transition (EMT) and metastasis (2-6). TWIST overexpression can promote metastasis in vivo by inducing EMT, causing gain of epithelial stem cell properties, and inducing invadopodia formation (1, 7, 8). Silencing TWIST prevents lung metastasis in mice (1, 6). Altogether, these suggest that TWIST plays an essential in tumor metastasis.
Distal-less homeobox gene 4 (DLX4) (also reported as BP1) (9) is a member of the DLX family of homeobox genes and was initially found to be a repressor of the β-globin gene in erythroid cells (10). DLX4 gene expression is upregulated in 63% of AML cases including 81% of pediatric and 47% of adult cases, and in 32% of T-ALL (9) and in breast cancer (11). DLX4 controls many aspects of embryonic development, including bone morphogenesis, skeletal patterning and cell differentiation. Although absent from most normal adult tissues, DLX4 is widely expressed in leukemias, lung, breast, ovarian and prostate cancers (12-16). DLX4 is located on chromosome 17q21-22, which is known to be amplified in breast cancer. Expression of DLX4 correlates with estrogen receptor status and also cancer progression and invasion in breast cancer (11, 14, 15, 17). DLX4 mRNA expression is also found to be significantly higher in tumors with lymph node metastasis and a high histological grade (16). Knockdown of DLX4 by RNA interference (RNAi) inhibits the motility and invasion ability in JEG-3 cells (18). In sporadic breast cancer, DLX4 negatively regulates BRCA1 (19). DLX4 counteracts key transcriptional control mechanisms of the TGF-beta cytostatic program and blocks the antiproliferative effect of TGF-beta through induction of p15(Ink4B) and p21(WAF1/Cip1) expression(20). In addition, misregulation of homeobox genes are generally correlated with tumorgenesis and cancer progression (21). For example, HOXB7 and HOXA10 are found to induce EMT in epithelial cell lines and promote metastasis (21-23). As a member of homeobox family, DLX4 may be involved in breast tumor progression, invasion and metastasis.
In this study, we sought to investigate the relationship between DLX4 and TWIST in cancer development and cancer metastasis. We found that DLX4 promotes TWIST gene expression by direct interaction with TWIST regulatory regions. Upregulation of TWIST results in increased migratory and invasive capacity. Knocking down DLX4 decreased TWIST expression and reduced the migration ability of cancer cell lines. Furthermore, we found a correlation between DLX4 and TWIST expression in breast tumors.
Materials and Methods
Plasmids, cell lines and antibodies
The pcDNA3-DLX4 construct was kindly provided by Dr. T Takahashi (Japan) (24). pcDNA5/FRT/TO-DLX4 vector (pcDNA5-DLX4) was constructed by inserting the EcoRV/NheI digested full-length DLX4 with PCR amplification into pcDNA5/FRT/TO vector (primers for PCR in Supplementary Material: Table S1). The Flp-In-T-REx system (Invitrogen) HEK293 cells with the pcDNA5/FRT/TO empty vector (293Vec) and the vector containing the full-length DLX4 cDNA (293DLX4) for induction were made according to our previously described method (2, 25-27). MDA-MD-231, MCF7, HeLa and HEK293 cell lines were purchased from ATCC. The pGL3-hTwist-Luc promoter/reporter plasmid with a 2,300-bp DNA fragment amplified by PCR from the proximal enhancer/promoter region of the human TWIST gene was a gift from Dr. J Xu (28). Anti-FLAG M2 monoclonal antibodies (F3165) and Tubulin (T9026) monoclonal antibodies were purchased from Sigma. Goat anti-DLX4 polyclonal antibodies purchased from Santa Cruz (sc-18147, sc-18146). TWIST polyclonal antibodies were purchased from Cell Signaling whereas TWIST monoclonal antibodies from Abcam.
ChIP assays
ChIP assays were performed as previously described (26). The HEK293-DLX4 and HEK293-vector control (Ctrl) cells were cultured in 15-cm plates to 65% confluence and treated with 0.1μg/ml doxycycline (DOX) for 16 hours. MDA-MB-435 cells were cultured in 10-cm plates to 100% confluence. Proteins bound to DNA in these cells were cross-linked in 1% formaldehyde. The samples were sonicated and supernatants were pre-cleaned. The pre-cleaned supernatants were diluted and mixed with M2 Flag antibody beads in HEK293 DLX4 and Vector control (Ctrl) cells, or DLX4 antibodies plus protein A/G agarose in MDA-MB-435 cells, respectively. The beads were sequentially washed, eluted and digested with proteinase K. The eluted DNA was extracted with phenol/chloroform, precipitated with ethanol and resuspended in 1×TE buffer (10 mM Tris pH 7.5, 1mM EDTA pH 8.0). PCR for ChIP assays were performed using this DNA. The PCR product size and sequences of primers used for ChIP assays are shown in Supplementary Material: Table S1.
Luciferase reporter assays
Luciferase reporter assays were performed as described previously (2, 26, 28). Specifically, 40%-confluent HeLa cells in 12-well plates were transfected with 40 ng of the pGL3-hTwist-Luc promoter/reporter plasmid with indicated pCDNA5-DLX4 and 30ng of β-Gal expression plasmids using Lipofectamine 2000. Luciferase and β-gal activities were measured 2 days after transfection. The luciferase activity of each sample was normalized to the β-gal activity of the same sample. The 293Vec or 293DLX4 inducible cells were transfected with 50ng of pGL3-hTwist-Luc promoter/reporter plasmid with/without DOX induction for 2 days. All experiments were performed in triplicates.
shRNA-based stable knockdown of DLX4
The GIPZ lentiviral shRNAmir-GFP system (Open Biosystems) was used to knock down human DLX4 in MDA-MB-231 cells. Lentiviruses that co-express GFP and shRNAs targeting DLX4 (clone V2LHS_202853, V2LHS_202987), and with expression of GFP and empty vector control were used to infect MDA-MB-231 cells. The detailed methods for making shRNA-based stable knockdown of DLX4 cell lines was described previously (26).
In vitro invasion and migration assays
In vitro invasion assays were performed using 24-well Transwell unit with polycarbonate filters (Corning Costar, Cambridge, MA). The 293DLX4 and 293vector cells with or without overnight DOX+/- treatments (0.5μg/ml) were placed in the upper part of the Transwell. HeLa cells transfected with DLX4 and empty vector for two days were placed in the upper part of the Transwell. All cells were incubated for 1-2 days, fixed with ethanol and stained with 0.05% Crystal Violet for 30 min. Cells in the upper chamber were removed with a cotton swab. Cells that invaded through the Matrigel (Matrigel™ Basement Membrane Matirx, BD Biosciences, USA) to the underside of the filter (5 fields/filter) were counted. Three invasion chambers were used per treated group. The values obtained were calculated by averaging the total number of cells from three filters. The experimental procedures for in vitro migration assays were the same as the in vitro invasion assay described above except that the filters were not coated with Matrigel. All experiments were performed in triplicates.
Cell Proliferation Assays
Cell proliferation assays were performed using a CCK-8 (Cell Counting Kit-8) from Beyotime according to the manufacturer's protocol. First, 5,000 cells/well of 293Vec or 293DLX4 cells (100 μl/well) were seeded in a 96-well plate and cultured for 24, 48 or 72 h. 10 μl of the CCK-8 solution was added to each well of the plate and incubated for another 1-4 hours. The absorbance at 450 nm was measured with a microplate reader. A calibration curve was prepared using the data obtained from the wells that contain known numbers of viable cells. Cell proliferation assay were performed in triplicates.
Western blot analysis of human breast tumor tissues
Twenty eight human breast invasive ductal carcinoma specimens were collected from surgically removed tumor tissues at Luzhou Medical College Affiliated Hospital in Luzhou, China. All patients were Chinese women from western China and aged from 33 to 72 years. The tumor tissues were immediately frozen in liquid nitrogen and stored at -80°C. Tumor tissue lysates were prepared after homogenizing the tissues in a lyses buffer containing a protease inhibitor cocktail. Fifty milligrams of each tissue lysate was analyzed by western blotting using antibodies against TWIST, DLX4 and tubulin.
Immunohistochemical staining
Immunohistochemistry was described previously (2). Sections (5 μm thick) were deparaffinized in xylene and rehydrated in graded alcohols and distilled water. After antigen retrieval, endogenous peroxidase activity was quenched with 0.3% hydrogen peroxide in methanol for 30 min followed by rehydration in 1×PBS and incubation with 5% rabbit serum for 30 min to block nonspecific antigens. The slides were then incubated overnight at 4°C with antibodies against TWIST (ab 50887, Abcam) or DLX4 (sc-18146, Santa Cruz) in Tris-buffered saline containing 2% serum and 1% bovine serum albumin (BSA). This was followed by incubation with secondary anti-IgG at dilution of 1:200 for 30 min at room temperature followed by peroxidase-conjugated avidin-biotin complexes and 3,3'-diaminobenzidine (DAKO, Glostrup, Denmark). The sections were then counterstained with Mayer's hematoxylin and analyzed by standard light microscopy. Sections were incubated with Tris-buffered saline containing 2% rabbit serum and 1% BSA without primary antibody as negative controls.
Statistical analysis
Paired Student's t-test was used to determine any significant differences. P-value ≤ 0.05 was considered to be significant.
Results
DLX4 upregulates TWIST expression
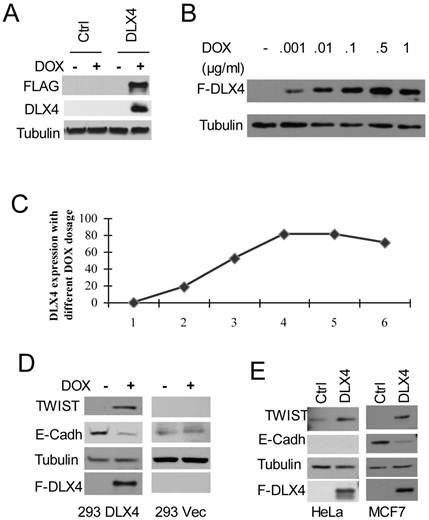
To test whether DLX4 upregulates TWIST expression, we generated stably inducible HEK293 cell lines expressing FLAG-tagged DLX4 (HEK293DLX4 or DLX4) or a control with the empty FLAG-tag vector (293Vec or Ctrl) (Figure 1A). To test the effect of DOX dosage in the HEK293DLX4 cell line, cells were treated overnight with DOX at 0, 0.001, 0.01, 0.1, 0.5 or 1 μg/ml. We observed that addition of DOX at 0.1μg/ml gave the highest DLX4 induction; further increasing the amount of DOX did not increase DLX4 induction any more (Fig. 1B, C). Therefore, 0.1μg/ml DOX was used for the following experiments. To test whether DLX4 upregulates TWIST expression in our established HEK293DLX4 cell line, we performed western blots for FLAG-DLX4 and TWIST and found that TWIST expression was increased upon induction of DLX4 by DOX in HEK293DLX4 cells (Figure 1D, left panel), but not in HEK293Vec cells with or without DOX addition (Figure 1D, right panel). We performed RT-PCR to determine whether the increase in TWIST protein was due to increased protein stability or increased mRNA expression. The RT-PCR analysis showed that the DLX4 mRNA level was increased in 293DLX4 cells, but not in HEK293Vec cells with addition of DOX (Supplementary Material: Figure S1 & S2). To further test whether DLX4 upregulates TWIST expression in general, other two cancer cell lines, the TWIST-negative breast cancer cell line MCF-7 and the TWIST-positive cell line HeLa, were transiently transfected with pCDNA5/DLX4 (DLX4) or the control vector (Ctrl). Two days later, whole cell extracts were prepared for western blotting. The results showed that TWIST expression was increased by DXL4 in both cell lines (Figure 1E, left panel: HeLa; right panel: MCF-7). Taken together, our results clearly demonstrate that DLX4 promotes expression of TWIST.
DLX4 upregulates TWIST expression, which correlates with E-Cadherin downregulation. A. Establishment of FLAG-tagged DLX4 inducible cell lines. Inducible HEK293 cells with F-DLX4(293DLX4) or empty vector control (Ctrl, 293Vec) were treated overnight with vehicle or Doxycyclin (DOX). Western blotting was performed with whole cell lysates and antibodies against FLAG, DLX4 or Tubulin. F-DLX4 protein was only detected in 293 DLX4 cells after DOX treatment. B. Induction effect at different dosages of DOX in the 293DLX4 cell line. DOX was added to the culture media at 0, 0.001, 0.01, 0.1, 0.5 or 1 μg/ml. Western blotting was performed with whole cell lysates and antibodies against FLAG or Tubulin. C. Induction curve. Band intensities were determined by densitometry and normalized to the tubulin band intensity. D. DLX4 upregulates TWIST protein expression. Expression of TWIST and DLX4 was assayed by western blotting in inducible 293DLX4 and 293 vector control cells. E. Western blotting for forced expressed of DLX4 in MCF7 breast cancer cells (left panel) or HeLa cells (right panel) with indicated antibodies (FLAG antibody as F-DLX4).
E-Cadherin suppression may be mediated by DLX4 through upregulation of TWIST
Previous reports that showed TWIST overexpression can induce mesenchymal markers, and repress epithelial markers, for example E-cadherin, expression (1, 6, 29, 30). Therefore, western blotting for E-Cadherin was also performed using the above mentioned three different cell lines with forced DLX4 expression. As we predicted, E-Cadherin expression was decreased in HEK293DLX4 cell line after addition of DOX, but not in HEK293 control cells (Figure 1D) or the TWIST-negative breast cancer cell line MCF-7 (Figure 1E, right panel). These data support the idea that E-Cadherin suppression is mediated by DLX4 through upregulation of TWIST. We should note that we didn't detect any E-cadherin expression in HeLa cells due to the genomic deletion (Figure 1D, data not shown). In HeLa cells, we can detect TWIST upregulation, but cannot study its effect on E-Cadherin (Figure 1E, left panel).
DLX4 binds to the promoter region and last exon of TWIST gene
We searched for putative DLX4 binding sites (concensus = 5'ATATATATG 3') in the exons and introns of the TWIST gene and up to 10 kb upstream of the transcriptional start site (TSS) (Fig. 2A). Although we found no consensus DLX4 binding sites, a similar sequence (AAATATATG) was located in the last exon of TWIST. We designed primers for chromosome immunoprecipitation (ChIP) spanning the AAATATATG site and six regions within 7.5 kb upstream of the TSS to experimentally search for DLX4 binding sites (Supplementary Material: Table S1). 293DLX4 and 293Vec cells were treated with DOX for 16 h and then processed for ChIP using an antibody against the FLAG-Tag. The results showed that 293DLX4 cells had a significantly stronger signal than 293Vec (Ctrl) at the sites of primer pairs 1 (Fig. 2B) and primer pairs 2 (Fig. 2C); no binding was detected at the sites of primer pairs 3 and 4 (Fig. 2D). These data suggest that DLX4 associates with a region 0.6 kb upstream of the transcriptional start site (promoter region) and the last exon (+1.6kb relative to the TSS) of the TWIST gene.
DLX4 binds to the regulatory region of the TWIST gene. A. TWIST gene structure and position of primers for ChIP assays. B, C &D. ChIP assays were performed with chromatin extracts from 293 DLX4 and control cells and anti-FLAG M2 beads for immunoprecipitation. PCR was performed with primer pair 1 (B), primer pair 2 (C) or primer pairs 3 and 4 (D). E. Endogenous DLX4 binds to the TWIST gene promoter region. Left panel: A ChIP assay was performed using the DMA-MB-435 cell line. 10% of input was used as a positive control. As a negative control, the immunoprecipitation was performed with normal mouse IgG. Right panel: Western blots to assay DLX4 and Tubulin protein expression in whole cell extracts from JEG-3, MDA-MB-435 (MDA-435) or HeLa cell lines. F. DLX4 expression enhances Twist reporter gene activity. 239Vec and 293DLX4 inducible cells were treated with DOX or vehicle. The activity of the pGL3-hTwist-Luc reporter gene was determined by luciferase assay. G. HeLa cells were cotransfected with the indicated amounts of DLX4 expression vector and a Twist promoter luciferase reporter vector. Luciferase activity of each sample was normalized to the β-gal activity. All experiments were performed in triplicates. **, p<0.001.
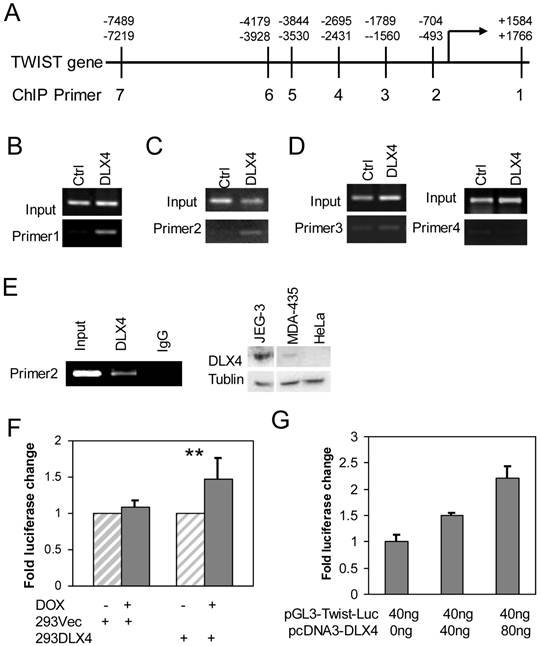
We next confirmed the direct binding of DLX4 to the TWIST gene by performing ChIP assays using a DLX4 antibody with extracts from MDA-MB-435 cells, which endogenously express both DLX4 and TWIST (Fig.2E, right panel and data not shown) (2). The results showed that endogenous DLX4 directly bound to the upstream regulatory region (-0.6kb). No signal was detected for the same region when the ChIP was performed with normal IgG (Fig.2E, left panel).
We next tested whether DLX4 could regulate a TWIST-Luciferase reporter vector containing 2.3-kb of the upstream promoter region (pGL3-hTwist-Luc) (28), including our ChIP-identified DLX4-binding site. The pGL3-hTwist-Luc reporter was transfected into 293Vec or 293DLX4 cells with or without DOX induction. The results showed that DOX-induced DLX4 expression enhanced reporter gene activity 1.5-fold in 293DLX4 cells. No effect was observed in 293Vec cells, which indicated that DOX did not non-specifically affect transcription from the TWIST promoter (Fig. 2F). Finally, we co-transfected HeLa cells, which are DLX4-negative, with the pGL3-hTwist-Luc reporter and increasing amounts of the pcDNA-DLX4 expression vector as indicated in Fig. 2F. The results showed that DLX4 expression enhanced reporter gene activity in a dose-dependent manner (Fig. 2G).
DLX4 promotes migratory and invasive ability in cancer cell lines
To examine whether overexpression of DLX4 promotes migratory and invasive activity in cancer cell lines, cell wound-healing, migration and invasion assays were performed using 293DLX4 and 293Vec cell lines. When cell motility was examined by the wound-healing assay, we observed that 293DLX4 (DOX+) cells more rapidly closed the wound than 293DLX4 (DOX-) cells (Fig. 3A). Overexpressing DLX4 by DOX treatment increased the migratory ability of 293DLX4 cells nearly twofold (Fig.3B) and stimulated invasive ability more than threefold (Fig.3C) in Transwell assays. DOX had no effect on cell migration or invasion in cells containing the empty vector (293Vec).
DLX4 promotes migratory and invasive activity in cancer cell lines. A. Wound-healing assay. HEK293-DLX4 cells were plated in a six-well dish. Then linear wounds were created by scraping confluent cell monolayers after the cells were treated with DOX or vehicle for 12 h. The “wounded” areas were examined at the indicated time points under 100× magnification. B. Cell migration assay. **, p<0.001. C. Cell invasion assay. **, p<0.001. D. Cell proliferation assay. Migration and invasion assays were performed using HEK293-DLX4 and the empty vector control cells with or without DOX induction using transwell plates described in the Materials & Methods section. Cell proliferation was assayed using a CCK-8 kit. D&E. Cell migration and invasion assays in HeLa cell over-expressing of pcDNA3-DLX4 or pcDNA3 empty vector. **, p<0.001. G. Representative migration assay image at Fig. 3D.
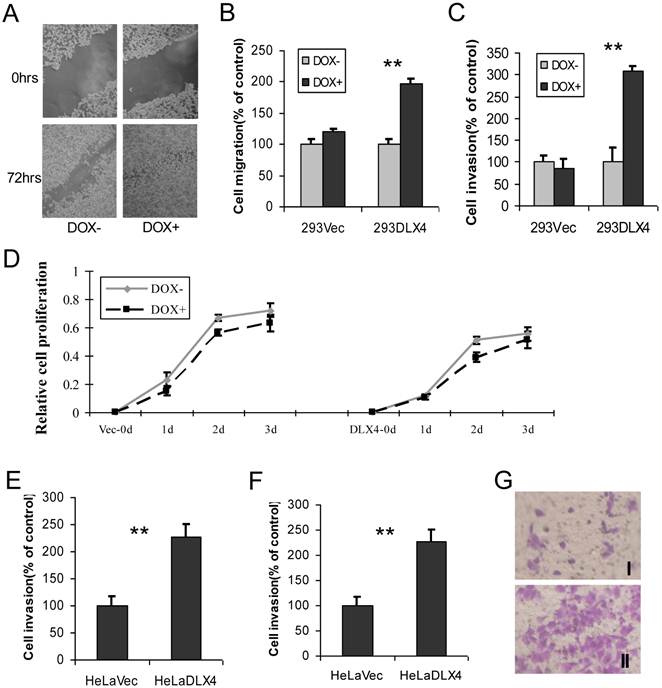
Next, cell migration and invasion assays were carried out using HeLa cells transfected with pcDNA3-DLX4 or empty vector control. Overexpression of DLX4 increased migratory activity 1.7-fold (Fig.3E) and stimulated invasive activity 2.4-fold (Fig.3F). The result at Fig.3G showed representative images of migration assay.
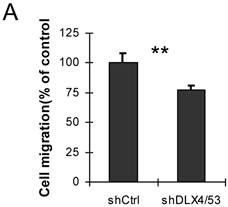
DLX4 was previously shown to play a role in tumor cell growth in some cells, for example, in ovarian cancer cells in low serum (12). To examine whether DLX4 expression in the 293DLX4 cell line also promotes cell growth, a cell proliferation assay was performed using a CCK-8 (Cell Counting Kit-8) from Beyotime according to the manufacturer's protocol. Surprisingly, the results showed that both 293Vec and 293 DLX4 cells had slightly reduced cell proliferation following DOX addition (Fig. 3D). On the other hand, knockdown DLX4 in the highly aggressive, TWIST- and DLX4-positive breast cancer cell line MDA-MB-231 (2, 19) reduced cell migratory ability (Fig. 4A, Supplementary Material: Figure S3 & S4). Taken together, these results support the idea that DLX4 promotes migratory and invasive activity in cancer cell lines.
Knockdown of DLX4 in MDA-MB231 cells reduces cell migration activity. A. Migration assay. The migration assays were performed using MDA-MB-231 cells with shRNA-mediated knockdown of human DLX4 (shDLX4/53) or the empty vector control (Ctrl). **, p<0.001.
The relationship between DLX4 and TWIST expression in human breast tumor tissues
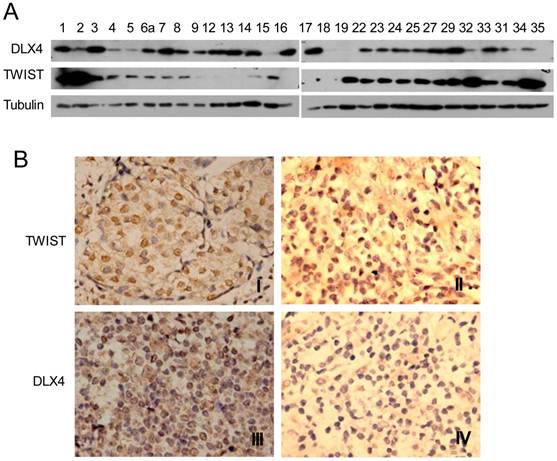
DLX4 is widely expressed in leukemias, lung, breast, ovarian and prostate cancers (12-16) and correlates with breast cancer progression and invasion (14, 15). However, the relationship between TWIST and DLX4 is poorly understood in human breast cancer tissues. In order to investigate the relationship between TWIST and DLX4 in human breast tumors, a total of 28 invasive ductal breast carcinomas from patients were collected and assayed for TWIST and DLX4 expression by western blotting and immunohistochemistry (IHC). The western blot results are shown in Fig.5A. Representative images from the IHC are shown in Fig.5B and summarized in Table 1. Of 28 patients, 24 patients (85.7%) had detectable or high levels of DLX4 expression in breast tumors, consistent with previous reports (11, 17). 16 tumors (57.14%) exhibited detectable or high levels of both TWIST and DLX4 proteins, 2 tumors (7.14%) exhibited undetectable levels of both TWIST and DLX4 proteins, 3 tumors (10.7%) exhibited a relatively low DLX4 and high TWIST, 5 tumors (17.86%) exhibited a high level of DLX4 and an undetectable level of TWIST, 2 tumors (7%) exhibited undetectable DLX4 and high TWIST. In summary, 64.3% of patients showed a highly positive correlation between expression of DLX4 and TWIST. 10.7% of patients exhibited low DLX4 and high TWIST expression. 25% of patients had a reverse correlation between expression of DLX4 and TWIST (Table 1). These data support the idea that the expression of TWIST and DLX4 are positively correlated in most (64.3%) of the invasive breast carcinomas.
DLX4 and TWIST expression analysis in human breast invasive ductal carcinomas
| DLX4 | TWIST | No. Cases | Percentage (%) |
|---|---|---|---|
| +++/++ | +++/++ | 16 | |
| - | - | 2 | 64.3 |
| -/+ | +++ | 3 | 10.7 |
| +++/++ | - | 5 | |
| - | +++/++ | 2 | 25 |
Expression of DLX4 and TWIST in human breast tumor tissues. A. DLX4 and TWIST expression in human breast tumor tissues. Tumor tissues from 28 patients with invasive ductal breast tumors were collected, whole tissue extracts were prepared and western blotting analysis was performed with the indicated antibodies. Note that 24 of 28 samples were DLX4 positive (85.7%). B. Representative immunohistochemical analysis of TWIST and DLX4 proteins in invasive ductal breast tumors. TWIST protein expression was positively correlated with DLX4 expression in most of the breast tumors. Note that I and III were TWIST and DLX4 expression positive in one tissue, whereas II and IV were TWIST and DLX4 negative respectively in others.
Discussion
TWIST is a class II member of the highly conserved family of basic helix-loop-helix (bHLH) transcription factors and its upregulation correlates with low E-cadherin expression, high cancer aggressiveness and poor survival rate (1, 3). TWIST can regulate EMT indirectly through other downstream target genes, e.g., TWIST can activate PDGFRalpha, Snail1 or Snail2 to promote EMT and metastasis (8, 31, 32). TWIST can regulate EMT indirectly through binding to the E-cadherin promoter and suppressing transcription (26, 33, 34). Overexpression of TWIST induces chromosomal instability in breast cancer (4). TWIST upregulates AKT2 expression in breast cancer through the PI3K/AKT pathway (35). We and others recently demonstrated that TWIST represses ERα expression in breast cancer cells through a TWIST/NuRD (6) or Twist1-HDAC1-DNMT3B repressor complex (2, 29), which contributes to hormone resistance (29). Expression of TWIST alone can only induce partial EMT in some cell lines, but expression of both TWIST and H-Rasv12 or ErbB2 can induce complete EMT and override premature senescence (36).
TWIST is regulated by several upstream regulators or pathways for tumor metastasis. TWIST is a direct target of NF-κB and regulated by a negative feedback mechanism (37). TWIST is directly regulated by HIF-1α, inducing metastasis through EMT (33, 38). The Wnt pathway regulates TWIST function by inhibiting mouse mammary cell differentiation in mouse cell lines (39). SRC-1/PEA3 regulates TWIST expression and promotes breast cancer metastasis (28). CHIP suppresses tumor progression in human breast cancer by inhibiting oncogenic pathways. SRC-3 positively regulates TWIST and SRC-3 is a direct target for ubiquitylation and degradation by CHIP (40). MiR-580, CPEB1 and CPEB2 act as negative regulators of TWIST1 expression translationally in a sequence-specific and additive/cooperative manner in MCF-10A cell lines (41). All these data suggest that TWIST is regulated in a complex manner by multiple pathways and highlight the importance of studying how the molecular regulation of TWIST gene expression contributes to tumor metastasis.
In this study, multiple lines of evidence suggested that DLX4 is an upstream regulator of TWIST. First, DLX4 upregulated TWIST expression in the MCF-7 breast cancer cell line as well as other cell lines, including HeLa cell and HEK293. Subsequently, repression of E-Cadherin expression enhanced migratory and invasive capacity. Second, the results of our ChIP experiments suggested that DLX4 positively regulates TWIST gene expression by binding to a region upstream of the TSS (-0.6kb). However, we cannot exclude the possibility that other DLX4 binding sites exist. In the future, ChIP-sequencing assays will provide us more comprehensive information by helping to map DLX4 binding across the whole genome. Third, over-expression of DLX4 promoted cell migration and invasion, whereas knockdown of DLX4 decreased the migratory ability of cancer cell lines.
Finally, TWIST expression is correlated with DLX4 expression, at least in part, in invasive ductal breast tumor samples. We collected invasive ductal breast carcinomas from 28 patients and examined the expression of both DLX4 and TWIST by western blotting and IHC. Consistent with previous reports, 24 samples (85.7%) had detectable or high levels of DLX4 expression (11, 17). 64.3% of samples showed a highly significant correlation between expression of DLX4 and TWIST protein, including 57.14% of tumors with detectable or high levels of both TWIST and DLX4 proteins, 7.14% tumors with undetectable level of both TWIST and DLX4 proteins. 10.7% of samples exhibited low DLX4 and very high TWIST expression. Only 25% of samples showed a reverse correlation between DLX4 and TWIST expression.
The pathological data from our tumor collection showed that all the tumors were larger than 2 cm in diameter (mainly from 2 to 5 cm, data not shown), which is bigger than that reported in western countries (< 2cm, data not shown). Thus, the samples we collected are more likely to represent late stage breast tumors. We would like to comment that it is very important for earlier diagnosis and earlier treatment for breast cancer in developing countries. Due to the small sample size, we cannot draw a definitive conclusion and we are in the process of collecting more such samples and conducting systematic research. Based on currently available information, we expect that either DLX4 or TWIST alone or in combination could be used as a diagnostic marker of cancer stages.
In summary, our findings support the idea that DLX4 can drive tumor metastasis through a pathway involving upregualtion of TWIST, which in turn drives EMT. The expression of TWIST and DLX4 proteins are positively correlated in most of the invasive breast carcinomas. Although further functional studies are necessary to confirm the relationship between DLX4 and TWIST, elucidating the role of DLX4 in regulating TWIST and metastasis is an important step toward understanding the molecular biology of cancer metastasis. These data have also opened a new line of research to explore whether DLX4 could be used as a novel pharmaceutical target for inhibiting breast cancer metastasis.
Supplementary Material
Figures S1-S4, Table S1, and Supplementary Methods.
Competing Interests
The authors have declared that no competing interest exists.
Acknowledgements
This work was supported by National Natural Science Foundation of China (81172049, 30371493), Scientific Research Foundation of Education Bureau of Sichuan Province (10ZA038), Key Research Project of Luzhou City (2011-S-30) and a Starting Package of Luzhou Medical College to J.F. We thank Drs. T Takahashi, J Xu, J Wong, W Long, Z Yang, Ms. J Liu and members of the Research Center for Preclinical Medicine of Luzhou Medical College for materials and experimental assistance.
References
1. Yang J, Mani SA, Donaher JL. et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927-39
2. Fu J, Zhang L, He T. et al. TWIST Represses Estrogen Receptor-alpha Expression by Recruiting the NuRD Protein Complex in Breast Cancer Cells. Int J Biol Sci. 2012;8:522-32
3. Qin Q, Xu Y, He T, Qin C, Xu J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012;22:90-106
4. Mironchik Y, Winnard PT Jr, Vesuna F. et al. Twist overexpression induces in vivo angiogenesis and correlates with chromosomal instability in breast cancer. Cancer Res. 2005;65:10801-9
5. Cheng GZ, Zhang W, Wang LH. Regulation of cancer cell survival, migration, and invasion by Twist: AKT2 comes to interplay. Cancer Res. 2008;68:957-60
6. Fu J, Qin L, He T. et al. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 2011;21:275-89
7. Mani SA, Guo W, Liao MJ. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704-15
8. Eckert MA, Lwin TM, Chang AT. et al. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell. 2011;19:372-86
9. Haga SB, Fu S, Karp JE. et al. BP1, a new homeobox gene, is frequently expressed in acute leukemias. Leukemia. 2000;14:1867-75
10. Chase MB, Fu S, Haga SB. et al. BP1, a homeodomain-containing isoform of DLX4, represses the beta-globin gene. Mol Cell Biol. 2002;22:2505-14
11. Fu Y, Lian Y, Kim KS. et al. BP1 Homeoprotein Enhances Metastatic Potential in ER-negative Breast Cancer. J Cancer. 2010;1:54-62
12. Hara F, Samuel S, Liu J, Rosen D, Langley RR, Naora H. A homeobox gene related to Drosophila distal-less promotes ovarian tumorigenicity by inducing expression of vascular endothelial growth factor and fibroblast growth factor-2. Am J Pathol. 2007;170:1594-606
13. Schwartz AM, Man YG, Rezaei MK, Simmens SJ, Berg PE. BP1, a homeoprotein, is significantly expressed in prostate adenocarcinoma and is concordant with prostatic intraepithelial neoplasia. Mod Pathol. 2009;22:1-6
14. Man YG, Schwartz A, Levine PH, Teal C, Berg PE. BP1, a putative signature marker for inflammatory breast cancer and tumor aggressiveness. Cancer Biomark. 2009;5:9-17
15. Man YG, Fu SW, Schwartz A, Pinzone JJ, Simmens SJ, Berg PE. Expression of BP1, a novel homeobox gene, correlates with breast cancer progression and invasion. Breast Cancer Res Treat. 2005;90:241-7
16. Yu M, Wan Y, Zou Q. Prognostic significance of BP1 mRNA expression level in patients with non-small cell lung cancer. Clin Biochem. 2008;41:824-30
17. Fu SW, Schwartz A, Stevenson H. et al. Correlation of expression of BP1, a homeobox gene, with estrogen receptor status in breast cancer. Breast Cancer Res. 2003;5:R82-7
18. Sun YY, Lu M, Xi XW. et al. Regulation of Epithelial-Mesenchymal Transition by Homeobox Gene DLX4 in JEG-3 Trophoblast Cells: A Role in Preeclampsia. Reprod Sci. 2011;18:1138-45
19. Kluk BJ, Fu Y, Formolo TA. et al. BP1, an isoform of DLX4 homeoprotein, negatively regulates BRCA1 in sporadic breast cancer. Int J Biol Sci. 2011;6:513-24
20. Trinh BQ, Barengo N, Naora H. Homeodomain protein DLX4 counteracts key transcriptional control mechanisms of the TGF-beta cytostatic program and blocks the antiproliferative effect of TGF-beta. Oncogene. 2011;30:2718-29
21. Castronovo V, Kusaka M, Chariot A, Gielen J, Sobel M. Homeobox genes: potential candidates for the transcriptional control of the transformed and invasive phenotype. Biochem Pharmacol. 1994;47:137-43
22. Wu X, Chen H, Parker B. et al. HOXB7, a homeodomain protein, is overexpressed in breast cancer and confers epithelial-mesenchymal transition. Cancer Res. 2006;66:9527-34
23. Yoshida H, Broaddus R, Cheng W, Xie S, Naora H. Deregulation of the HOXA10 homeobox gene in endometrial carcinoma: role in epithelial-mesenchymal transition. Cancer Res. 2006;66:889-97
24. Tomida S, Yanagisawa K, Koshikawa K. et al. Identification of a metastasis signature and the DLX4 homeobox protein as a regulator of metastasis by combined transcriptome approach. Oncogene. 2007;26:4600-8
25. Fu J, Jiang J, Li J. et al. Deleted in breast cancer 1, a novel androgen receptor (AR) coactivator that promotes AR DNA-binding activity. J Biol Chem. 2009;284:6832-40
26. Fu J, Qin L, He T. et al. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 2011;21:275-89
27. Fu J, Yoon HG, Qin J, Wong J. Regulation of P-TEFb elongation complex activity by CDK9 acetylation. Mol Cell Biol. 2007;27:4641-51
28. Qin L, Liu Z, Chen H, Xu J. The steroid receptor coactivator-1 regulates twist expression and promotes breast cancer metastasis. Cancer Res. 2009;69:3819-27
29. Vesuna F, Lisok A, Kimble B. et al. Twist contributes to hormone resistance in breast cancer by downregulating estrogen receptor-alpha. Oncogene. 2012;31:3323-34
30. Hong J, Zhou J, Fu J. et al. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011;71:3980-90
31. Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res. 2011;71:245-54
32. Smit MA, Geiger TR, Song JY, Gitelman I, Peeper DS. A Twist-Snail axis critical for TrkB-induced epithelial-mesenchymal transition-like transformation, anoikis resistance, and metastasis. Mol Cell Biol. 2009;29:3722-37
33. Yang MH, Hsu DS, Wang HW. et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol. 2010;12:982-92
34. Vesuna F, van Diest P, Chen JH, Raman V. Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochem Biophys Res Commun. 2008;367:235-41
35. Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007;67:1979-87
36. Ansieau S, Bastid J, Doreau A. et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell. 2008;14:79-89
37. Sosic D, Richardson JA, Yu K, Ornitz DM, Olson EN. Twist regulates cytokine gene expression through a negative feedback loop that represses NF-kappaB activity. Cell. 2003;112:169-80
38. Yang MH, Wu MZ, Chiou SH. et al. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008;10:295-305
39. Howe LR, Watanabe O, Leonard J, Brown AM. Twist is up-regulated in response to Wnt1 and inhibits mouse mammary cell differentiation. Cancer Res. 2003;63:1906-13
40. Kajiro M, Hirota R, Nakajima Y. et al. The ubiquitin ligase CHIP acts as an upstream regulator of oncogenic pathways. Nat Cell Biol. 2009;11:312-9
41. Nairismagi ML, Vislovukh A, Meng Q. et al. Translational control of TWIST1 expression in MCF-10A cell lines recapitulating breast cancer progression. Oncogene. 2012 [Epub ahead of print]
Author contact
![]() Corresponding author: Dr. Junjiang Fu, Key Laboratory of Epigenetics and Oncology, The Research Center for Preclinical Medicine, Luzhou Medical College, Luzhou, Zhongshan Rd 3-319, Luzhou City, China, Tell: 86-830-3160283, Email: fujunjiangcom. Or to: Dr. Yongxiang Zhao, yongxiang_zhaocom
Corresponding author: Dr. Junjiang Fu, Key Laboratory of Epigenetics and Oncology, The Research Center for Preclinical Medicine, Luzhou Medical College, Luzhou, Zhongshan Rd 3-319, Luzhou City, China, Tell: 86-830-3160283, Email: fujunjiangcom. Or to: Dr. Yongxiang Zhao, yongxiang_zhaocom