Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Summary
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(1):1-15. doi:10.7150/ijbs.5249 This issue Cite
Research Paper
Identification of MicroRNAs in Helicoverpa armigera and Spodoptera litura Based on Deep Sequencing and Homology Analysis
Xie Ge1, 2, Yong Zhang1, Jianhao Jiang1,2, Yi Zhong1, Xiaonan Yang1, Zhiqian Li1,2, Yongping Huang1, Anjiang Tan1, ![]()
1. Key Laboratory of Insect Developmental and Evolutionary Biology, Institute of Plant Physiology and Ecology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, 200032, P.R. China;
2. Graduate School of Chinese Academy of Sciences, Shanghai, 200032, P.R. China.
Received 2012-9-18; Accepted 2012-11-21; Published 2012-12-1
Abstract
The current identification of microRNAs (miRNAs) in insects is largely dependent on genome sequences. However, the lack of available genome sequences inhibits the identification of miRNAs in various insect species. In this study, we used a miRNA database of the silkworm Bombyx mori as a reference to identify miRNAs in Helicoverpa armigera and Spodoptera litura using deep sequencing and homology analysis. Because all three species belong to the Lepidoptera, the experiment produced reliable results. Our study identified 97 and 91 conserved miRNAs in H. armigera and S. litura, respectively. Using the genome of B. mori and BAC sequences of H. armigera as references, 1 novel miRNA and 8 novel miRNA candidates were identified in H. armigera, and 4 novel miRNA candidates were identified in S. litura. An evolutionary analysis revealed that most of the identified miRNAs were insect-specific, and more than 20 miRNAs were Lepidoptera-specific. The investigation of the expression patterns of miR-2a, miR-34, miR-2796-3p and miR-11 revealed their potential roles in insect development. miRNA target prediction revealed that conserved miRNA target sites exist in various genes in the 3 species. Conserved miRNA target sites for the Hsp90 gene among the 3 species were validated in the mammalian 293T cell line using a dual-luciferase reporter assay. Our study provides a new approach with which to identify miRNAs in insects lacking genome information and contributes to the functional analysis of insect miRNAs.
Keywords: microRNA, deep sequencing, homolog analysis, Helicoverpa armigera, Spodoptera litura, Bombyx mori.
Introduction
MicroRNAs (miRNAs) are approximately 22 nucleotide (nt) long, single-stranded, endogenous, small non-coding RNAs. miRNAs are processed into double-stranded complexes by Drosha and Dicer from hairpin precursors transcribed from the genome by RNA polymerase II. One of the strands binds to Argonaute to form an RNA-induced silencing complex (RISC), which guides the complex to target mRNAs to direct translational silencing or mRNA degradation [1, 2]. The remaining strand, called the miRNA-star strand (miR*), either is degraded or accumulates at low levels in most cases [3].
In recent years, numerous miRNAs have been identified in plants, animals, and viruses [4-6]. In miRBase Release 18 (http://www.mirbase.org/, released on November, 2011), the miRNAs of 168 species were published, including 24 insect species. Although miRNAs such as cel-let-7 and cel-lin-4 were identified by a forward genetic method [7-9], most miRNAs have been identified by a combination of RNA sequencing and in silico prediction methods [10, 11]. However, the lack of sufficient genome information in most species, especially in non-model species, has limited the further identification of a wider range of miRNAs.
The cotton bollworm (Helicoverpa armigera) and the cotton leafworm (Spodoptera litura) are serious lepidopteran pests of various crops. Because miRNAs are involved in regulating development and metamorphosis in insects [4, 12-18], the identification of miRNAs in these two pests will help to uncover their physiological roles and provide potential targets for pest control. In S. litura, many miRNAs have been identified by computational prediction or stem-loop polymerase chain reaction (PCR) [19, 20]. However, deficient genome sequences have restricted the further identification of miRNAs. Additionally, no miRNAs have been identified through small RNA sequencing in H. armigera and S. litura. In contrast, the silkworm Bombyx mori, a model lepidopteran insect, has a previously released genome sequence with more than 500 mature miRNA sequences published. Although miRNAs are frequently acquired and lost during evolution, functional miRNAs evolve slowly, maintaining a particularly high homology among closely related species [21-23]. The relatively close relationship among H. armigera, S. litura and B. mori increases the likelihood of identifying conserved miRNAs in H. armigera and S. litura based on B. mori miRNA and genomic sequences.
In this study, we sequenced small RNAs (sRNAs) of H. armigera and S. litura, and identified 97 and 91 conserved miRNAs, respectively, using B. mori miRNAs as references (named har-miRNAs for miRNAs from H. armigera and sli-miRNAs for miRNAs from S. litura). We also identified 1 novel har-miRNA, 8 har-miRNA candidates and 4 sli-miRNA candidates by computational prediction based on the B. mori genome and H. armigera BAC sequences. A comparison of these miRNAs with those from other species indicated that most of the identified miRNAs were insect-specific, with many being Lepidoptera-specific. Quantitative reverse transcription PCR (qRT-PCR) was performed to investigate the expression profiles of 4 miRNAs, and the results revealed their potential roles in insect development. miRNA target prediction revealed that conserved miRNA target sites exist in various genes among the 3 species. In addition, 3 conserved miRNA targets of the Hsp90 gene were validated in a mammalian 293T cell line using a dual-luciferase reporter assay. The present study not only provides a new strategy for miRNA identification in insect species lacking genome information, but also presents insights into the conservation and functions of lepidopteran miRNAs.
Materials and Methods
Insect strains
H. armigera and S. litura strains were maintained in our laboratory at the Shanghai Institute of Plant Physiology and Ecology and were reared on an artificial diet under a 28°C temperature and a Light-Dark 14:10 photoperiod.
Small RNA sequencing
Total RNA was isolated from whole-body homogenates of different developmental stages of H. armigera and S. litura using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions.
Small RNAs were sequenced with an Illumina Genome Analyzer (Illumina, SanDiego, CA, USA) at the Beijing Genomics Institute (BGI, Shenzhen, Guangdong, China). First, total RNA was size-fractionated and 10-30 nt sRNAs were isolated. Subsequently, 5' and 3' adaptors were ligated to the sRNAs, and double-stranded nucleic acids were acquired after reverse transcription PCR (RT-PCR). The PCR products were sequenced using Solexa's sequencing-by-synthesis (SBS) technology, which is a high-throughput sequencing method. The 35 nt sequence tags from the Solexa sequencing were subjected to a primary analysis in which low-quality tags and adaptor contaminants were discarded.
miRNA prediction
Using the B. mori miRNAs presented in miRBase Release 18 (http://www.mirbase.org/) as references, sequences that aligned perfectly with precursor miRNAs and aligned to mature miRNAs with forward matching were annotated as conserved miRNAs in H. armigera and S. litura. To identify other types of sRNAs, we used sequences from SilkDB (SilkDB, http://silkworm.genomics.org.cn/) [24, 25], NCBI (http://www.ncbi.nlm.nih.gov/), Rfam (http://rfam.sanger.ac.uk/) and RepBase (http://www.girinst.org/repbase/) as references to annotate the non-coding RNAs (rRNAs, tRNAs, snRNAs, snoRNAs and repeat-associated small RNAs) and degraded fragments of expressed genes (exons and introns) in the remaining sequences. The BLASTn program was used as the alignment method, and the e-value was set to 10-5. Using the miRNA prediction software Mireap (http://sourceforge.net/projects/mireap/), we predicted novel miRNAs from the remaining sRNAs. B. mori genomic sequences and H. armigera BAC sequences were used as references to provide flanking sequences of the sRNAs for fold-back structure prediction. sRNAs that could not be annotated were classified as unannotated.
Homology analysis of miRNAs
The miRNA sequences in the other species were downloaded from miRBase (Release 18) and the relevant published studies [26-28]. BLASTn was used to compare the H. armigera and S. litura miRNAs with other species. Because all species published in miRBase were analyzed, including species with long evolutionary distances, the e-value was set to 10-3 so that more miRNA homologs could be identified. Reverse-matched sequences were discarded from the BLAST results. The alignments of miRNA sequences were conducted by Mega 4 [29], using IUB as the DNA weigh matrix.
Expression profiles of miRNAs
The expression profiles of miR-2a, miR-34, miR-2796-3p and miR-11 were investigated by qRT-PCR with the miScript Reverse Transcription Kit and a miScript SYBR Green PCR Kit (Qiagen, Valencia, CA, USA) according to the manufacturers' instructions. The monitoring and analysis of the qRT-PCR were performed on an ABI 7900 real-time PCR system (Applied Biosystems, Foster City, CA, USA), and the PCR conditions were as follows: 95°C for 15 minutes for denaturation and 40 cycles of 94°C for 15 seconds, 55°C for 30 seconds and 70°C for 30 seconds. The primers for each miRNA are listed in Supplementary Material: Table S1. The small nuclear RNA U6 of B. mori was used as an internal control.
Homologous miRNA target prediction
The mRNA sequences for H. armigera and S. litura genes were downloaded from NCBI. The 3' untranslated regions (UTRs) were extracted for homology analysis. BLASTn was used to find homologous regions in these 3' UTRs, and the e-value threshold was set to 10-5. The miRNA targets were then predicted in the homologous sequences. 3 miRNA target-prediction software programs were used: miRanda (http://www.microrna.org/microrna/getDownloads.do) [30, 31], PITA (http://genie.weizmann.ac.il/pubs/mir07/mir07_exe.html), and microTar (http://tiger.dbs.nus.edu.sg/microtar/) [32]. The thresholds were set to a score of ≥ 140 for miRanda (default), ddG ≤ 0 for PITA, and energy ≥ 0.5 for microTar. The miRNA targets predicted by at least two of the software programs were selected. Homologous regions in different species targeted by common miRNAs were considered to contain conserved miRNA target sites.
Plasmid construction and dual-luciferase reporters assay
Precursors of bmo-miR-14, bmo-miR-2766 and bmo-miR-9a were amplified (primers listed in Supplementary Material: Table S1) from the B. mori genome DNA with 150-300 bp (base pair) flanking sequences and inserted into the multi-cloning site of pmR-mCherry (Clontech, Mountain View, CA, USA) for miRNA expression. We only cloned B. mori miRNAs because they have high homology with the corresponding miRNAs of H. armigera and S. litura. To estimate the regulation of miRNAs at their target sites, the plasmid phRL-TK (Promega, Madison, WI, USA) was used as a reporter. We amplified the 3' UTRs of BmoHsp90, HarHsp90 and SliHsp90 (primers listed in Supplementary Material: Table S1) and inserted the sequences behind the Renilla luciferase reporter gene in phRL-TK. pGL3 (Promega, Madison, WI, USA) with the Firefly luciferase reporter gene was used as a control to indicate the transfection efficiency.
The mammalian 293T cell line used for the dual-luciferase reporter assay was kindly provided by Dr. Mofang Liu of the Institute of Biochemistry and Cell Biology, Shanghai Institute for Biological Sciences. The cells were cultured in DMEM (high glucose, Gibco, Life Technologies, Grand Island, NY, USA) with 10% FBS. Before transfection, the cells were plated in a 96-well tissue-culture plate. After 24 h (hours), two reporter plasmids (in total 10 ng per well) and the miRNA-expressing plasmids (100 ng per well) were co-transfected using Lipofectamine™ 2000 Transfection Reagent (Invitrogen, Life Technologies, Grand Island, NY, USA). Approximately 24-48 h after the transfection, the Renilla and Firefly luciferase activities were quantified using the Luciferase Assay Reagent II (LAR II) (Promega, Madison, WI, USA) on a Modulus™ single-tube multimode reader (Turner Biosystems, Sunnyvale, CA, USA) according to the manufacturers' instructions. The transfections were performed in triplicate. A paired t-test was used to analyze the significance of the differences.
Results
Identification of conserved miRNAs
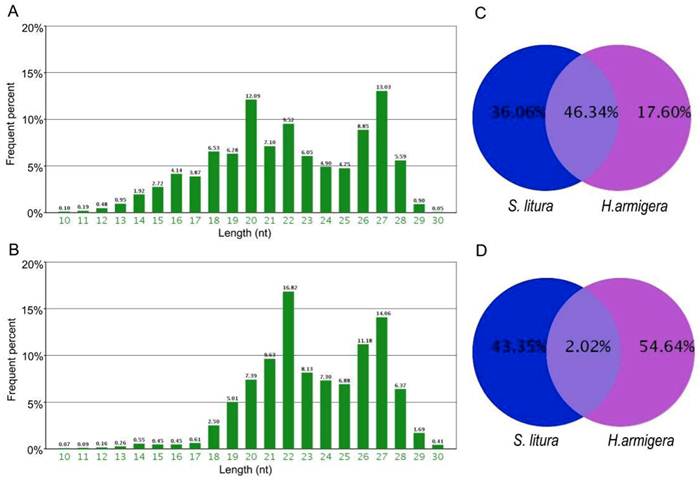
Using high-throughput sequencing, we obtained 5,266,540 total sRNA sequences in H. armigera, and after eliminating the redundant sequences, we obtained 1,125,767 unique sequences. In S. litura, 11,443,493 total sRNA sequences were acquired, corresponding to 901,369 unique sequences. The length of these sRNAs ranged from 10 nt to 30 nt. Their length distributions in both H. armigera and S. litura showed a bimodal distribution. One of the peaks was at approximately 20-22 nt, representing the miRNAs [33], and the other was at approximately 27 nt, which may be the piRNA-like sRNAs [34] (Fig. 1A, 1B). A comparison between the two species showed that only 2.02% of the unique sRNAs are common between these two organisms; however, these shared sRNAs represent 46.34% of the total sRNAs (Fig. 1C and 1D), which means their average number of reads are higher than the sRNAs specific in each species. Such a redundancy of common sRNAs indicates that those sRNAs occurring simultaneously in both insects were expressed much more abundantly than the species-specific sRNAs.
Length distribution and comparison of small RNA tags sequenced in H. armigera and S. litura. The two bar diagrams show the length distribution of the small RNAs in H.armigera (A) and S. litura (B). Comparison of the total (C) and unique (D) small RNAs are presented.
The acquired sRNAs were aligned with known miRNAs, rRNAs, tRNAs, snRNAs, snoRNAs, repeat associated RNAs, introns, and exons. The sRNAs aligned with B. mori miRNA precursors and mature sequences in miRBase Release 18 [35-38] were annotated as miRNAs. The distributions of the sRNAs among different categories are shown in Supplementary Material: Table S2. After alignment, we obtained 923 and 818 unique sRNAs in H. armigera and S. litura, respectively, aligned to 75 known miRs and 25 known miR*s in B. mori (Table 1). Among the sequences aligned to each B. mori miRNA, only the one with the most reads was retained as the final version. We finally acquired 97 conserved miRNAs in H. armigera and 91 in S. litura (Supplementary Material: Table S3).
Known miRNA alignment.
| miRs | miR*s | miRNA precursors | |
|---|---|---|---|
| In Total | 75 | 25 | 78 |
| H. armigera | 74 | 23 | 77 |
| S. litura | 71 | 20 | 74 |
All the B. mori miRNAs in miRBase Release 18 were used as the references. The row “In Total” shows the information about the alignment of all the miRNAs in both H. armigera and S. litura. The three columns summarized the numbers of miRNA sequences from each species aligned to B.mori miRs, miR*s, or miRNA precursors.
Novel miRNA prediction
Here we used BAC sequences of H. armigera and the genomic sequence of B. mori to perform the novel miRNA prediction because no genomic information for H. armigera or S. litura was available and no S. litura BAC sequences were found in NCBI. Using mireap, we identified 10 possible mature miRNAs in H. armigera and 4 in S. litura (Table 2). Among them, har-miR-m1, har-miR-m2 and the 4 S. litura candidate miRNAs were identified from the B. mori genomic sequences, and the other 8 sequences of H. armigera were identified from the H. armigera BAC sequences. Only har-miR-m7 had more than 5 reads, and its complementary miR* was also identified, indicating that it was a novel miRNA. All the other sRNA sequences did not have corresponding miR*s that were identified and thus were classified as miRNA candidates. However, har-miR-m1 and sli-miR-m1 were homologous to each other and thus they may be a common miRNA in these 2 species, with their miR* sequences most likely being expressed at a level too low for detection. In addition, har-miR-m2 can be aligned to miR-193 in some insects other than B. mori, including Heliconius melpomene, which is a lepidopteran species, indicating that this miRNA may also be conserved in insects.
Novel miRNA sequences and numbers of reads in H. armigera and S. litura.
| miRNA | Sequences | Length (nt) | Number of reads |
|---|---|---|---|
| har-miR-m1 | GUGGGGCAAAUUGCGAAAGC | 20 | 16 |
| har-miR-m2 | UACUGGCCUGCUAAGUCCCAA | 21 | 43 |
| har-miR-m3 | GAUCAACUUUCGACUGAUUUUUC | 23 | 6 |
| har-miR-m4 | UCCACAGUACCUGGCGUGUCCGCU | 24 | 9 |
| har-miR-m5 | UGACAUCUCUUUGAGGAGCGCGUG | 24 | 5 |
| har-miR-m6 | AGCAGUGGGACGAUAAAAAGG | 21 | 5 |
| har-miR-m7 | UCUCACGGAUAUAAAAUUUAAGA | 23 | 7 |
| har-miR-m7* | UGUGUCCGUUUUUACAACAAGC | 22 | 1 |
| har-miR-m8 | ACAAAGCGAUUUCGGCCCGAC | 21 | 4 |
| har-miR-m9 | UGUCCGCACAAUGGUGACACCCG | 23 | 3 |
| sli-miR-m1 | GUGGGGCAAAUUGCGAAAGCUG | 22 | 14 |
| sli-miR-m3 | CUCUCACCCAGGAGGCUCGG | 20 | 20 |
| sli-miR-m4 | UACAGCUAGGAUACCAAAGAGA | 22 | 14 |
| sli-miR-m5 | CGGGAGAUGUGGUGUUCGGGA | 21 | 336 |
Nucleotide bias analysis of identified miRNAs
The nucleotide bias at the first position of the identified miRNAs with a certain length was analyzed (Fig. 2 A, B). In both H. armigera and S. litura, the miRNAs showed a dominant bias to uracil (U) at the first nucleotide, especially the miRNAs with a length of 19-23 nt. This observation agrees with the characteristic that miRNAs usually begin with a U at the 5' terminus [39].
First nucleotide bias and position nucleotide bias. The first nucleotide bias of the miRNAs in H. armigera (A) and S. litura (B) was analyzed in combination with the length distribution. In addition, the miRNA nucleotide bias at each position in H.armigera (C) and S. litura (D) was examined.
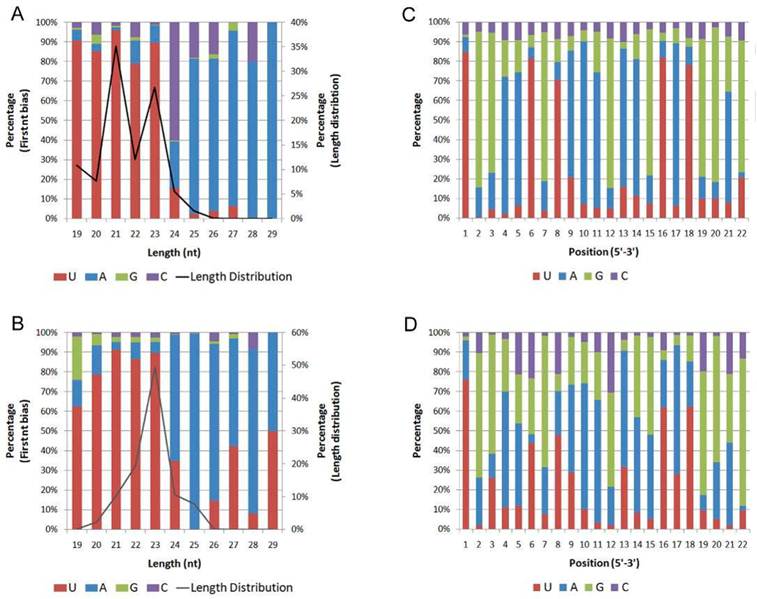
We also analyzed the percentage of the four nucleotides appearing at each position (Fig. 2 C, D). The five positions showing the most dominant bias to U are the 1st, 6th, 8th, 16th, and 18th nucleotides. In canonical cases, the 2nd to the 8th nucleotides of miRNAs, called the "seed region," pair perfectly with their target sites. Some nucleotides in the 3' portion of many miRNAs also show supplementary pairing [40]. Thus, the 1st and 8th nucleotides are at the edges of the “seed region,” the 6th nucleotide is within the “seed region,” and the 16th and 18th nucleotides are near the 5' terminus of the miRNAs. The bias to U at these positions may contribute to the miRNA regulatory mechanism. The nucleotide bias of these miRNAs was quite similar to that described in a previous report on miRNAs [33].
Homology analysis of identified miRNAs
In miRBase Release 18, 562 B. mori miRNAs are included. Among these, only 100 miRNAs were identified in H. armigera and/or S. litura, with 88 being common to all three species (Fig. 3A). The homology of these miRNAs in insects and other animal classes was analyzed. We used BLASTn to align the H. armigera miRNAs (har-miRNAs) and S. litura miRNAs (sli-miRNAs) to all miRNA sequences of other species in miRBase (Release 18), and to the published miRNA sequences in other lepidopteran insects [26-28]. Only metazoan species contain homologs of these miRNAs. We divided the species into four categories: insects, other arthropods (arthropods other than insects), other invertebrates (invertebrates other than arthropods) and vertebrates. Based on the BLAST results, the miRNAs were classified into four types: highly conserved (with homologs in vertebrates), invertebrate-specific (with homologs only in invertebrates), arthropod-specific (with homologs only in arthropods) or insect-specific (with homologs only in insects). 16 miRNAs were highly conserved: let-7, miR-10, miR-100, miR-124, miR-133, miR-137, miR-184, miR-190, miR-1a, miR-210, miR-281, miR-33, miR-7, miR-993a*, miR-993b*, and miR-9a. However, most of the miRNAs were insect-specific. Among those, 29 har-miRNAs and 20 sli-miRNAs had homologs only in lepidopteran species, and thus were considered possible Lepidoptera-specific miRNAs (Supplementary Material: Tables S4 and S5).
Homology analysis of the identified miRNAs. (A) Comparison of identified miRNAs of H. armigera (red) and S. litura (blue) as well as those of B. mori (green). (B) Homology analysis of miRNA sequences homologous to lepidopteran miR-993a* and miR-993b*. The alignment was conducted for all the homologs of har-miR-993a*, har-miR-993b*, sli-miR-993a*, and sli-miR-993b*. The nucleotides are marked in different colors. The names of the miRNA sequences are listed on the left, labeled with their taxonomic order (for insects, marked in red) or phylum (for non-insects, marked in black). The abbreviations represent the species as follows: aae, Aedes aegypti; cqu, Culex quinquefasciatus; dme, Drosophila melanogaster; dps, Drosophila pseudoobscura; dsi, Drosophila simulans; har, Helicoverpa armigera; sli, Spodoptera litura; bmo, Bombyx mori; tca, Tribolium castaneum; dre, Danio rerio; fru, Fugu rubripes; tni, Tetraodon nigroviridis. The dotted line in the middle of the figure separates the miR-993 family members from the miR-10 family members.
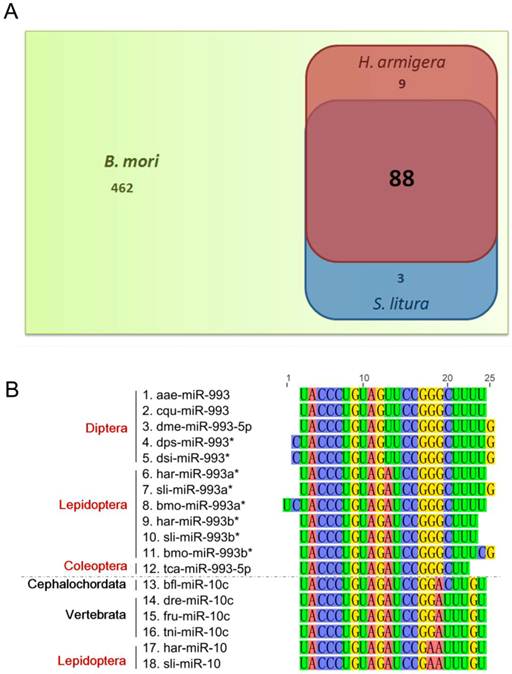
Although occasionally expressed abundantly, miR*s are often degraded or show a low expression level [3, 41, 42]. Therefore, we assumed that miR*s vary more dramatically than miRs among different species. Consistent with our hypothesis, most miR*s were insect-specific (Supplementary Material: Tables S4 and S5). Surprisingly, there were two miR*s in the highly conserved miRNAs, namely, miR-993a* and miR-993b*. However, in the BLAST results of these two miR*s, only miR-10 family members in vertebrates were homologous (Fig. 3B). In fact, there was no miR-993 in vertebrates. In H. armigera and S. litura, miR-10s were also homologous to miR-993a*s and miR-993b*s. Therefore, the miR-993s in invertebrates may be mimics of miR-10s and are most likely in the same miRNA family as miR-10s.
Expression analysis of identified miRNAs
qRT-PCR was used to validate and analyze the expression of two conserved miRNAs - miR-2a and miR-34 - and two insect-specific miRNAs - miR-2796-3p and miR-11. Because the latter 2 miRNAs are expressed remarkably differently between the 2 species, we only chose the species with the higher expression level to analyze the expression profile. Although the expression levels were quite different, all 4 miRNAs could be detected in the relevant species, validating the sequencing results (Fig. 4).
Expression levels of miRNAs at different developmental stages. qRT-PCR was used to analyze the expression levels of miR-2a (A, B), miR-34 (C, D), har-miR-2796-3p (E) and sli-miR-11 (F). The transcript levels were calculated relative to the amount of U6 after normalization. 3 technical replications were performed. The values are the average±SD of the 3 repeats.
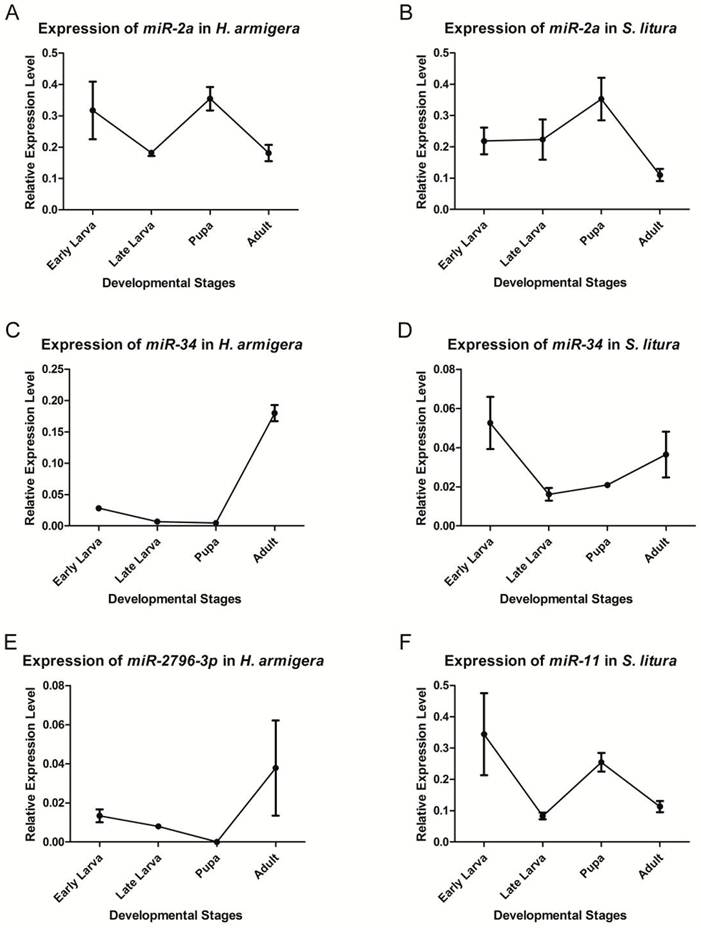
We selected 4 developmental stages - early larva, late larva, pupa and adult - in which to analyze the expression profile. The miRNAs could be detected during every stage of the corresponding species, except that har-miR-2796-3p was not detected in the H. armigera pupal stage, which was most likely due to its low expression level at this stage. All of these miRNAs showed stage-specific expression profiles during development. miR-2a showed a higher expression at the pupal stage in both species, indicating its possible functions at this stage. The expression of miR-34 was more abundant during the early larval and adult stages in both species; however, its level was highest in adults in H. armigera, whereas its expression reached its peak in early larvae in S. litura. har-miR-2796-3p was expressed most abundantly in adults, and sli-miR-11 was highly expressed in the early larval and pupal stages.
Conserved miRNA targets
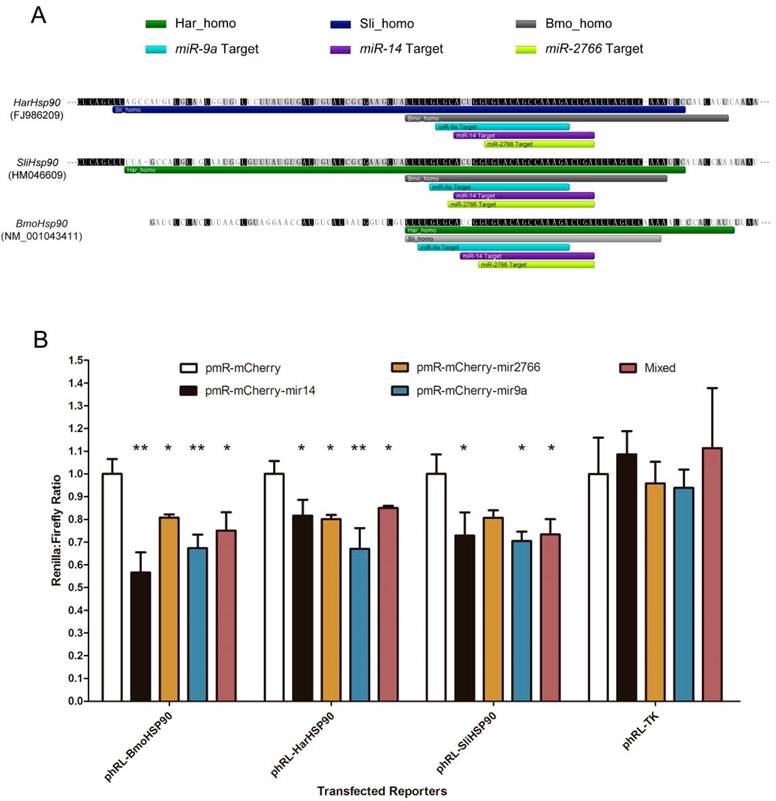
Using 3 miRNA target prediction software programs - miRanda, PITA, and microTar - we predicted the targets of conserved miRNAs in homologous regions in the 3' UTRs of corresponding genes in B. mori, H. armigera and S. litura. The common miRNA targets are listed in Table 3. 19 different genes are involved, including transcription factors, heat shock proteins, and genes involved in hormone pathways and metabolism. The targets predicted by all 3 programs are considered most reliable. Moreover, the miRNA targets can be regarded as highly conserved if they exist in the homologous gene regions of all 3 species. The results show that 3 miRNA targets are highly conserved. All the 3 targets exist in the 3' UTR of Hsp90, and the targeting miRNAs are miR-9a, miR-14 and miR-2766 (Fig. 5A). Using dual-luciferase reporter plasmids to transfect 293T cells, we validated these target sites in the Hsp90 3' UTR. When we over-expressed bmo-miR-9a, bmo-miR-14 and bmo-miR-2766, the relative expression of the reporter gene (Renilla luciferase) exhibited a significant decrease, with a suppression efficiency from approximately 20% to 45% (Fig. 5B). Suppression of bmo-miR-2766 on SliHsp90 is slightly less significant (p-value=0.05104), probably due to the subtle differences between sli-miR-2766 and bmo-miR-2766. Because most miRNAs in animals function in the fine tuning of their target genes [43], a gene down-regulated by miRNAs even at an efficiency of less than 20% can be regarded as a target [44]. Therefore, our results demonstrate the regulation of Hsp90expression by these miRNAs.
Common miRNAs targeting the homologous regions in 3' UTRs of B. mori, H. armigera, and S. litura.
| Gene | ID_har* | Homologous Region In 3'UTR (har)§ | ID_sli* | Homologous Region In 3'UTR (sli)§ | ID_bmo* | Homologous Region In 3'UTR (bmo)§ | Common miRNA Targets¶ |
|---|---|---|---|---|---|---|---|
| 14-3-3zeta | GQ131301 | 6..249 | AB378097 | 6..243 | miR-263a | ||
| 6..296 | DQ311235 | 6..291 | miR-278* | ||||
| EF210316 | 6..291 | ||||||
| NM_001046699 | 6..291 | ||||||
| actin | EU527017 | 58..141 | AB701689 | 50..132 | bantam, miR-965* | ||
| NM_001126254 | 50..132 | ||||||
| ADP/ATP translocase | AY253868 | 1..28 | AY227000 | 1..28 | miR-2765 | ||
| NM_001043607 | 1..28 | ||||||
| ArgK | EF600057 | 1..211 | HQ840714 | 1..211 | miR-8 | ||
| beta-tubulin | JF767013 | 100..152 | AB003287 | 105..157 | miR-2796-5p | ||
| AB072307 | 106..158 | ||||||
| NM_001043422 | 106..158 | ||||||
| CCE | FJ997310 | 496..541 | AB521203 | 162..207 | miR-87 | ||
| chitinase | AY325496 | 780..976 | AB032107 | 860..1055 | miR-87 | ||
| AY325497 | 861..1056 | ||||||
| E75 | JQ266225 | 22..319 | AB024904 | 21..314 | miR-33, miR-278*, miR-87 | ||
| AB024905 | 21..314 | ||||||
| NM_001112609 | 21..314 | ||||||
| NM_001112610 | 21..314 | ||||||
| 3..319 | AF332550 | 3..316 | miR-33, miR-278* | ||||
| AF332551 | 3..316 | ||||||
| NM_001043577 | 3..316 | ||||||
| EcR | EU180021 | 14..51 | BMOBMECRB1 | 14..51 | miR-14, miR-9a* | ||
| D49476 | 14..51 | ||||||
| D49478 | 14..51 | ||||||
| HM046618 | 117..154 | NM_001043866 | 14..51 | ||||
| NM_001173375 | 14..51 | ||||||
| BMOECDYREC | 695..732 | ||||||
| FF1 | HQ260326 | 77..175 | AB649122 | 82..180 | miR-305* | ||
| AF426830 | 82..180 | ||||||
| NM_001044063 | 82..180 | ||||||
| HQ260326 | 77..275 | BMOFTZF1P | 15..203 | miR-305*, miR-970 | |||
| Hsp21.4 | HM046612 | 106..148 | AB195972 | 109..154 | miR-1175-3p | ||
| NM_001043520 | 109..154 | ||||||
| HM046612 | 581..662 | AB195972 | 568..647 | miR-305 | |||
| NM_001043520 | 568..647 | ||||||
| Hsp70 | FJ432703 | 25..85 | HM046611 | 27..87 | miR-8 | ||
| JF417984 | 497..624 | AB016836 | 370..500 | miR-1a*, miR-3338 | |||
| FJ573459 | 370..500 | ||||||
| NM_001043372 | 370..500 | ||||||
| Hsp90 | FJ986209 | 9..101 | HM046609 | 12..101 | miR-14, miR-2766, miR-285, miR-998, miR-9a, miR-9c* | ||
| HM593517 | 9..101 | ||||||
| FJ986209 | 57..108 | AB060275 | 43..96 | miR-14, miR-2766, miR-9a, miR-9c* | |||
| HM593517 | 57..108 | NM_001043411 | 43..96 | ||||
| HM046609 | 57..98 | AB060275 | 43..84 | miR-14, miR-2766, miR-9a, miR-9c* | |||
| NM_001043411 | 43..84 | ||||||
| LpR | GU433377 | 30..69 | AB211594 | 30..69 | miR-13a | ||
| GU433377 | 1304..1375 | AB211594 | 1048..1116 | miR-iab-8 | |||
| GU433377 | 1343..1375 | AB201471 | 1079..1111 | miR-iab-8 | |||
| AB201472 | 1079..1111 | ||||||
| AB201473 | 1079..1111 | ||||||
| AB201474 | 1654..1686 | ||||||
| NM_001111338 | 1079..1111 | ||||||
| NM_001111339 | 1079..1111 | ||||||
| NM_001111340 | 1079..1111 | ||||||
| PBANR | JN228350 | 479..596 | JN228346 | 843..960 | miR-375* | ||
| TCTP | GU969276 | 33..152 | HQ896486 | 31..149 | miR-2766, miR-305*, miR-989 | ||
| GU969276 | 121..154 | DQ003481 | 99..132 | miR-989, miR-14, miR-2766, miR-375*, miR-9c | |||
| EF210320 | 99..132 | ||||||
| NM_001044107 | 99..132 | ||||||
| HQ896486 | 118..149 | DQ003481 | 99..130 | miR-989, miR-14, miR-2766, miR-9c | |||
| EF210320 | 99..130 | ||||||
| NM_001044107 | 99..130 | ||||||
| TF AP-4 | DQ224406 | 20..91 | EU534187 | 21..92 | miR-9a* | ||
| NM_001123344 | 21..92 | ||||||
| TF POU | AY513764 | 549..675 | BMOPOUM1 | 494..619 | miR-2745, miR-316* | ||
| USP | EU526832 | 467..552 | EU180022 | 125..212 | miR-79, miR-9a* |
*: "ID" indicates the "LOCUS" field in NCBI. Abbriviation: har: H. armigera; sli: S. litura; bmo: B. mori.
§: BLASTn was used to identify homologous regions in 3' UTRs. E-value threshold was set to 10-5.
¶: miRanda, PITA, and microTar were all used for miRNA target prediction. Only the miRNA targets being predicted by more than two software programs are included in this table.
Conserved miRNA target sites in 3' UTRs of Hsp90 genes. (A) Homologous regions and miRNA target sites in the 3' UTRs of Hsp90 genes. The bars indicate regions homologous to H. armigera (dark green), S. litura (dark blue) or B. mori (grey) and predicted miRNA target sites of miR-9a (light blue), miR-14 (purple) or miR-2766 (light green). (B) Down-regulation of reporter gene expression with the 3' UTRs of Hsp90 genes. miRNA precursors were inserted into pmR-mCherry for miRNA over-expression. These miRNA-expressing plasmids were co-transfected into 293T cells with the pGL3-promoter and pRL-TK parental (Renilla control) or pRL-TK-3'UTR plasmids into which the Hsp90 3' UTRs were respectively cloned. The y-axis shows the Renilla luciferase activity (relative luminescence units [RLU]) normalized to the firefly luciferase activity and compared with the vector control, which was set to 1.0 within each experiment. The values are the average±SD of 3 biological repeats. The abbreviations represent the species as follows: Bmo, B. mori; Har, H. armigera; Sli, S.litura. A paired t-test was used to analyze the significance of the differences. A difference with p-value less than 0.05 was considered significant (marked with "*" in the figure) and less than 0.01 was considered highly significant (marked with "**").
Discussion
Homology-based predictions of lepidopteran miRNA using the B. mori genome sequence is a valid approach
The identification of miRNAs based on genomic informations has been reported in several lepidopteran insects, such as silkworm (B. mori) [45, 46], tobacco hornworm (Manduca sexta) [28] and monarch butterfly (Danaus plexippus) [27]. Additionally, in two lepidopteran insects - Heliconius melpomene and Bicyclus anynana - miRNAs have been identified using BAC sequences [26, 47]. In the present study, we report the identification of miRNAs in H. armigera and S. litura based on small RNA sequencing. Because complete H. armigera or S. litura genomes are not available, we used the B. mori miRNA and genome sequences as references. As a result, 33.66% and 33.34% of the sRNA sequences in H. armigera and S. litura, respectively, could be mapped to the B. mori genome. Although the percentage is lower than that of mapped B. mori sRNA sequences (which is greater than 60%) [45, 46], the homology is still significant. The lower mapping ratio may be because B. mori belongs to Bombycidae, whereas H. armigera and S. litura belong to Noctuidae. The miRNAs identified in this study are believed to be highly conserved among lepidopteran insects.
Most of the conclusions of the present study are based on the assumption that the mature miRNA sequences are conserved among related species. To evaluate the reliability of this assumption, we chose two highly conserved har-miRNAs - har-let-7 and har-miR-133 - and two insect-specific har-miRNAs - har-miR-1000 and har-miR-14 - to analyze the conservation among the homolog precursors (Supplementary Material: Fig. S1). Insects from different orders published in miRBase were considered in this analysis. The results demonstrated that the precursors were conserved in the mature sequence region and that two nucleotides flanking this region were also relatively conserved, particularly among species belonging to the same order. The precursors of the miRNA homologs from species in the same genus were more analogous. These results are in accordance with a previously published study on highly conserved miRNAs in S. litura [19]. Thus, when B. mori pre-miRNAs and mature miRNAs are used as references for lepidopteran miRNA identification, conserved miRNAs in H. armigera and S. litura can be obtained.
A minor portion of the identified miRNAs are highly conserved from insects to vertebrates
To understand the homology of miRNAs, we compared the conserved miRNAs of H. armigera and S. litura with the miRNA sequences of all species published in miRBase (Release 18) and other published lepidopteran miRNAs [26-28]. 16 miRNAs - including let-7, miR-10, miR-100, miR-124, miR-133, miR-137, miR-184, miR-190, miR-1a, miR-210, miR-281, miR-33, miR-7, miR-993a*, miR-993b*, and miR-9a - were conserved from insects to vertebrates (Supplementary Material: Tables S4 and S5). These miRNAs are thought to have homologous functions in diverse species. Studies on their functions in other species, such as in flies and mice, can provide clues to their functions in lepidopteran insects.
For example, let-7 has been demonstrated to be involved in the development of diverse animals, including nematode, fly, mouse, and human [48]. In addition, some of the target genes of let-7 appear to be conserved among these animals, such as the conservation of the interaction of let-7 and two genes - RAS (a homolog of let-60) and TRIM71 (a homolog of lin-41) - from nematodes to vertebrates [48]. Moreover, let-7 is induced by ecdysone [49] and is required for neuromusculature remodeling during the metamorphosis of flies [50]. Therefore, let-7 in lepidopteran insects may also play a role in developmental regulation. These data provide a hint for the functional analysis of let-7.
It was surprising to find two miR*s, miR-993a* and miR-993b*, in this highly conserved set of miRNAs. These miR*s are homologous to miR-10s in vertebrates. With regard to miRBase, we found that mir-993s have only been identified in invertebrates, most of which also expresses miR-10s (except the nematodes). As previously reported, miR-993 belongs to the miR-100/10 family, and both miR-993 and miR-10 are derived from the ancient miR-100 through duplication and arm-switching [51]. The higher expression of miR-993 (miR-993-3p) compared with miR-993* (miR-993-5p) was found in Drosophila melanogaster [52]. However, in many other insects, including Tribolium castaneum and B. mori, miR-993*s are expressed at higher levels than miR-993s [46, 53]. Therefore, the relative expression of mature miRNAs in the opposite arms of one precursor differs from species to species. Arm-switching allows some miR*s to become functional miRNAs. Because the miR-100/10 family members are located within Hox complexes and can regulate the expression of the relevant Hox genes [54], and Hox genes are involved in development regulation, miR-993s may play a role in insect development by targeting Hox genes.
Most identified miRNAs are insect-specific
Most of the miRNAs identified in this study were insect-specific. As there are many insect-specific features, such as metamorphosis and pheromones [55], these miRNAs may specifically regulate insect genes and relevant pathways. A typical example is miR-14, which is found only in insects. Previous studies have demonstrated its involvement in a positive autoregulatory loop controlling ecdysone signaling in Drosophila [16], which plays a key role in metamorphosis. We believe the other insect-specific miRNAs may also be involved in insect-specific features. Because insects constitute the most diverse group of animals, studies on these miRNAs can provide cues on insects' extensive adaptivity.
Among insect-specific miRNAs, 29 har-miRNAs and 20 sli-miRNAs were regarded as Lepidoptera-specific (Supplementary Material: Tables S4 and S5), and we hypothesize that they have specific functions in lepidopteran insects and could possibly be used as targets for lepidopteran pest control. In both species, nine Lepidoptera-specific miRNAs, including miR-9c, miR-92b, miR-306a, miR-308, miR-745, miR-2755, miR-2766, miR-2767 and miR-2768, have homologs in all the 6 analyzed lepidopteran insects, indicating that they have conserved functions in lepidopteran species. Another Lepidoptera-specific miRNA, miR-1175-5p, has homologs in only the 3 types of moths, but not in butterflies, and thus it may have specific functions in moths, most likely regulating moth-specific genes. Although we cannot be sure of their inexistence in other species due to the absence of genomic information for most insects, particular attention should be paid to these miRNAs with respect to their specific functions in lepidopteran insects or in moths. These miRNAs may be involved in some special biological processes that distinguish Lepidoptera (or moths) from other species.
Some conserved miRNAs are involved in Lepidoptera development
The developmental expression profiles of the miRNAs miR-2a, miR-34, miR-2796-3p and miR-11 were investigated by qRT-PCR. These miRNAs showed different expression patterns (Fig. 4). According to the rationale that the high expression of an miRNA at a specific stage suggests its possible regulatory role in that stage [56], the different patterns of the miRNAs indicate their diverse functions during development. The high expression of miR-2a and miR-11 in pupae reflects their possible involvement in metamorphosis, whereas the high level of miR-34 and miR-2796-3p in the adult stage may indicate their roles in adult development or reproduction. In a comparison with the expression of relevant miRNAs in B. mori reported by Zhang et al [57], miR-34 showed a higher expression in the adult stage in all three species, indicating its important and possibly conserved role in adult development. Interestingly, the low expression of miR-34 in pupae is similar to that observed in Drosophila, in which miR-34 was found to be under the control of two key hormones regulating metamorphosis. In detail, Drosophila miR-34 is repressed by ecdysone and induced by juvenile hormone [15]. Thus, we infer that the hormonal regulation of miR-34 and its involvement in metamorphosis may be conserved in these insects. However, miR-2a, which was called miR-2 by Zhang et al [57], was expressed at the highest levels at the embryonic and adult stages of B. mori but at the pupal stage in H. armigera and S. litura, indicating that some conserved miRNAs likely have distinct functions in different species.
The conserved regions in the 3' UTRs of B. mori, H. armigera, and S. litura genes implied that they may have conserved miRNA target sites. Through the target prediction of homologous regions in the 3' UTRs, we identified a number of genes that could potentially be targeted by common miRNAs (Table 3). These targeted genes included transcription factors, heat shock proteins, and genes involved in hormone pathways or metabolism. The diverse genes indicate the various roles miRNAs can play in different species. However, most of the genes play a role in development. For example, genes involved in hormone pathways, such as EcR, USP and PBANR, are related to metamorphosis. Metabolism is also related to insect growth because aberrant metabolism may lead to abnormal development. Transcription factors are regulators of most genes involved in all types of biological processes, including development. Moreover, heat shock proteins are involved in stress adaptation and development. Therefore, although the conserved miRNAs may play a role in diverse biological processes, they affirmably participate in the regulation of development.
Among these genes, Hsp90 was selected to validate its regulation by the miRNAs because its 3' UTR was predicted to contain 3 conserved miRNA target sites. HSP90 is a conserved protein playing a role not only in development, but also in cancer progression, evolutionary diversification, and the regulation of coding and non-coding genes including miRNAs [58]. We used a dual-luciferase reporter assay for the target validation because this is a commonly used method to visualize miRNA-dependent gene silencing [59]. According to our preliminary experiments, lepidopteran cell lines showed low transfection efficiency, which impaired the down-regulation of reporters by the corresponding miRNAs (data not shown) and may lead to the neglect of some weak effects of miRNAs on gene regulation. Therefore, the mammalian 293T cell line was used instead for this assay because of its high transfection efficiency. According to the results (Fig. 5B), the 3 miRNA target sites in the Hsp90 3' UTRs were proven to be authentic. The results indicated that conserved genes in different insect species could be targeted by common miRNAs and that such conserved regulation may play roles in various biological processes. The regulation of Hsp90 by miRNAs indicates that these conserved miRNAs are most likely involved in the regulation of development, and that miRNAs may regulate coding and non-coding genes through Hsp genes.
Summary
In summary, this study provides a new method for miRNA identification in non-model insects lacking genome information. Moreover, the analysis of expression patterns and the validation of conserved miRNA targets revealed the diversity and conservation of miRNA functions in different insect species. The results extend our understanding of insect miRNA biological functions and will shed light on the identification of insect-specific miRNA targets that can be used in the future for lepidopteran pest control.
Supplementary Material
Table S1~S5, and Fig.S1.
Abbreviations
miRNA: microRNA; nt: nucleotides; RISC: RNA-induced silencing complex; miR*: miRNA-star strand; PCR: polymerase chain reaction; qRT-PCR: quantitative reverse transcription polymerase chain reaction; sRNA: small RNA; SBS: sequencing-by-synthesis; UTR: untranslated region; har-miRNA: Helicoverpa armigera miRNA; sli-miRNA: Spodoptera litura miRNA.
Acknowledgements
This work was supported by grants from National Basic Research Program of China (2012CB114101) and National Science Foundation of China (30825007 and 31030060).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lee I, Ajay SS, Yook JI. et al. New class of microRNA targets containing simultaneous 5'-UTR and 3'-UTR interaction sites. Genome Res. 2009;19:1175-83
2. Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102-14
3. Kato M, de Lencastre A, Pincus Z. et al. Dynamic expression of small non-coding RNAs, including novel microRNAs and piRNAs/21U-RNAs, during Caenorhabditis elegans development. Genome Biol. 2009;10:R54
4. Brennecke J, Hipfner DR, Stark A. et al. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003;113:25-36
5. Llave C, Xie Z, Kasschau KD. et al. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science. 2002;297:2053-6
6. Pfeffer S, Sewer A, Lagos-Quintana M. et al. Identification of microRNAs of the herpesvirus family. Nat Methods. 2005;2:269-76
7. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843-54
8. Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855-62
9. Reinhart BJ, Slack FJ, Basson M. et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901-6
10. Weaver DB, Anzola JM, Evans JD. et al. Computational and transcriptional evidence for microRNAs in the honey bee genome. Genome Biol. 2007;8:R97
11. He PA, Nie Z, Chen J. et al. Identification and characteristics of microRNAs from Bombyx mori. BMC Genomics. 2008;9:248
12. Kloosterman WP, Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11:441-50
13. Ambros V. The functions of animal microRNAs. Nature. 2004;431:350-5
14. Carrington JC, Ambros V. Role of microRNAs in plant and animal development. Science. 2003;301:336-8
15. Sempere LF, Sokol NS, Dubrovsky EB. et al. Temporal regulation of microRNA expression in Drosophila melanogaster mediated by hormonal signals and broad-Complex gene activity. Dev Biol. 2003;259:9-18
16. Varghese J, Cohen SM. microRNA miR-14 acts to modulate a positive autoregulatory loop controlling steroid hormone signaling in Drosophila. Genes Dev. 2007;21:2277-82
17. Sokol NS, Ambros V. Mesodermally expressed Drosophila microRNA-1 is regulated by Twist and is required in muscles during larval growth. Genes Dev. 2005;19:2343-54
18. Leaman D, Chen PY, Fak J. et al. Antisense-mediated depletion reveals essential and specific functions of microRNAs in Drosophila development. Cell. 2005;121:1097-108
19. Gao L, Zuo H, Liu K. et al. A New Strategy for Identification of Highly Conserved microRNAs in Non-Model Insect, Spodoptera litura. Int J Mol Sci. 2012;13:612-27
20. Rao Z, He W, Liu L. et al. Identification, expression and target gene analyses of micrornas in Spodoptera litura. PLoS One. 2012;7:e37730
21. Willmann MR, Poethig RS. Conservation and evolution of miRNA regulatory programs in plant development. Curr Opin Plant Biol. 2007;10:503-11
22. de Wit E, Linsen SE, Cuppen E. et al. Repertoire and evolution of miRNA genes in four divergent nematode species. Genome Res. 2009;19:2064-74
23. Nozawa M, Miura S, Nei M. Origins and Evolution of MicroRNA Genes in Drosophila Species. Genome Biol Evol. 2010;2:180-9
24. Wang J, Xia Q, He X. et al. SilkDB: a knowledgebase for silkworm biology and genomics. Nucleic Acids Res. 2005;33:D399-402
25. Xia Q, Zhou Z, Lu C. et al. A draft sequence for the genome of the domesticated silkworm (Bombyx mori). Science. 2004;306:1937-40
26. Surridge AK, Lopez-Gomollon S, Moxon S. et al. Characterisation and expression of microRNAs in developing wings of the neotropical butterfly Heliconius melpomene. BMC Genomics. 2011;12:62
27. Zhan S, Merlin C, Boore JL. et al. The monarch butterfly genome yields insights into long-distance migration. Cell. 2011;147:1171-85
28. Zhang X, Zheng Y, Jagadeeswaran G. et al. Identification and developmental profiling of conserved and novel microRNAs in Manduca sexta. Insect Biochem Mol Biol. 2012;42:381-95
29. Tamura K, Dudley J, Nei M. et al. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596-9
30. Betel D, Wilson M, Gabow A. et al. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149-53
31. Enright AJ, John B, Gaul U. et al. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1
32. Thadani R, Tammi MT. MicroTar: predicting microRNA targets from RNA duplexes. BMC Bioinformatics. 2006;7(Suppl 5):S20
33. Zhang B, Stellwag EJ, Pan X. Large-scale genome analysis reveals unique features of microRNAs. Gene. 2009;443:100-9
34. Sai Lakshmi S, Agrawal S. piRNABank: a web resource on classified and clustered Piwi-interacting RNAs. Nucleic Acids Res. 2008;36:D173-7
35. Griffiths-Jones S, Saini HK, van Dongen S. et al. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154-8
36. Griffiths-Jones S, Grocock RJ, van Dongen S. et al. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140-4
37. Griffiths-Jones S. The microRNA Registry. Nucleic Acids Res. 2004;32:D109-11
38. Ambros V, Bartel B, Bartel DP. et al. A uniform system for microRNA annotation. RNA. 2003;9:277-9
39. Lau NC, Lim LP, Weinstein EG. et al. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858-62
40. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215-33
41. Jagadeeswaran G, Zheng Y, Sumathipala N. et al. Deep sequencing of small RNA libraries reveals dynamic regulation of conserved and novel microRNAs and microRNA-stars during silkworm development. BMC Genomics. 2010;11:52
42. Shin C. Cleavage of the star strand facilitates assembly of some microRNAs into Ago2-containing silencing complexes in mammals. Mol Cells. 2008;26:308-13
43. Plasterk RH. Micro RNAs in animal development. Cell. 2006;124:877-81
44. Dolken L, Malterer G, Erhard F. et al. Systematic analysis of viral and cellular microRNA targets in cells latently infected with human gamma-herpesviruses by RISC immunoprecipitation assay. Cell Host Microbe. 2010;7:324-34
45. Jagadeeswaran G, Zheng Y, Sumathipala N. et al. Deep sequencing of small RNA libraries reveals dynamic regulation of conserved and novel microRNAs and microRNA-stars during silkworm development. BMC Genomics. 2010;11:52
46. Liu S, Li D, Li Q. et al. MicroRNAs of Bombyx mori identified by Solexa sequencing. BMC Genomics. 2010;11:148
47. Conceicao IC, Long AD, Gruber JD. et al. Genomic sequence around butterfly wing development genes: annotation and comparative analysis. PLoS One. 2011;6:e23778
48. Bussing I, Slack FJ, Grosshans H. let-7 microRNAs in development, stem cells and cancer. Trends Mol Med. 2008;14:400-9
49. Chawla G, Sokol NS. Hormonal activation of let-7-C microRNAs via EcR is required for adult Drosophila melanogaster morphology and function. Development. 2012;139:1788-97
50. Sokol NS, Xu P, Jan YN. et al. Drosophila let-7 microRNA is required for remodeling of the neuromusculature during metamorphosis. Genes & development. 2008;22:1591-6
51. Griffiths-Jones S, Hui JH, Marco A. et al. MicroRNA evolution by arm switching. EMBO Rep. 2011;12:172-7
52. Ruby JG, Stark A, Johnston WK. et al. Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs. Genome Res. 2007;17:1850-64
53. Marco A, Hui JH, Ronshaugen M. et al. Functional shifts in insect microRNA evolution. Genome Biol Evol. 2010;2:686-96
54. Woltering JM, Durston AJ. MiR-10 represses HoxB1a and HoxB3a in zebrafish. PLoS One. 2008;3:e1396
55. Zhang G, Wang H, Shi J. et al. Identification and characterization of insect-specific proteins by genome data analysis. BMC Genomics. 2007;8:93
56. Du T, Zamore PD. microPrimer: the biogenesis and function of microRNA. Development. 2005;132:4645-52
57. Zhang Y, Zhou X, Ge X. et al. Insect-Specific microRNA Involved in the Development of the Silkworm Bombyx mori. PLoS One. 2009;4:e4677
58. Sawarkar R, Sievers C, Paro R. Hsp90 globally targets paused RNA polymerase to regulate gene expression in response to environmental stimuli. Cell. 2012;149:807-18
59. Wang B, Doench JG, Novina CD. Analysis of microRNA effector functions in vitro. Methods. 2007;43:91-104
Author contact
![]() Corresponding author: Dr. Anjiang Tan, E-mail: tananjiangac.cn.
Corresponding author: Dr. Anjiang Tan, E-mail: tananjiangac.cn.