Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(1):45-54. doi:10.7150/ijbs.5194 This issue Cite
Research Paper
Tuning of Alternative Splicing - Switch From Proto-Oncogene to Tumor Suppressor
Aleksandra Shchelkunova1, Boris Ermolinsky1, Meghan Boyle1, Ivan Mendez1, Michael Lehker1, Karen S. Martirosyan2, Alexander V. Kazansky1,3 ![]()
1. Department of Biomedicine, The University of Texas at Brownsville, Brownsville, TX 78520, USA;
2. Department of Physics and Astronomy, The University of Texas at Brownsville, Brownsville, TX 78520, USA;
3. Department of Cellular and Molecular Biology, Baylor College of Medicine, Houston, TX 77030, USA.
Received 2012-9-9; Accepted 2012-12-7; Published 2012-12-19
Abstract
STAT5B, a specific member of the STAT family, is intimately associated with prostate tumor progression. While the full form of STAT5B is thought to promote tumor progression, a naturally occurring truncated isoform acts as a tumor suppressor. We previously demonstrated that truncated STAT5 is generated by insertion of an alternatively spliced exon and results in the introduction of an early termination codon. Present approaches targeting STAT proteins based on inhibition of functional domains of STAT's, such as DNA-binding, cooperative binding (protein-protein interaction), dimerization and phosphorylation will halt the action of the entire gene, both the proto-oncogenic and tumor suppressor functions of Stat5B.
In this report we develop a new approach aimed at inhibiting the expression of full-length STAT5B (a proto-oncogene) while simultaneously enhancing the expression of STAT5∆B (a tumor suppressor). We have demonstrated the feasibility of using steric-blocking splice-switching oligonucleotides (SSOs) with a complimentary sequence to the targeted exon-intron boundary to enhance alternative intron/exon retention (up to 10%). The functional effect of the intron/exon proportional tuning was validated by cell proliferation and clonogenic assays. The new scheme applies specific steric-blocking splice-switching oligonucleotides and opens an opportunity for anti-tumor treatment as well as for the alteration of functional abilities of other STAT proteins.
Keywords: STAT proteins, RNA splicing, tumor suppressor, splice-switching oligonucleotides, cell-cycle progression.
INTRODUCTION
The STAT (Signal Transducer and Activator of Transcription) proteins are a family of cytoplasmic transcription factors that were originally identified in the mid-1990's [1, 2]. To date, two predominant STAT isoforms have been identified: a full-length transcription factor and a C-terminal truncated isoform lacking the transactivation domain [3, 4]. We previously determined that the truncated STAT isoform is generated by retention of an alternatively spliced intron (Figure 1), resulting in the insertion of an early termination codon [4]. The resultant STAT5∆ protein, which lacks a transactivation domain, is then expressed. Phosphorylated STAT5∆ can translocate to the nucleus and bind to specific promoter sequences, thereby blocking the transcription of downstream genes due to lack of the transactivation domain. These truncated isoforms have been identified for STATs 1, 3, 4, STAT 5a and 5b, and have been shown to be the product of alternative splicing [4-6]. While at least one group showed that STAT isoforms are a result of post-transcriptional proteolytic cleavage, it was noted that this event does not occur in vivo [6].
Modulation of STAT5B isoforms by antisense oligonucleotides. 18m-anti and 19-anti bars above the transcripts indicate antisense splice-switching oligonucleotides. Blue arrows represent oligonucleotides for semi-quantitative RT-PCR analysis of splice-blocking.
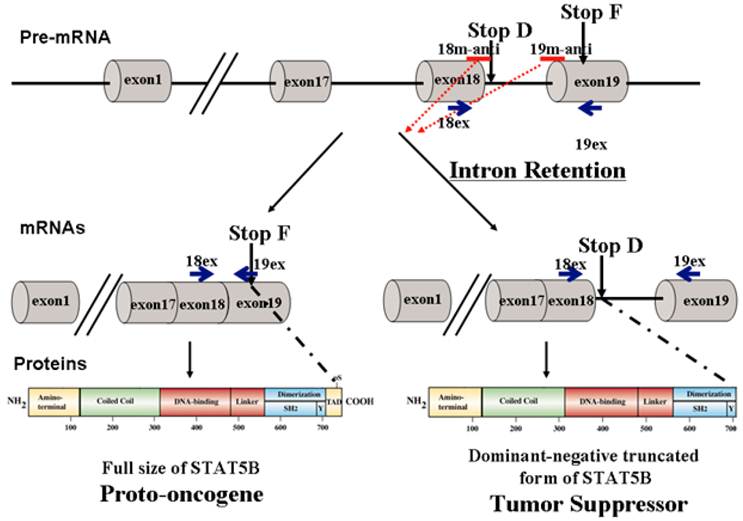
Constitutive activation of STAT5 proteins has been demonstrated in many diverse human cancer cell lines and clinical tumors including those of the prostate [7-14]. A clear understanding of STAT5 function in vivo has been greatly hindered by the existence of two nearly identical genes, STAT5A and STAT5B, which share 93% homology at the amino acid level. An additional complication stems from the existence of naturally occurring C-terminal truncated dominant-negative isoforms of STAT5 [4].
Previous reports have suggested potentially diverse functions for STAT5 isoforms. Although, recent studies provide clear evidence that STAT5B, contributes to tumor progression in epithelial cancers [11, 13, 15, 16], especially those of the prostate [11]. Specific activation of full-length STAT5B in epithelial cells representing invasive and metastatic prostate cancer has been previously demonstrated [11, 13]. This finding is consistent with STAT5 being highly activated in high-grade human prostate cancers [9]. Increased activation of STAT5 was also associated with increasingly aggressive behavior of prostate cancer [9, 17]. In contrast, the naturally occurring dominant-negative truncated isoform STAT5∆B can block cell cycle progression and inhibit growth, invasive potential and clonogenic ability (hallmark of transformed and malignant potential [18]) of cancer cell lines [11, 17, 19, 20]. Furthermore, we demonstrated that STAT5∆B could inhibit the growth of cancer cells in grafting studies in vivo [10, 11]. Thus, the previous research demonstrated role of dominant-negative form of STAT5B as tumor suppressor [11, 17, 19, 20], which blocks cell-cycle progression.
Current therapeutic strategies against STAT5B and other STAT proteins are predominantly based on inhibiting STAT functional domains such as DNA binding, cooperative binding (protein-protein interactions), and dimerization/phosphorylation domains. Inhibition of these key domains not only halts proto-oncogenic functions, but also inhibits tumor suppressor actions of dominant-negative STAT5∆B, therefore diminishing the effectiveness of the therapy. To overcome therapeutic challenges, we developed a novel strategy based on endogenous mechanisms regulating STAT5B isoforms that utilizes a pro-drug capable of converting full length STAT5B (proto-oncogenic) into the dominant negative truncated form of STATB (tumor suppressive). Therefore we applied specific steric-blocking splice-switching oligonucleotides (SSOs) with a complimentary sequence to the targeted exon-intron boundary in the pre-mRNA of Stat5B to induce alternative intron/exon retention enhancing production of tumor suppressor protein. The SSOs complementary to exon/intron boundary sequences can block access of the spliceosomal machinery and stimulate retention of the associated exon [21-23]. They were used to alter splicing for therapeutic purposes in cell line models of human disease [21]. Splice-blocking oligonucleotides should bind specifically to target sequence while at the same time contain extra- or intra-molecular complementarity, have suitable solubility, and do not bind too tightly (favoring off-target effects) or too loosely (causing low efficacy). The SSOs must be stable in serum, hybridize at a high melting temperature, and form duplexes with the targeted RNA, which are not processed by RNase H and/or other nucleases. This is usually achieved by 2' modification of the sugar with O-methyl, O-methoxyethyl or O-aminopropyl groups or by backbone modifications such as in N3' → P5' phosphoroamidate, peptide nucleic acids (PNAs), phosphorodiamidate morpholino (PMO), or locked nucleic acids (LNAs) [24-26]. PNAs offer specificity similar to Morpholinos, but their low water solubility makes PNA experiments difficult. Morpholino oligos are usually soluble at several millimolar aqueous concentrations. We have chosen to use Morpholino oligonucleotides, which form stable base pairs with complementary nucleic acid sequences but do not bind proteins to a significant extent, and they mediate RNA cleavage by RNase H or the RISC (RNA induced silencing complex).
In this work, we also developed quantitative PCR methodology to detect splice variants of STAT5B, providing a feasible solution to the significant challenge of separating nearly identical truncated forms of STAT5A and STAT5B. Here, we report evidence of the feasibility of implementing steric-blocking splice-switching morpholino oligonucleotides for the modulation of STAT5B isoforms.
METHODS
Tissue Culture
Human PC-3 prostate cancer cells were obtained from American Type Culture Collection (Manassas, VA) and maintained in RPMI 1640 supplemented with 10% fetal bovine serum.
Morpholino oligonucleotides and transfection
Morpholino antisense oligonucleotides (m-anti) were purchased from Gene Tools, LLC (Philomath, OR, USA). They were delivered into PC-3 prostate cancer cells using Endo-Porter reagent (Gene Tools, LLC) as per manufacturer protocol.
cDNA production
Total RNA was isolated 24 hr post-treatment and reverse transcribed using an RNeasy Mini Kit (QIAGEN, CA) and SuperScript®IIIRT Reverse Transcriptase (Invitrogen, CA) according to recommended manufacturer protocols.
Sequence and analysis of DNA fragment
Sequence of RT-PCR fragments has been performing in both directions by SeqWright DNA technology Services (Houston, TX). Alignment of sequences has been done by BLASTN 2.2.27 against NCBI nucleotide collection.
TagMan probes for SPLICE and NONSPLICE variants of Stat5B
Real-time PCR was performed via StepOne Real-Time PCR System (ABI) utilizing self-designed TaqMan hydrolysis probes (SPLICE and NONSPLICE) and TaqMan-based ABI gene expression assays (Hs99999905_m1, Gapdh). All qPCR experiments were performed in duplicate. Briefly, cDNA samples were introduced to a MicroAmp Optical 48-well reaction plate (ABI), which was covered with MicroAmp 48-well Optical Adhesive Film (ABI). Each experimental well contained 20 µl total reaction volume and included 10 µl of TaqMan Universal PCR Master Mix (ABI), 1 µl of 20x TaqMan assay, 8 µl of water and 1µl of analyzed sample. Self-designed 20x assays (SPLICE and NONSPLICE) contained 16µМ each of forward and reverse primers and 5 µМ of TaqMan probe. The final PCR conditions were as follows: pre-heating at 50 °C for 2 min, denaturation at 95°C for 10 min and 40 cycles of amplification and quantification (15 sec at 95 °C and 60 sec at 60 °C). Samples were normalized using Taqman based ABI gene expression assay Hs99999905_m1 for human Glyceraldehyde 3-phosphate dehydrogenase (Gapdh, amplicon size = 122 bp).
Sequence information for designing SPLICE and NONSPLICE splice variant primers and probes was obtained from NCBI NG_007271.1. For this project, SPLICE is considered a transcript variant without introns between exons 18 and 19 while NONSPLICE indicates the presence of the intron between exons 18 and 19. TaqManTM probes, labeled at the 5' end with FAM (6-carboxyfluorescein) and at the 3' end with TAMRA (6-carboxytetramethylrhodamine), were synthesized via IDT. Primers and probes for TaqMan hydrolysis qPCR assays specific for each splice variant were designed using the Primer Express v3.0 program (ABI). For SPLICE (Figure 1), the forward (5'- CAGCGCCACGTACATGGAC -3') and reverse (5'- CGAAGTCCCCATCGGTGTC -3') primer sequences amplify a 75 bp region and probe (5'-FAM- AGGACTGAGTCAGGGTTCTGTGGGTACATGTTATAGT-TAMRA-3') were designed to span the junctions between exons 18-19. For NONSPLICE (Figure 1), the forward (5'- GGCTTTCCCCTTCCCTTCA -3') and reverse (5'- GAAGTCCCCATCGGTGTCAA -3') primer sequences amplify a 75 bp region, and probe (5'-FAM- CTTTCCCTCCCAAGCCCTGACTCAGTC -TAMRA-3') was designed to span intron-19 exon junctions. Primers and probes are specific to the input template as no other targets were found in selected NCBI Transcript Reference Sequences database. Duplex formation and secondary nucleic acid structures of primer sequences were also evaluated using OligoAnalyzer 3.0 (Integrated DNA Technologies). The analysis of primers was performed by Primer-Blast, encompassed all homo sapien DNA sequences in the NCBI database, and showed only one specific product (http://blast.ncbi.nlm.nih.gov/).
To assess the efficiency of assays and assure validity of the ΔΔCT method, standard curves were prepared for serial dilutions of input cDNA for SPLICE, NONSPLICE and Gapdh (ABI, Hs 999999016_m1) assays. All serial dilutions of human cDNA were prepared and run in triplicate. From the amplification curves, CT (Cq) values for each concentration were determined and the slope of CT versus nanograms of cDNA was used to calculate the efficiency of amplification (using StepOne software, ABI). TaqManTM assays were linear at 6 serial dilutions of cDNA (from 1 to 40 ng) and the efficiencies and correlation coefficients (R2) were determined by the average of 2 independent experiments. For SPLICE, the slope was −3.16, R2 = 0.997, efficiency = 107.255, and for NONSPLICE slope was −3.223, R2 = 0.989, and efficiency = 104.305. For the gapdh assay (ABI, Hs 99999905_m1) the slope =−3.246, R2 = 0.989, efficiency = 103.256.
In a separate validation experiment to confirm the validity of the ΔΔCT method, standard curves were analyzed for serial dilutions of input cDNA for SPLICE, NONSPLICE and Gapdh. The dilution series of cDNA containing the target gene of interest were analyzed by qPCR. The slopes of the dilution series were calculated by determining ΔCT between each point on the dilution series plotted against the log of sample (cDNA) input (mass). The rationale is that in performing ΔΔCT analysis, no or very minimal difference in efficiencies should exist between the gene of interest and the endogenous control (i.e. Gapdh) [27]. For each of these assays, the efficiency and R2 values were appropriate and the slope of the line for ΔCT of Gapdh and targets (SPLICE and NONSPLICE) was less than 0.1. For qPCR analysis, each sample was run in triplicate. Each run included a control lacking template to test for contamination in assay reagents. After a 94 0C denaturation for 10 min, reactions were cycled 40 times with a 94 0C denaturation for 15 sec, and a 60 0C annealing for 1 min. Three types of controls aimed at detecting genomic DNA contamination in the RNA sample or during the RT or qPCR reactions were included: a RT mixture without reverse transcriptase, a RT mixture including the enzyme but no RNA, and a negative control (reaction mixture without cDNA template). The data were collected and analyzed using OneStep Software (ABI).
Preparation of Whole Cell Extracts
Cells were rinsed twice with ice-cold PBS (Invitrogen, Grand Island, NY), collected via cell scraper, and suspended in radioimmunoprecipitation assay buffer [50 mM NaF, 10 mM Na4P2O7, 0.5% sodium deoxycholate, 0.1% SDS, 1% NP40, 150 mM NaCl, 9.1 mM Na2HPO4, and 1.7 mM NaH2PO4 (pH 7.4)] containing the following protease and phosphatase inhibitors: 1 mM Na3VO4; 1 mM phenylmethylsulfonyl fluoride; 2 µg/ml aprotinin; 2 µg/ml antipain; 2 µg/ml leupeptin; and 2 µg/ml benzamidine. After incubation at 4 °C for 30 min, cell extracts were isolated by centrifugation and supernatants were collected. Protein concentrations were measured using the Bio-Rad protein assay reagent (Bio-Rad, Hercules, CA).
Western Blot Analysis
Protein samples were separated by 7.5 % SDS-PAGE and transferred overnight to an Odyssey Nitrocellulose Membrane (LI-COR Biosciences). We routinely analyzed 60 µg of total cell lysate/lane. An antibody recognizing the amino terminus of STAT5 (N-20) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and ß-actin (AC-74) antibody was obtained from Sigma (St. Louis, MO, USA). After incubation with primary antibodies, the membranes were washed with PBS containing 0.1 % Tween 20 (PBST) thrice. Membranes were further incubated for 1 hr with IRDye800CW-conjugated goat anti-rabbit IgG and IRDye680-conjugated goat anti-mouse IgG secondary antibodies (LI-COR Biosciences) diluted in Odyssey Blocking Buffer. The blots were then washed three times with PBS-T and rinsed with PBS. Proteins were visualized by scanning the membrane using Odyssey Infrared Imaging System (LI-COR Biosciences) for both 700- and 800-nm wavelengths.
Cell Proliferation Assay
Cell proliferation assays were performed using the colorimetric 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfonyl)-2H-tetrazolium assay (CellTiter 96 AQ; Promega, Madison, WI) according to the manufacturer's protocol. Cells were plated in 96-well plates at a density of 2 x 103 cells/well. After transfection with SSOs, cells were grown for 12 hr in complete media followed by 36 hr in serum-free media.
Clonogenic Assay
24 hr after SSOs transfection in six-well plates, PC-3 cells were counted and re-plated in 100 mm dishes (200 cells/dish) for 10 days in complete medium. Dishes were washed once with 0.85 M NaCl, stained with crystal violet (Sigma, St. Louis, MO, USA) dissolved in ethanol for 15 min, and washed well to remove excess crystal violet. Colonies containing >50 cells were scored as positive. Each experiment was performed twice in triplicates.
Statistical Analysis
Values were expressed as the mean ± SE. Statistical analyses were performed by Student's t test for paired comparisons using StatView program (Abacus Concepts, Inc., Berkeley, CA, USA). Differences were considered statistically significant at P<0.05 and all experiments were performed independently twice or thrice.
RESULTS
Specific intron retention in Stat5B pre-mRNA by morpholino oligonucleotides in PC-3 cells
PC-3 cells were treated with 2'-O-methyl-modified oligoribonucleoside phosphorothioate with complimentary sequence to the targeted 5' exon-intron boundary (18m-anti) and to the 3' intron-exon boundary (19m-anti) in order to shift splicing patterns as shown in Figure 1 at concentration of 6 uM each. The sequences of 18m-anti and 19m-anti oligos are TGCCACCTAC/TTCTGTGGGT and GAGTCAGGG/CTTGGGAGGGAAAGAA, respectively. Underlined is the 9 bp sequence of the 5'splice site, the slash indicates the exon-intron boundary, and conserved nucleotides of splicing consensus are highlighted in bold. Scrambled mispared oligonucleotide (m-mis) RyrCACrryCTrCTyrGyyr was used as a negative control; where lower-case letters indicate mismatches, y - pyrimidine nucleotide; and r - purine nucleotide. Antisense sequences with mismatches were appropriately distributed along the sequence to provide a stringent and realistic assessment of sequence specificity.
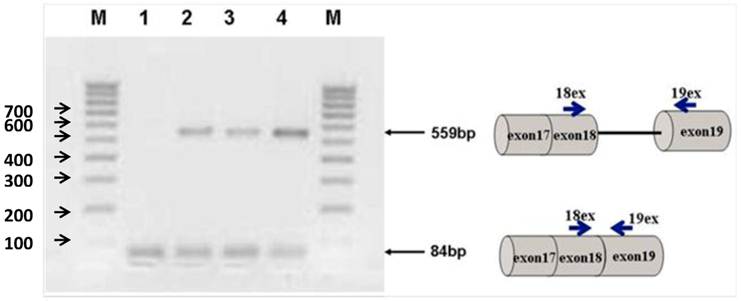
Figure 2 shows the transfection of PC-3 cells with steric-blocking splice-switching oligonucleotides. An induced shift in splicing of Stat5b isoforms is present (Figure 2). All targeted Morpholino oligonucleotides generated measurable amounts of predictable product at 559 bp confirmed by sequence analysis. These results demonstrate that the designed oligos are capable of enhancing intron retention. Furthermore, simultaneous introduction of both 18m-anti and 19m-anti oligos resulted in an increase of unspliced RT-PCR products (Figure 2, lane 4), suggesting that targeting splice-junctions blocked splicing events. Additionally, we observed that at high concentrations, even non-specific oligonucleotides (m-mis) cause a decrease in cell viability. RNA was isolated 24 hr post-treatment following reverse transcription. To validate the targeting procedure, the level and nature of transcript alteration was characterized by reverse transcriptase (RT)-PCR analysis of splice modification (18ex and 19ex, Figure 1).
Agarose gel electrophoresis of semi-quantitative RT-PCR splice-blocking products. Splice-junctions were targeted with two splice-blocking Morpholino oligonucleotides as described. Lane 1: m-mis; 2: 18m-anti; 3: 19m-anti; 4: combination of 18m-anti and 19m-anti. M - 100 bp ladder. All targeted Morpholino oligonucleotides generated measurable amounts of predictable products indicating splice retention effect. (559bp).
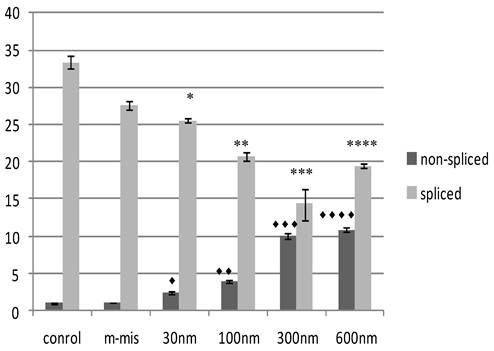
To validate the targeting procedure at lower concentration of SSO treatment, we designed a protocol utilizing Quantitative Reverse Transcriptase (RT) PCR analysis of splice modification using custom made TaqMan probes to detect splice variants of STAT5B (refer to Materials and Methods). This procedure detects measurable dose-dependent shift in splicing leading to the retention of specific introns. Next, we treated PC-3 cells with increased concentrations of an equimolar mix of 18m-anti and 19-anti. Analysis of treated cells was performed on the total RNA isolated 24 hr post-treatment. Figure 3 shows the results of quantitative examination of total RNA by RT-PCR. Results demonstrate tuning of the ratio of non-spliced and spliced forms produced post-incubation with morpholino oligonucleotides. The quantity of STAT5B isoform expression and proportion of non-spliced vs. spliced forms increased after treatment by morpholino oligonucleotides. We observed a dose-dependent increase in splicing retention as indicated by a shift in the ratios of the corresponding mRNA's while mismatched oligo (m-mis) had no effect on splicing (see Figure 3).
Effect of SSO's treatment on quantity of transcription of Stat5B isoforms. The results of the analysis of total RNA by quantitative RT-PCR. Data demonstrate tuning of the ratio of non-spliced and spliced forms produced after treatment with morpholino oligonucleotides. Control (untreated) cells, m-mis- treatment with 600 nM mispared oligonucleotide, 30 nM, 100 nM, 300 nM, 600 nM - treatment with corresponding concentration of the mix of 18m-anti and 19m-anti. Error bars indicate mean ±SD (n=3). ♦, P = 0.002; ♦♦, P = 0.0008; ♦♦♦, P = 0.008; ♦♦♦♦, P = 0.0014; *, P = 0.0016; **, P =0.003; ***, P =0.008; ****, P =0.0003.
Splice regulation by the SSOs induce production of truncated dominant-negative isoform of STAT5 protein
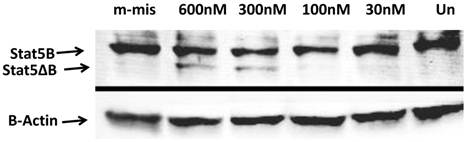
To analyze functional ability of splice blocking effects on protein expression, we performed Western blot analysis. Total protein levels from steric-blocking splice-switching oligonucleotide treated cells revealed an SSO-dependent increase in STAT5∆B protein production (Figure 4). These data are consistent with RT-PCR results and confirm that splice retention produces a functional transcript that can be efficiently translated into corresponding STAT5∆B protein.
Western Blot analyses of the splice blocking affect of morpholino oligonucleotides. Splice-junctions were targeted with two splice-blocking morpholino oligonucleotides as described above. m-mis - 600 nM of mis-pared oligonucleotide; 600 nM 300 nM, 100 nM, and 30 nM corresponding concentration of combination of 18m-anti and 19m-anti; Un -untreated cells.
Inhibition of PC-3 cells proliferation and viability by shifting STAT5B isoform toward truncated dominant-negative isoform
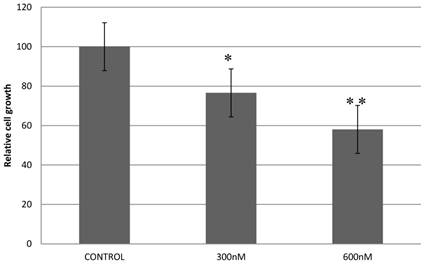
Previously, we reported that expression of a dominant-negative form of STAT5B reduces cell proliferation and survival by blocking cell cycle progression [10, 11]. Our aim in manipulating splice switching to create the dominant-negative truncated form from the full form of STAT5B is to produce a potential tumor suppressor. We next analyzed effects of induced splice switching by SSOs on cell proliferation and survival. We have observed dose dependent decrease of cell proliferation rate by SSOs treatment. Quantitative analysis of cell growth was determined by the application of different concentrations of SSOs to tumor cells (Figure 5) and indicated that splice-blocking oligonucleotides decrease the rate of cell proliferation. Colony formation assays (a hallmark of transformed and malignant cell potential [28]) revealed concentration dependent reduction of PC-3 cell viability following SSOs treatment (Figure 6). To distinguish the toxicity of oligos to the cells from STAT5ΔB-mediated cell growth retardation, we treated cells with a mismatch morpholino oligonucleotide similar to SSOs but which not cause splice retention.
Treatment with splice-blocking oligonucleotides decreases cell proliferation. Control - 600 nM of mispared oligonucleotide; 600 nM, 300 nM corresponding concentration of combination of 18m-anti and 19m-anti. Error bars indicate mean ±SD (n=3). *, P = 0.009; **, P =0.0115.
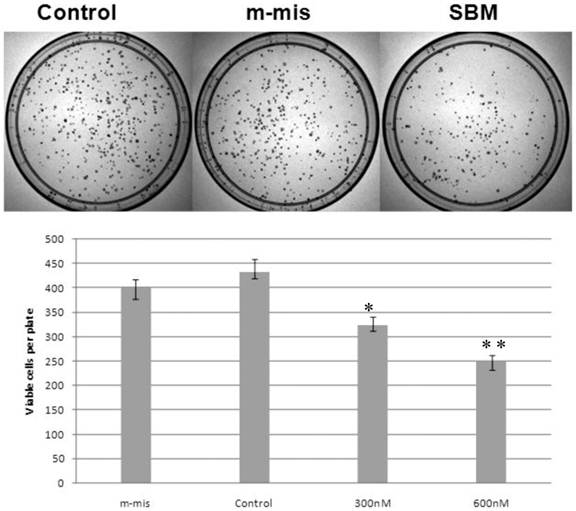
Reduction of cell survival by splice-blocking morpholino-oligonucleotides. Effect of splice blocking on cell viability as determined via clonogenic assay. Top panel shows images of representative plates treated with 600 nM morpholino oligonucleotides while the bottom panel quantitatively represents cell viability of SBM treated cells compared with untreated cells. M-mis - scrambled mispared oligonucleotide was used as a negative control for cell toxicity. Error bars indicate mean ±SD (n=3). *, P = 0.0075; **, P =0.007.
DISCUSSION AND CONCLUDING REMARKS
Alternative splicing is quickly gaining momentum as a viable target for molecular therapies [29]. STAT family of proteins is involved in many physiological processes and participates in inflammatory responses and cancer progression. The problem with current approaches of therapeutic targeting of STAT proteins is simultaneous inhibition of opposite functions of STAT isoforms. In this work we are presenting the design of the new approach to overcome this problem.
Splice-switching oligonucleotides are considered a novel class of therapeutics intended to induce therapeutically favorable splice variants of targeted genes [6]. The potential therapeutic benefit of antisense oligonucleotides has been demonstrated by several reports [30-32]. Antisense Morpholino oligonucleotides complementary to exon/intron boundary sequences can block access of the spliceosomal machinery and enhance alternative exon inclusion [23, 33]. The demonstrated approach focused on induction of the specific splice retention that can occur naturally [4]. We have demonstrated that application of Morpholino oligonucleotides to target exon-intron boundary in the pre-mRNA of Stat5B are able to induce intron/exon retention, which was confirmed by semi-quantitative RT-PCR. . While limited delivery efficiency caused incomplete modification of splicing, all targeted Morpholino oligos generated measurable amounts of predicted target products at 559 bp (Figure 2). These results demonstrate that the designed oligonucleotides are capable of enhancing intron retention (Figure 2). Additionally, quantitative RT-PCR analysis confirmed a dose-dependent shift in splicing leading to the specific intron retention (Figure 3) and our results indicate that a modified transcript with the additional exon can be efficiently translated into STAT5∆B protein (Figure 4).
Expression levels of STAT5∆B protein after enhancement was still significantly lower than the full length form. Although relative protein levels are lower, studies have previously demonstrated that the dominant-negative form has a significantly stronger impact on transcriptional regulation due to a slower rate of deactivation [4, 15]. Our cell culture data are consistent with previously reported observation regarding the tumor suppressor activity of STAT5∆B [10, 11]. Cell proliferation assays revealed that generation of the dominant-negative isoform of STAT5B by splice switching SSOs leads to a dose-dependent decrease in cell proliferation (Figure 5). In addition a clonogenic assay, which reflects the malignant potential of cancer cell lines, indicated a specific and significant reduction in prostate cancer cell viability as induced by SSOs treatment (Figure 6).
These data demonstrate the feasibility of using specific steric-blocking splice-switching oligonucleotides with a complimentary sequence to induce alternative intron/exon retention to convert a proto-oncogene (full length STAT5B) into a tumor suppressor (dominant-negative truncated form of STAT5B) as a possible anti-tumor therapeutic strategy. Moreover, alternative splice-switching oligonucleotides inducing a switch from proto-oncogene to tumor suppressor functions could potentially be used for therapeutic tuning of other STAT proteins.
A common feature of most STATs is alternative splicing, which leads to generation of a dominant-negative isoform. STAT proteins are involved in wide variety of physiological processes including immune response and tumor progression. Ability to modulate their actions and specifically switch function from tumor activating to tumor suppressing would be highly beneficial in many areas of biomedical research. In conclusion we developed and confirmed a novel method to implement steric-blocking splice-switching oligonucleotides for targeted delivery towards the development of novel therapeutic strategies.
AKNOWLEDGEMENTS
This work was supported by National Institute of Health (SC3GM087201) and by Frances Rusteberg Faculty Fellowship at The University of Texas at Brownsville. The authors thank Dr. Kari Brewer-Savannah and Dr. Daniele Provenzano from UTB for useful suggestions and critical reviewing of the manuscript.
COMPLETING INTERESTS
The authors declare no competing interests.
References
1. Darnell J.EJr, Kerr I.M, Stark G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415-21
2. Ihle J.N, Kerr I.M. Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet. 1995;11(2):69-74
3. Schindler C, Strehlow I. Cytokines and signaling STAT. Adv Pharmacol. 2000;47:113-74
4. Kazansky A.V. et al. Regulation of mammary gland factor/Stat5a during mammary gland development. Mol Endocrinol. 1995;9(11):1598-609
5. Lim C.P, Cao X. Structure, function, and regulation of STAT proteins. Mol Biosyst. 2006;2(11):536-50
6. Ramos H.L, O'Shea J.J, Watford W.T. STAT5 isoforms: controversies and clarifications. Biochem J. 2007;404(1):e1-2
7. Chen H. et al. VEGF, VEGFRs expressions and activated STATs in ovarian epithelial carcinoma. Gynecol Oncol. 2004;94(3):630-5
8. Debierre-Grockiego F. Anti-apoptotic role of STAT5 in haematopoietic cells and in the pathogenesis of malignancies. Apoptosis. 2004;9(6):717-28
9. Li H. et al. Activation of signal transducer and activator of transcription 5 in human prostate cancer is associated with high histological grade. Cancer Res. 2004;64(14):4774-82
10. Kazansky A.V, Greenberg N.M. Role STAT5B in Prostate Cancer Progression. (manuscript in preparation). 2005
11. Kazansky A.V, Spencer D.M, Greenberg N.M. Activation of signal transducer and activator of transcription 5 is required for progression of autochthonous prostate cancer: evidence from the transgenic adenocarcinoma of the mouse prostate system. Cancer Res. 2003;63(24):8757-62
12. Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4(2):97-105
13. Xi S. et al. Constitutive activation of Stat5b contributes to carcinogenesis in vivo. Cancer Res. 2003;63(20):6763-71
14. Morcinek J.C. et al. Activation of STAT5 triggers proliferation and contributes to anti-apoptotic signalling mediated by the oncogenic Xmrk kinase. Oncogene. 2002;21(11):1668-78
15. Kazansky A.V, Rosen J.M. Signal Transducer and Activator of Transcription 5B Potentiates v-Src-mediated Transformation of NIH-3T3 Cells. Cell Growth & Differentiation. 2001;12:1-7
16. Kazansky A. et al. Differential effects of prolactin and src/abl kinases on the nuclear translocation of STAT5B and STAT5A. The Jornal of Biological Chemistry. 1999;274(32):22484-92
17. Dagvadorj A. et al. Transcription factor signal transducer and activator of transcription 5 promotes growth of human prostate cancer cells in vivo. Clin Cancer Res. 2008;14(5):1317-24
18. Fiebig H.H, Maier A, Burger A.M. Clonogenic assay with established human tumour xenografts: correlation of in vitro to in vivo activity as a basis for anticancer drug discovery. Eur J Cancer. 2004;40(6):802-20
19. Gu L. et al. Stat5 promotes metastatic behavior of human prostate cancer cells in vitro and in vivo. Endocr Relat Cancer. 2010;17(2):481-93
20. Santos C.I, Costa-Pereira A.P. Signal transducers and activators of transcription-from cytokine signalling to cancer biology. Biochim Biophys Acta. 2011;1816(1):38-49
21. Bauman J, Jearawiriyapaisarn N, Kole R. Therapeutic potential of splice-switching oligonucleotides. Oligonucleotides. 2009;19(1):1-13
22. Singh N.K. et al. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. 2006;26(4):1333-46
23. Hua Y. et al. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007;5(4):e73
24. Sazani P, Kole R. Therapeutic potential of antisense oligonucleotides as modulators of alternative splicing. J Clin Invest. 2003;112(4):481-6
25. Sazani P. et al. Nuclear antisense effects of neutral, anionic and cationic oligonucleotide analogs. Nucleic Acids Res. 2001;29(19):3965-74
26. Roberts J. et al. Efficient and persistent splice switching by systemically delivered LNA oligonucleotides in mice. Mol Ther. 2006;14(4):471-5
27. Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45
28. Cao X. et al. Activation and association of Stat3 with Src in v-Src-transformed cell lines. Mol Cell Biol. 1996;16(4):1595-603
29. Bauman J.A. et al. Anti-tumor activity of splice-switching oligonucleotides. Nucleic Acids Res. 2010;38(22):8348-56
30. Montgomery R.L. et al. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation. 2011;124(14):1537-47
31. Muntoni F, Wood M.J. Targeting RNA to treat neuromuscular disease. Nat Rev Drug Discov. 2011;10(8):621-37
32. Feng J. et al. Antisense oligodeoxynucleotides targeting ATM strengthen apoptosis of laryngeal squamous cell carcinoma grown in nude mice. J Exp Clin Cancer Res. 2011;30:43
33. Singh N.N. et al. A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol. 2009;6(3):341-50
Author contact
![]() Corresponding author: Department of Biomedicine, BRHB 1.122, The University of Texas at Brownsville, 80 Fort Brown, Brownsville, TX 78520, USA. Tel: 1 (956) 882-5780; Email: Alexander.Kazanskyedu.
Corresponding author: Department of Biomedicine, BRHB 1.122, The University of Texas at Brownsville, 80 Fort Brown, Brownsville, TX 78520, USA. Tel: 1 (956) 882-5780; Email: Alexander.Kazanskyedu.