Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Material and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(1):67-77. doi:10.7150/ijbs.5577 This issue Cite
Research Paper
Inhibition of Filamin-A Reduces Cancer Metastatic Potential
Xi Jiang1, 2, *, Jingyin Yue2,*, Huimei Lu2, Neil Campbell2, Qifeng Yang2,3, Shijie Lan1,2, Bruce G. Haffty2, Changji Yuan1, ![]() , Zhiyuan Shen2,
, Zhiyuan Shen2, ![]()
1. Cancer Center, The First Hospital of Jilin University, Changchun, Jilin Province, China.
2. The Cancer Institute of New Jersey, New Brunswick, NJ 08903, USA.
3. Current address: Qilu Hospital, Shandong University, Department of Breast Surgery, Jinan, Shandong Province, China.
* Authors with equal contribution.
Received 2012-11-21; Accepted 2012-12-16; Published 2012-12-21
Abstract
Filamin-A cross-links actin filaments into dynamic orthogonal networks, and interacts with an array of proteins of diverse cellular functions. Because several filamin-A interaction partners are implicated in signaling of cell mobility regulation, we tested the hypothesis that filamin-A plays a role in cancer metastasis. Using four pairs of filamin-A proficient and deficient isogenic cell lines, we found that filamin-A deficiency in cancer cells significantly reduces their migration and invasion. Using a xenograft tumor model with subcutaneous and intracardiac injections of tumor cells, we found that the filamin-A deficiency causes significant reduction of lung, splenic and systemic metastasis in nude mice. We evaluated the expression of filamin-A in breast cancer tissues by immunohistochemical staining, and found that low levels of filamin-A expression in cancer cells of the tumor tissues are associated with a better distant metastasis-free survival than those with normal levels of filamin-A. These data not only validate filamin-A as a prognostic marker for cancer metastasis, but also suggest that inhibition of filamin-A in cancer cells may reduce metastasis and that filamin-A can be used as a therapeutic target for filamin-A positive cancer.
Keywords: Filamin-A, ABP-280, migration, invasiveness, metastasis, biomarker.
Introduction
Among all cancer mortalities, 90% exhibit systemic disease with metastases [1, 2]. Because metastasis is a complex process, involving detachment of tumor cells from the primary sites, invasion, migration, intravasation, survival in the vasculature, extravasation, and colonization at the secondary sites, it is regulated by many intra-cellular and extra-cellular factors. The identification and characterization of these biological molecules that play roles in cancer metastasis will not only provide valuable diagnostic and prognostic markers but also may serve as therapeutic targets.
The human Filamin-A, also known as Actin Binding Protein 280 [3, 4], is a protein of 2647 amino acids. It forms a homo-dimer and cross-links cortical actin filaments into a dynamic three-dimensional structure [5-7]. Filamin-A interacts with more than 60 functionally diverse proteins, serving as the scaffold in various signaling networks [8, 9]. Among these filamin-A interacting proteins, many are implicated in cancer metastasis, such as cell membrane receptor integrins, small GTPases, and others [10-15]. However, the direct role of filamin-A in cancer metastasis remains elusive. In this study, we validate filamin-A as a cancer prognostic marker and therapeutic target. We show that inhibition of filamin-A significantly reduces migration and invasion of cancer cells, and alleviates both spontaneous and systemic metastases of xenograft cancer cells in nude mice. Furthermore, low levels of filamin-A expression in breast cancer tissues are associated with a better outcome in term of distant metastasis-free survival. These data suggest that filamin-A is a valuable biomarker for cancer metastasis, and filamin-A also may be used as a target to inhibit cancer metastasis.
Material and methods
Cell lines and cell cultures
Four pairs of isogenic cell lines were grown in a 37°C incubator supplied with 5% CO2, and with the same culture media as described previously[16, 17]. The M2 melanoma cell spontaneously lost filamin-A expression, and the A7 cell is a derivative of M2 with exogenous filamin-A expression. Melanoma cell line C8161, breast cancer cell line MDA-MB-231 (MB231) and MDA-MB-436 (MB436) have normal filamin-A expression, and were stably transfected with vector expressing shRNA against filamin-A as previously described[16-18], resulting in C8161-KD, MB231-KD and MB436-KD. Another shRNA targeting a different region of filamin-A was used to knockdown filamin-A in C8161 cells and labeled as C8161-KD1. The corresponding control cells (C8161-con, MB231-con and MB436-con) were transfected with vectors expressing control shRNA. The knockdown efficiency of filamin-A by shRNA has been demonstrated previously [16]. It has also been shown that the deficiency of filamin-A in these cells does not affect in vitro growth and tumor xenograft growth at the inoculation sites in the nude mice[16-18].
Wound healing assay
Log-phase cells were plated into 10cm dishes. When the cell culture reached 70-80% confluence, the cells were cultured in serum-deprived medium that contains 0.2% bovine serum albumin (BSA) for 24 hours, and then gaps of 1mm open spaces (wound) were generated manually by scratching the monolayer of cell culture. The “healing” effect was monitored microscopically periodically as the cells migrate to cover the blank surface in complete growth media with 10% serum. Photographs were taken at 0, 12 and 24 hours after the wound was generated.
Time-elapse microscopy
Cells were plated into 24-well plate 8 hours prior to time-lapse migration experiments. Time-lapse microscopy experiments were performed using a Carl Zeiss fluorescent microscope (Axiovert-200M) equipped with a Carl Zeiss digital camera (AxioCam MRC), an automated stage controller and an environmental chamber that maintains temperature, humidity, and CO2 levels. Images of individual cells were captured using 10× objective lens at 6-min time intervals for 10 hours. AxioVision software with tracking module (Carl Zeiss MicroImaging GmbH) was used to calculate velocity. About 15 cells were analyzed for each experiment, and the experiment was conducted three times. The results are presented as mean±SD and student's t-test was used for the analysis of statistical significance.
Trans-well (Boyden Chamber) cell invasion assay
Log-phase cells were cultured in serum-deprived medium for 24 hours. The cells were then harvested, washed twice in PBS, re-suspended in culture medium with 0.2 % BSA, and then adjusted to a final concentration of 4×105/mL. BD Falcon™ cell culture inserts (with 8.0 µm pore size coated with Matrigel) were used. Then, 0.9 mL of complete growth media (with 10% serum) were added into each well of cell culture plate as chemo-attractant solution in the outer chamber. In the culture inserts (inner chamber), 0.3mL of cell suspension were added. Cell cultures were maintained in incubator. After 4, 12 and 24 hours of incubation, cells inside the culture inner chamber were removed with cotton swab and cells migrated through membrane were trypsinized and counted. Each time-point has triplicate culture inserts, and experiment repeated twice. Results are expressed as mean±SD and statistical significance was analyzed using the Student's t-test.
In vivo xenograft model for melanoma metastasis
The animal protocols used in this study were approved by the Robert Wood Johnson Medical School Institutional Animal Care and Use Committee (IACUC). The C8161 cells reproducibly form xenograft tumors at the inoculation sites, and were therefore used in this assay. Log-phase EGFP-labeled C8161 cells were inoculated subcutaneously in nude mice (1×106 cells per mouse) to generate tumors, 8 mice for each cell line, with normal or reduced level of filamin-A. Five weeks after inoculation, mice were sacrificed and dissected to collect lung and spleen specimens. After recording the weight, the organs were fixed in formalin, and used to generate paraffin-embedded tissue sections. Lung surface metastases were visualized after fixation in 10% Bouin's solution. Spontaneous metastases in organs were confirmed by immunohistochemistry (IHC) staining with antibody against GFP. The incidence of metastasis was calculated and statistical significance was analyzed using the Chi-Square Test.
Systemic metastasis resulting from intracardiac injection of tumor cells
Log-phase EGFP-labeled melanoma cells C8161 and breast cancer cells MDA-MB-231 with normal or reduced levels of filamin-A were inoculated in nude mice by intracardiac injection (1×106 cells per injection and 4 mice for each group). Four and eight weeks after injection with C8161 and MB-231 cells, tumor metastases were detected using the EGFP marker with Kodak 2000MM Image Station (Eastman Kodak Company, New Haven, USA). Then the animals were sacrificed and femurs were collected for bone microarchitecture analysis using Micro-computed tomography (µCT) analysis. Three-dimensional μCT studies were performed by the Preclinical Imaging Shared Resource at the Cancer Institute of New Jersey. Femurs were fixed in 10% neutralized formalin and dehydrated in 70% alcohol. Femur samples were scanned using the INVEON PET/CT (Siemens Healthcare). Images were acquired at the highest resolution and without CCD binning, providing a voxel size of 9.44 μm. A 1° rotation step through a 360° angular range with 6500msec exposure was used. CT scans were reconstructed with Beam Hardening Correction and Hounsfield Calibration before being analyzed using Inveon Research Workplace (IRW) software (Siemens Healthcare). After processing with a 3D Gaussian Filter, segmentation of regions of interest were conducted and measurements of cortical thickness were made using IRW. A 3mm shaft region of cortical bone 4mm proximal to the distal tip of the femur and a 94μm section immediately proximal to the distal physis was analyzed for each sample.
Immunohistochemical analysis of tissue microarray for the expression of filamin-A
A tissue microarray was constructed for this analysis as described previously [19]. The study was approved by our institutional IRB. In brief, this paraffin-embedded tissue micro-array contains a duplicated set of breast cancer tissues of 158 cases of stage I/II breast cancers. A similar procedure as reported previously[20] was adapted for IHC staining. Briefly, the 5 μm-thick tissue sections were de-paraffinized with xylene. The antigen was retrieved in citrate acid buffer (pH 6.5) by steaming in a rice cooker for 20 min. Slides were blocked with 5% milk in TBS-T (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% Tween 20) for 30 min at room temperature, and incubated with mouse monocolonial anti-filamin-A antibody (MAB1678, EMD Millipore Corporation, Billerica, MA, 1:100 dilution) for 3 hours at room temperature. Following three washes with TBS-T, the slides were incubated for 1 hour at room temperature with anti-Mouse IgG secondary antibody (1:100 dilution) that was conjugated with horseradish peroxidase. The filamin-A positive stain (in brown color) was visualized by incubating the slides with chromogenic substrate diaminobenzidine (DAB), followed by 10-second contrast staining (in blue color) with hematoxylin for the nuclei. The slides were then dehydrated and mounted.
Assessment of filamin-A staining among the breast cancer epithelial cells other than the stroma tissues was quantitative and done by two independent pathologists who were blinded to patient outcome (HL and QY). For each duplicate core, the region of predominant staining intensity was scored. A case was scored as filamin-A negative only when the positively stained cancer cell area(s) were less than 25% of the total cancer epithelial tissue area on both of the duplicate tissue cores, otherwise the case was counted as filamin-A positive.
Statistical analysis
Filamin-A status was assembled in a database and analyzed using SAS User's Guide, Version 9.1 (SAS Institute, Cary, NC). The study endpoint was distant metastasis-free survival and calculated from the date of radiation therapy completion, as this represents the start of continuous risk for relapse. Distant metastases were defined as clinical evidence of distant disease based on clinical and/or radiographic evidence. The test of statistical significance was two-sided and value of p<0.05 was considered statistically significant.
Results
Inhibition of filamin-A impairs migration of cancer cells
It has been reported that filamin-A interacts with Rho GTPases [12-14] to reorganize the actin network, which may be required to generate forces for cell local motion. Filamin-A also regulates the trafficking of cell adhesion receptor integrins back to cell surface, which is critical for cell spreading and migration [10, 21]. We hypothesized that inhibition of filamin-A in the cancer cells reduces cell motility and subsequently cancer metastasis. To test this hypothesis, we used sets of previously established isogenic melanoma and breast cancer cells[16-18], including melanoma cell line C8161, breast cancer cell line MDA-MB-231 (MB231), and MDA-MB-436 (MB436). In those cells the filamin-A expression was inhibited by filamin-A shRNA[16-18]. The M2 melanoma cells that had spontaneously lost filamin-A expression, and the A7 cells derived by re-expressing filamin-A in M2 cells were also used.
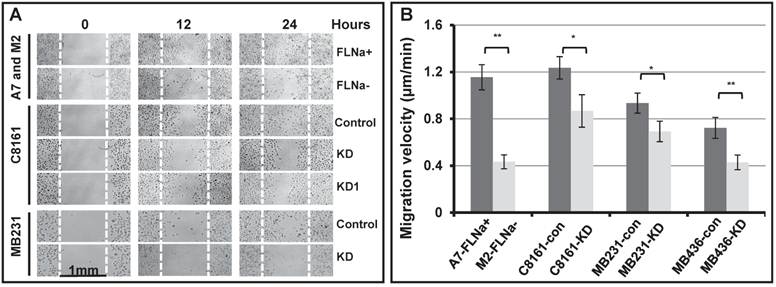
To test the hypothesis, we first applied the wound healing assay (Figure 1A). The gaps of 1 mm wide open space (wounds) were manually generated on sub-confluent monolayer of cell culture with serum starvation pre-treatment. The bright field images of the gaps were taken at various times after the gaps were generated. As shown in Figure 1A, the filamin-A deficient M2, C8161 (FLNa-KD and FLNa-KD1), MB231 (FLNa-KD) are much less efficient to migrate to the scratch culture surface than the filamin-A proficient A7, C8161 (control-shRNA), and MB231 (control-shRNA) cells within 24 hours after the gaps were generated. These data suggest that the filamin-A deficient cells are not as mobile as the proficient cells. To confirm this, we then applied time-lapse microscopy to monitor the cell migration. Cell migration velocity was calculated using series images (see Material and Methods). As shown in Figure 1B, a significant decrease in migration speed was observed in filamin-A deficient cells compared with filamin-A wild type cells (p<0.05 or p<0.01).
Reduced cell migration ability in filamin-A deficient cells. Four pairs of isogenetic filamin-A proficient and deficient cell lines were used (see Material and Methods for details): A7 (FLNa+) and M2 (FLNa-); C8161-con (filamin-A control) and C8161-KD (filamin-A knockdown); MB231-con (filamin-A control) and MB231-KD (filamin-A knockdown); MB436-con (filamin-A control) and MB436-KD (filamin-A knockdown). Wound healing assay and time-lapse microscopy were used to assess the ability of cell migration. Panel A shows the wound healing assay from 3 paired cell lines as indicated in the panel. The gaps of 1mm open space (wounds) were generated on semi-confluent monolayer cell culture. Images shown figure were taken at 0, 12 and 24 hours after wounds generated. White dotted line was used to indicate the edge of original wound generated on the surface of cell culture. Panel B shows the migration velocity for each cell line, which was calculated using time-lapse series images (see Material and Methods for details). * p<0.05 and ** p<0.01.
Inhibition of filamin-A impairs invasive potential of cancer cells
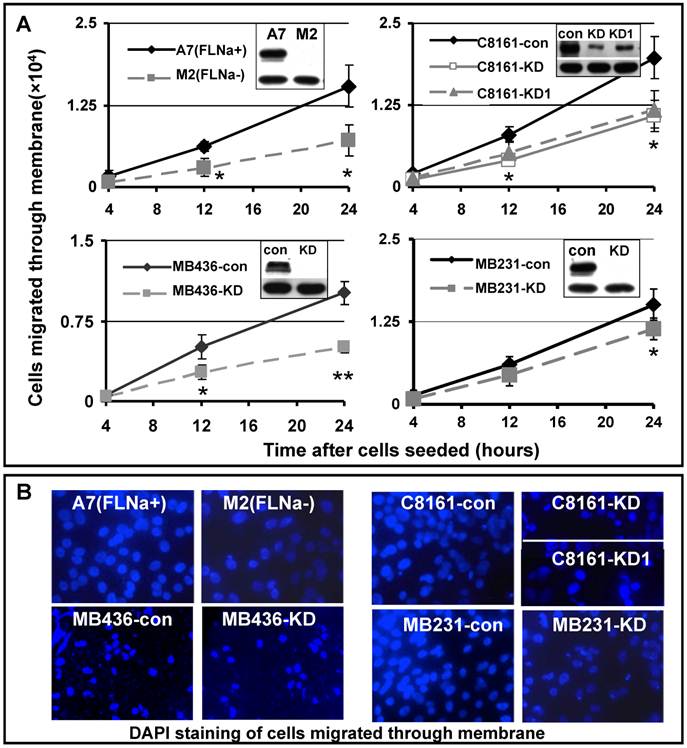
Cell migration is a process of directional movement [22], and loss of filamin-A compromises cell motility in culture as shown in Figure 1. Another critical step in cancer metastasis is the detachment of tumor cells from the primary sites, and invading the surrounding tissue. To test whether inhibition of filamin-A reduces cell invasion, we applied the Boyden chamber assay, which mimics the micro-environment at primary tumor sites. We inoculated the cells into the inner chamber, which is separated from the outer chamber by an 8 μM pored membrane on the bottom of the inner chamber. In order to migrate out of the inner chamber, the cells need to sense the chemo-attractant in the outer chamber (provided by serum), digest extra-cellular matrix (Matrigel) covering the pores, and change cell shape to migrate through the 8μM sized pores on the membrane, and reattach to the outer chamber side of the membrane. At various times after the cells were inoculated to the inner chamber, the cells migrated through the membrane were counted. As shown in Figure 2A, expression of filamin-A in A7 cells significantly increased the numbers of cells migrated through the membrane, when compared with the M2 cells that spontaneously lost filamin-A expression. In agreement with this, filamin-A knockdown in the filamin-A proficient C8161, MB436, and MB231 cells significantly reduced the number of cells migrated through the membranes. Fewer numbers of filamin-A deficient cells attached on the outer side of the membrane than filamin-A proficient cells were illustrated by DAPI staining after 24 hours of incubation (Figure 2B). The expression levels of filamin-A in each paired cell lines were detected by Western-Blot as shown in the top panels of insertions of Figure 2A. β-actin (lower panel in the inserts in Figure 2A) was used as loading control. Thus, Figures 1 and 2 clearly suggest that filamin-A plays an important role in cell migration and invasion, two critical properties associated with cancer metastasis.
Reduced cell invasiveness in filamin-A deficient cells. Boyden chamber trans-well assay was used to assess the cell invasiveness. Serum deprived cells were seeded in cell culture inner chamber coated with Matrigel, and complete growth medium was added into outer chamber as chemoattractant. Panel A shows the numbers of cells migrated from the inner chamber side of the membrane to outer chamber side of the membrane after 4, 12 and 24 hours incubation. The insertions in panel A are showing the expression levels of filamin-A in paired cell lines detected by Western-Blot. β-actin was used as loading control. Panel B shows the DAPI staining of cells attached to outer side of the membrane after 24 hours incubation (*p<0.05, and ** p<0.01).
Filamin-A deficiency reduces organ metastasis in nude mice xenograft model
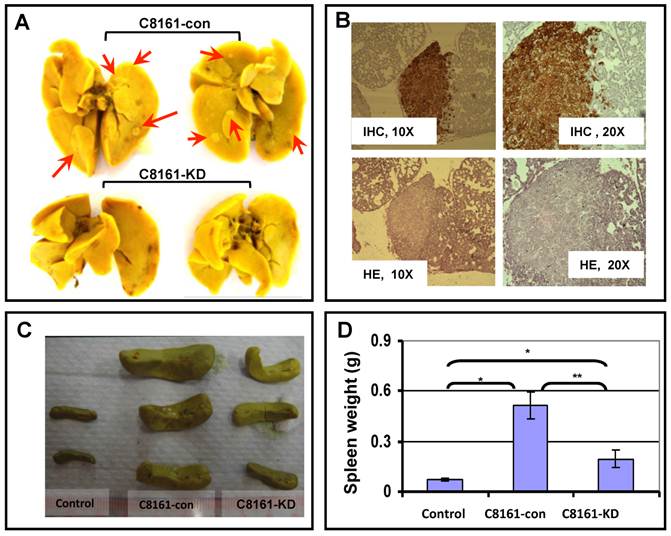
To directly test the role of filamin-A in cancer metastasis, we generated tumor xenograft using human melanoma cell line C8161. The C8161 cells were used for this experiment because they can form solid tumor reproducibly in nude mice and have high potential of metastasis. EGFP expressing C8161-con cells (with filamin-A) or C8161-KD cells (filamin-A knockdown) were injected into mice subcutaneously. Five weeks after inoculation, mice were sacrificed for dissection. As we reported previously, the filamin-A expression status does not affect the growth of EGFP expressing C8161 tumors at the inoculation sites[17, 18]. However, metastatic foci were visible on the surface of lungs collected from C8161-con tumor bearing mice after staining with Bouin's solution (Figure 3A). Routine H&E staining and immunohistochemistry staining with GFP antibody of the lung tissue sections further confirmed that the tumor metastases originated from the EGFP-expressing C8161 cells (Figure 3B). The incidence of lung metastasis was calculated, and is shown in Table 1 based on IHC staining with GFP antibody using lung tissue sections. Significant higher incidence was observed in lungs from filamin-A expressing tumor xenograft than filamin-A deficient ones, 87.5% vs 12.5% (p<0.01). The tumor metastasis in spleen was also assessed. As shown in Figure 3C and 3D, there are significant increases of spleen weights in mice injected with C8161-con cells than the C8161-KD cells, indicating more severe metastatic infiltration occurs in mice injected with filamin-A expressing tumor cells. These data collectively suggested that filamin-A positive cells have a stronger capability to metastasis into organs after subcutaneous inoculation, and inhibition of filamin-A expression significantly decreased the metastatic potential when tumor are locally formed by subcutaneous inoculation (Figure 3 and Table 1).
The incidence of lung metastasis in nude mouse tumor xenograft. IHC staining was performed with antibody against EGFP using lung tissue sections. Lung metastases were visualized under and counted under microscope, and then the incidence of lung metastasis was calculated. The Chi-Square Test was used for statistical analysis and p<0.05 was considered significant.
| Group | Number of mice inoculated | Number of mice with lung metastasis | Lung metastasis incidence (%) | Average number of lung metastases |
|---|---|---|---|---|
| C8161-con | 8 | 7 | 87.5 (7/8)** | 12.14±3.24 |
| C8161-KD | 8 | 1 | 12.5 (1/8)** | 2 (from 1 lung) |
** p<0.01.
Effect of filamin-A on organ metastasis in nude mouse xenograft model. Tumor bearing mouse was generated by injecting EGFP-expressing C8161-con or C8161-KD melanoma cells into nude mice subcutaneously. Mice were sacrificed to collect lungs and spleens at 5-week after the inoculation. Panel A shows the lung metastases after Bouin's solution staining (Red arrows). Panel B shows the IHC staining with GFP antibody and HE staining of lung tissue section from mouse with C8161-con tumor, which indicates the lung metastasis focus. Panel C shows the image of representative spleens collected from non-injecting mice, C8161-con tumor bearing mice, and C8161-KD tumor bearing mice. Panel D shows the average weight of spleens from three groups of mice.
Lack of filamin-A decreases systemic metastasis in nude mice
A complementary approach to assess metastatic potential is to inoculate tumor cells in the blood stream directly, via intracardiac injection, to assess systemic metastasis of cancer cells in an animal model. We injected EGFP labeled MB231 breast cancer cells and C8161 melanoma cells into nude mice through left ventricle injection, and monitored the systemic metastasis. As shown in Figures 4A, mice injected with MB231-con or C8161-con cells showed dramatic weight loss at 8 or 4 weeks after injection respectively, when compared to mice injected with MB231-KD or C8161-KD cells. In addition, there is more widespread metastasis in the filamin-A proficient MB231 and C8161 cells than the deficient counterparts, based on results acquired with animal live imaging system (Figure 4B). These data again demonstrate that filamin-A proficient tumor cells have high metastatic potential than the filamin-A deficient cells.
Systemic metastasis of cancer cells in nude mouse model. EGFP labeled breast cancer cell line MB231 and melanoma cell line C8161 were transfected with vectors expressing control shRNA or filamin-A targeting shRNA. Stable clones with filamin-A expression or filamin-A knock-down were isolated. Log-phase cells were injected into left ventricles of nude mice and body weight of injected mice were monitored weekly as shown in panel A. Four and eight weeks after injection, the mice inoculated with MB231-Con and C8161-Con displayed significant body weight loss (*p<0.05). Live images with EGFP marker were taken from anesthetic mice 8 weeks after injection for MB231 cells and 4 weeks for C8161 cells as shown in panel B. Hot spots of suspect tumor metastases were labeled with arrows and arrow heads for mouse injected with MB231-con and C8161-con cells respectively. Panel C shows the representative images of 94 μm sections proximal to distal physis. Panel D shows the quantitative analysis of physis wall thickness, which reduced significantly in mice with MB231-con tumor cells inoculation than in those injected with MB231-KD cells (p=0.022).
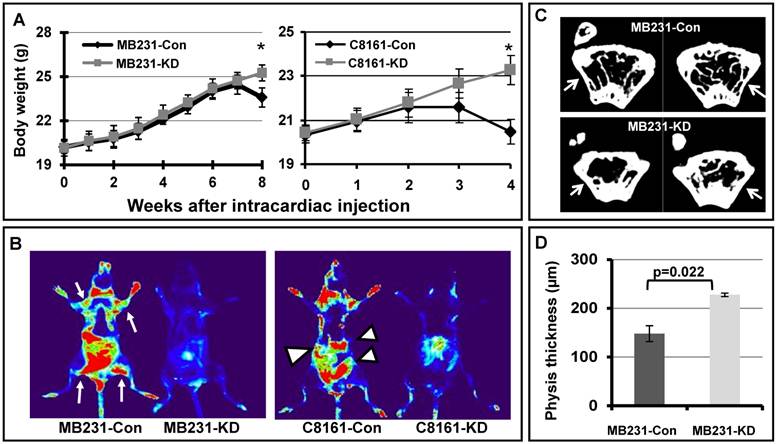
The bone marrow is one of the homing sites after intracardiac inoculation of metastatic breast cancer MDA-MB231. Since the growth of metastatic tumor cells and osteolytic lesions were more easily observed in the regions close to growth plate of distal physis [23], we collected femurs from sacrificed mice and measured the wall thickness of distal femur after 3D image reconstruction. Shown in Figure 4C are the representative images of 94 μm sections proximal to distal physis, and a significant decrease in wall thickness can be observed in femurs from mice injected with MB231-con cells as indicated by arrow in the panel, when compared with that of the MB231-KD cells. We further measured the distal physis wall thickness from 20 continuous images for each femur and quantitative data are shown in Figure 4D. The average physis wall thickness is 147.93µm and 227.55µm for mice injected with MB231-con and MB231-KD cells, respectively. Thus, a significant wall thickness reduction (~35%) was observed in mice with filamin-A wild type cells (MB231-con) injection compared with those inoculated with filamin-A deficient cells (MB231-KD) (p=0.022). However, there was no significant difference in femur wall thickness at regions away from growth plate in mice injected with different cells (Supplementary Material: Figure S1), suggesting these animals were in similar age and metastatic tumor cells were less likely homing at these regions of the femur. Therefore, in agreement with in vivo fluorescent imaging data (Figure 4A and 4B), bone µCT scanning results further demonstrate that tumor cells with filamin-A expression have stronger potential to form metastases in nude mice xenograft model.
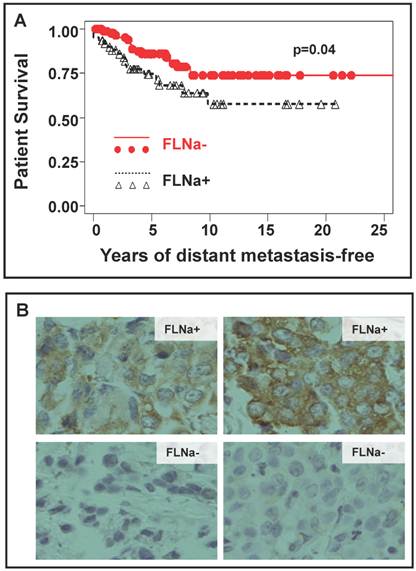
Reduction of filamin-A is associated with a better clinical survival of distant metastasis
Figures 1-4 have suggested that inhibition of filamin-A reduces cancer cell mobility and invasiveness, and decreases the metastatic potential. To directly validate this conclusion with clinical evidence, we performed IHC staining of filamin-A on a panel of paraffin-embedded breast cancer tissue-array. This array contains a duplicated set of breast cancer tissues of 158 cases of stage I/II breast cancers. The details of patient recruitment, treatment history, and follow-up of these patients were described previously [19]. Of the tissue array slides stained, 153 cases provided reliable information on filamin-A expression status. A case was scored as filamin-A negative only when the positively stained cancer epithelial cells account for less than 25% of the total cancer tissue area excluding the stromal regions (examples shown in Figure 5B). Based on the scoring by two independent pathologists, 92 out of 153 cases were filamin-A negative, and 61 were positive. When number of cases was plotted against the years of patient survival without distant metastasis as shown in Figure 5A, filamin-A negative breast cancer patients have significantly better survival than the filamin-A positive ones (p<0.05). This observation suggests that lack of filamin-A expression is associated with better survival due to lack of distant metastasis. It further supports the concept that lack-of-filamin-A may be used as a prognostic marker for lower risk of breast cancer metastasis, and inhibition of filamin-A may reduce cancer metastasis for filamin-A positive cancers.
Correlation of filamin-A expression status in tumor cells with patient survival. The paraffin-embedded tissue array with breast cancer tissues was performed immunohistochemistry staining with filamin-A antibody and filamin-A expression status was scored following the criteria stated in Material and Method. Panel A shows the plot of survival against years of distant metastasis-free in patients with filamin-A negative tumor and filamin-A positive tumor (p=0.04). Panel B shows the representative images of filamin-A negative and filamin-A positive tumor cell staining.
Discussion
Filamin-A may transform extra cellular signals into dynamic remodeling of actin cytoskeleton. Since reorganization of the actin cytoskeleton is essential for cell migration [24, 25], we hypothesized that inhibition of filamin-A may reduce cell migration and cancer metastasis, thus can be used as a therapeutic target for cancer metastasis inhibition. We show that down-regulation of filamin-A expression in filamin-A proficient cancer cells can effectively reduce their mobility and invasiveness, and inhibit metastasis on xenograft models. Consistent with this is that when filamin-A is re-expressed in filamin-A deficient M2 cells, it can restore the cell mobility. These data support a notion that for filamin-A proficient cancer, it is possible to inhibit filamin-A to reduce metastatic potential. Thus filamin-A may be used as a therapeutic target for cancer metastasis inhibition. We also show that lack of filamin-A expression in breast cancer tissues is correlated with better outcome in term of distant metastasis-free survival, thus filamin-A status in cancer may be used as a prognostic marker for metastasis.
The mechanism by which filamin-A regulates cell mobility and cancer metastasis is yet to be fully elucidated. However, the ability of filamin-A to re-organize the orthogonal actin cytoskeleton network may be an essential factor. It is known that filamin-A plays a role in the orthogonal actin cytoskeleton network reorganization that generates forces for cell locomotion [3, 7]. In addition to actin network, filamin-A also interacts with Rho family small GTPases[10-14] and several cell membrane proteins including integrins and migfilin [11, 26, 27]. During directional migration, the cells generate protrusion at the leading edge to spread cell components forward, but the rear edge retracts so that cells can move forward. This process relies on the integrity of three polymer systems: actin filaments [28], microtubules [29] and intermediate filaments[30]. During this process, filamin-A may act as an integrator to receive signals from extra-cellular matrix and transmit into actin network reorganization to generate forces for cell motility through the interactions with its partners.
It has also been shown that filamin-A plays a role for the cells to withstand mechanical stress [31], which may also contribute to cancer metastasis. Filamin-A deficient cells may be vulnerable in the blood stream and dock to metastatic site less efficiently. To form metastases, invaded cells must survive in the blood stream, then attach and proliferate at secondary sites. As shown in Figure 4, with inoculation of EGFP labeled C8161 or MB231 cells into nude mice, we detected strong EGFP signals by fluorescent live imaging in mice injected with C8161-con or MB231-con cells, but not in mice with C8161-KD or MB231-KD cells. This difference could be attributed to the mechanoprotection effect of filamin-A [31] and more filamin-A proficient cells survived exogenous physical forces in the blood stream, or more filamin-A proficient cells attached and dock to new sites.
By using the isogenic cell pairs, A7 and M2, the role of filamin-A in cell spreading and migration was well demonstrated[32], and similar results were achieved from other model systems[33-35]. In current study, we employed four pairs of isogenic cell lines with various expression of filamin-A, including human melanoma and breast cancer cell lines. Our data further confirmed that loss of filamin-A significantly reduces the capabilities of migration and invasiveness of serum-starved (quiescent) cells, while the reduction in cell motility can be compensated by the expression of filamin-A in the cells (Figs 1 and 2). These findings are in strong agreement with many previous reports that implied a role of filamin-A in cell mobility[32-41].
To evaluate the impact of filamin-A status in cancer prognosis, Ai et al. employed the comparative proteomics and identified filamin-A as a potential marker for metastatic human hepatocellular carcinoma (HCC)[36]. Filamin-A is readily undergone proteolysis at the two hinge regions to generate cleaved fragments with various molecular weight, such as 170, 110 and 90 kDa fragments[5, 6], which is regulated by its phosphorylation on Ser2152 [5, 42]. By using the antibody against C-terminus of filamin-A that can detect full-length and the 90 kDa (repeats 16-23) proteins, Bedolla et al. performed IHC staining on human prostate tissue microarray. They found that filamin-A proteolysis, resulting in a nuclear localization of the 90 kDa fragment, is associated with a reduction of metastatic potential of prostate cancer [37]. They further found that the levels of cytoplasmic full-length filamin-A is significantly elevated in metastatic prostate cancer when compared with the clinically localized cancers, prostatic intraepithelial neoplasia (PIN), or benign prostate tissues, while stromal cells displayed stronger nuclear filamin-A staining than cytosol across all types of tissues [37]. In agreement with these findings, our IHC staining of breast cancer tissue microarray with antibody against N-terminus, which recognizes full-length filamin-A but not the 90 kDa fragment, showed that increased levels of filamin-A in cancer cells (not the stromal) are associated with high risk of distant metastasis in patients (Figure 5A). It is worth pointing out that the majority our IHC staining signals of filamin-A are localized in the cytoplasm of cancer cells (Figure 5B). Interestingly, the same study by Bedolla et al also reported that the nuclear filamin-A fragment levels become progressively reduced as the benign prostate epithelium gradually progress to intraepithelial neoplasia, clinically localized cancer, and metastatic cancer [37]. Therefore, the loss of nuclear localized fragment of filamin-A appears to be associated with prostate progression, but the increased cytoplasmic full-length filamin-A is associated with prostate cancer metastasis. This signifies a complex role of filamin-A in cancer progression.
While these studies clearly support a role of filamin-A in cancer metastasis and perhaps at a later stage of metastasis, a potential role of filamin-A in cancer cell focal adhesion to the extra cellular matrix thus resisting the initial escape of cancer cells from the original sites have been recently proposed[43]. In a few breast cancer cell lines that overexpress ErbB2 or with PI3K mutations, Xu et al reported an enhanced metastatic potential with filamin-A knockdown[43]. They also showed that the benign and in situ carcinoma were associated with a positive staining of filamin-A in the basal membrane of the mammary epithelial cells or the edge of the in situ carcinomas areas[43]. In our studies, we focused on the staining of filamin-A among the cancer cells of clinically developed breast cancers that does not include the early stage of the breast cancer (Figure 5B). Our scores were not based on the filamin-A staining in the matrix in the early stage of cancer development, and we used an antibody (MAB1678) that recognizes the cytoplasmic full-length filamin-A. Furthermore, the breast cancer cell lines we used in the study have low levels of ErbB2 and we did not over-express exogenous ErbB2 in these cells. Human metastatic melanoma cell line C8161 was also employed in addition to A7 and M2 melanoma cells, and further used in the xenograft study.
In additional to its roles in cell mobility control, filamin-A has been reported to interact with BRCA1[44] and BRCA2 [45], modulating DNA damage responses[16-18, 44-46]. Filamin-A deficient tumor xenografts have been shown to be more sensitive to ionizing radiation, bleomycin, and cisplatin[17]. In the current study, we observed a correlation between filamin-A negativity in breast cancer tumors and patients living free of distant metastasis (Figure 5). This raises a possibility that filamin-A deficient cancer will not only be more sensitive to some DNA damage based chemotherapy, but also will be less metastatic. Thus filamin-A deficient cancer will likely be more responsive to radiation and relevant chemotherapy and less metastatic at the same time. On the other hand, the filamin-A expressing cancers are likely to be more metastatic and less sensitive to the bleomycin and cisplatin type of treatments. Thus filamin-A may be a biomarker of high value for individualized treatment planning. Our data clearly suggest that, for filamin-A proficient cancer cells, inhibition of filamin-A can reduce the metastatic potential. Considering that inhibition of filamin-A can sensitize the cancer cells to some DNA damage based therapy[16, 17], we propose that filamin-A is a promising target for molecular based therapy as targeting filamin-A will not only reduce metastasis but also sensitize cancer to certain DNA damage based therapy.
In summary, our study demonstrates filamin-A plays an important role in cancer metastasis. Due to its role in regulating cancer response to therapeutic DNA damage[16, 17, 44], filamin-A can be used as a prognostic marker to predict cancer metastasis and cancer response to certain DNA damage based therapy, and filamin-A can also be used as an inhibition target to reduce metastasis and concurrently sensitize filamin-A proficient cancer to some DNA damage based therapy.
Supplementary Material
Fig. S1.
Acknowledgements
X. Jiang is a recipient of a scholarship under the State Scholarship Fund from China Scholarship Council. This research was supported by NIH R01CA156706 (ZS), the Breast Cancer Research Foundation (BGH), and the Preclinical Imaging Shared Resource(s) of The Cancer Institute of New Jersey (P30CA072720). We thank Dr. Michele Reiss (Cancer Institute of New Jersey) for the instruction during mouse intracardiac injection.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Sporn MB. The war on cancer. Lancet. 1996;347:1377-81
2. Weigelt B, Peterse JL, van 't Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005;5:591-602
3. Wang K. Filamin, a new high-molecular-weight protein found in smooth muscle and nonmuscle cells. Purification and properties of chicken gizzard filamin. Biochemistry. 1977;16:1857-65
4. Wang K, Ash JF, Singer SJ. Filamin, a new high-molecular-weight protein found in smooth muscle and non-muscle cells. Proc Natl Acad Sci U S A. 1975;72:4483-6
5. Gorlin JB, Yamin R, Egan S, Stewart M, Stossel TP, Kwiatkowski DJ. et al. Human endothelial actin-binding protein (ABP-280, nonmuscle filamin): a molecular leaf spring. J Cell Biol. 1990;111:1089-105
6. van der Flier A, Sonnenberg A. Structural and functional aspects of filamins. Biochim Biophys Acta. 2001;1538:99-117
7. Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M. et al. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol. 2001;2:138-45
8. Feng Y, Walsh CA. The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat Cell Biol. 2004;6:1034-8
9. Popowicz GM, Schleicher M, Noegel AA, Holak TA. Filamins: promiscuous organizers of the cytoskeleton. Trends Biochem Sci. 2006;31:411-9
10. Byfield FJ, Wen Q, Levental I, Nordstrom K, Arratia PE, Miller RT. et al. Absence of filamin A prevents cells from responding to stiffness gradients on gels coated with collagen but not fibronectin. Biophys J. 2009;96:5095-102
11. Kim H, Sengupta A, Glogauer M, McCulloch CA. Filamin A regulates cell spreading and survival via beta1 integrins. Exp Cell Res. 2008;314:834-46
12. Leung R, Wang Y, Cuddy K, Sun C, Magalhaes J, Grynpas M. et al. Filamin A regulates monocyte migration through Rho small GTPases during osteoclastogenesis. J Bone Miner Res. 2010;25:1077-91
13. Ohta Y, Hartwig JH, Stossel TP. FilGAP, a Rho- and ROCK-regulated GAP for Rac binds filamin A to control actin remodelling. Nat Cell Biol. 2006;8:803-14
14. Ohta Y, Suzuki N, Nakamura S, Hartwig JH, Stossel TP. The small GTPase RalA targets filamin to induce filopodia. Proc Natl Acad Sci U S A. 1999;96:2122-8
15. Kim H, Nakamura F, Lee W, Hong C, Perez-Sala D, McCulloch CA. Regulation of cell adhesion to collagen via beta1 integrins is dependent on interactions of filamin A with vimentin and protein kinase C epsilon. Exp Cell Res. 2010;316:1829-44
16. Yue J, Wang Q, Lu H, Brenneman M, Fan F, Shen Z. The cytoskeleton protein filamin-A is required for an efficient recombinational DNA double strand break repair. Cancer Res. 2009;69:7978-85
17. Yue J, Lu H, Liu J, Berwick M, Shen Z. Filamin-A as a marker and target for DNA damage based cancer therapy. DNA Repair (Amst). 2012;11:192-200
18. Yue J, Lan S, Yuan C, Shen Z. Prognostic values of filamin-A status for topoisomerase II poison chemotherapy. Int J Biol Sci. 2012;8:442-50
19. Parikh RR, Yang Q, Higgins SA, Haffty BG. Outcomes in young women with breast cancer of triple-negative phenotype: the prognostic significance of CK19 expression. Int J Radiat Oncol Biol Phys. 2008;70:35-42
20. Liu J, Lu H, Ohgaki H, Merlo A, Shen Z. Alterations of BCCIP, a BRCA2 interacting protein, in astrocytomas. BMC Cancer. 2009;9:268
21. Kim H, Nakamura F, Lee W, Shifrin Y, Arora P, McCulloch CA. Filamin A is required for vimentin-mediated cell adhesion and spreading. Am J Physiol Cell Physiol. 2010;298:C221-36
22. Lauffenburger DA. Cell motility. Making connections count. Nature. 1996;383:390-1
23. Daubine F, Le Gall C, Gasser J, Green J, Clezardin P. Antitumor effects of clinical dosing regimens of bisphosphonates in experimental breast cancer bone metastasis. J Natl Cancer Inst. 2007;99:322-30
24. Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G. et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704-9
25. Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453-65
26. Gehler S, Baldassarre M, Lad Y, Leight JL, Wozniak MA, Riching KM. et al. Filamin A-beta1 integrin complex tunes epithelial cell response to matrix tension. Mol Biol Cell. 2009;20:3224-38
27. Lad Y, Jiang P, Ruskamo S, Harburger DS, Ylanne J, Campbell ID. et al. Structural basis of the migfilin-filamin interaction and competition with integrin beta tails. J Biol Chem. 2008;283:35154-63
28. Mooney DJ, Langer R, Ingber DE. Cytoskeletal filament assembly and the control of cell spreading and function by extracellular matrix. J Cell Sci. 1995;108( Pt 6):2311-20
29. Mikhailov A, Gundersen GG. Relationship between microtubule dynamics and lamellipodium formation revealed by direct imaging of microtubules in cells treated with nocodazole or taxol. Cell Motil Cytoskeleton. 1998;41:325-40
30. Arocena M. Effect of acrylamide on the cytoskeleton and apoptosis of bovine lens epithelial cells. Cell Biol Int. 2006;30:1007-12
31. Glogauer M, Arora P, Chou D, Janmey PA, Downey GP, McCulloch CA. The role of actin-binding protein 280 in integrin-dependent mechanoprotection. J Biol Chem. 1998;273:1689-98
32. Cunningham CC, Gorlin JB, Kwiatkowski DJ, Hartwig JH, Janmey PA, Byers HR. et al. Actin-binding protein requirement for cortical stability and efficient locomotion. Science. 1992;255:325-7
33. Castoria G, D'Amato L, Ciociola A, Giovannelli P, Giraldi T, Sepe L. et al. Androgen-induced cell migration: role of androgen receptor/filamin A association. PLoS One. 2011;6:e17218
34. Guiet R, Verollet C, Lamsoul I, Cougoule C, Poincloux R, Labrousse A. et al. Macrophage mesenchymal migration requires podosome stabilization by filamin A. J Biol Chem. 2012;287:13051-62
35. Li C, Yu S, Nakamura F, Pentikainen OT, Singh N, Yin S. et al. Pro-prion binds filamin A, facilitating its interaction with integrin beta1, and contributes to melanomagenesis. J Biol Chem. 2010;285:30328-39
36. Ai J, Huang H, Lv X, Tang Z, Chen M, Chen T. et al. FLNA and PGK1 are two potential markers for progression in hepatocellular carcinoma. Cell Physiol Biochem. 2011;27:207-16
37. Bedolla RG, Wang Y, Asuncion A, Chamie K, Siddiqui S, Mudryj MM. et al. Nuclear versus cytoplasmic localization of filamin A in prostate cancer: immunohistochemical correlation with metastases. Clin Cancer Res. 2009;15:788-96
38. Fox JW, Lamperti ED, Eksioglu YZ, Hong SE, Feng Y, Graham DA. et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21:1315-25
39. Lange M, Winner B, Muller JL, Marienhagen J, Schroder M, Aigner L. et al. Functional imaging in PNH caused by a new FilaminA mutation. Neurology. 2004;62:151-2
40. Sato M, Nagano T. Involvement of filamin A and filamin A-interacting protein (FILIP) in controlling the start and cell shape of radially migrating cortical neurons. Anat Sci Int. 2005;80:19-29
41. Gawecka JE, Griffiths GS, Ek-Rylander B, Ramos JW, Matter ML. R-Ras regulates migration through an interaction with filamin A in melanoma cells. PLoS One. 2010;5:e11269
42. Jay D, Garcia EJ, Lara JE, Medina MA, de la Luz Ibarra M. Determination of a cAMP-dependent protein kinase phosphorylation site in the C-terminal region of human endothelial actin-binding protein. Arch Biochem Biophys. 2000;377:80-4
43. Xu Y, Bismar TA, Su J, Xu B, Kristiansen G, Varga Z. et al. Filamin A regulates focal adhesion disassembly and suppresses breast cancer cell migration and invasion. J Exp Med. 2010;207:2421-37
44. Velkova A, Carvalho MA, Johnson JO, Tavtigian SV, Monteiro AN. Identification of Filamin A as a BRCA1-interacting protein required for efficient DNA repair. Cell Cycle. 2010;9:1421-33
45. Yuan Y, Shen Z. Interaction with BRCA2 suggests a role for filamin-1 (hsFLNa) in DNA damage response. J Biol Chem. 2001;276:48318-24
46. Meng X, Yuan Y, Maestas A, Shen Z. Recovery from DNA damage-induced G2 arrest requires actin-binding protein filamin-A/actin-binding protein 280. J Biol Chem. 2004;279:6098-105
Author contact
![]() Corresponding author: yuanchangjicom or shenzhedu.
Corresponding author: yuanchangjicom or shenzhedu.