Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(1):119-133. doi:10.7150/ijbs.5441 This issue Cite
Research Paper
Cell Budding from Normal Appearing Epithelia: A Predictor of Colorectal Cancer Metastasis?
Bin Jiang1 ![]() , Jeffrey Mason2, Anahid Jewett3, Jun Qian1, Yijiang Ding1, William CS Cho4, Xichen Zhang5, Yan-gao Man5,6
, Jeffrey Mason2, Anahid Jewett3, Jun Qian1, Yijiang Ding1, William CS Cho4, Xichen Zhang5, Yan-gao Man5,6
1. National Medical Centre of Colorectal Disease, The Third Affiliated Hospital, Nanjing University of Chinese Medicine, Nanjing, China.
2. Laboratory of Proteomics and Protein Science, Veterans Affair Medical Center, Washington, DC, USA.
3. Tumor Immunology Laboratory, Division of Oral Biology and Medicine, Jonson Comprehensive Cancer Center, UCLA School of Dentistry, Los Angeles, CA. USA.
4. Department of Clinical Oncology, Queen Elizabeth Hospital, Hong Kong, China.
5. Animal Science and Veterinary Medicine, Jilin University, Changchun, Jilin, China.
6. Diagnostic and Translational Research Center, Henry Jackson Foundation, MD, USA.
Received 2012-10-23; Accepted 2012-12-21; Published 2013-1-11
Abstract
Background: Colorectal carcinogenesis is believed to be a multi-stage process that originates with a localized adenoma, which linearly progresses to an intra-mucosal carcinoma, to an invasive lesion, and finally to metastatic cancer. This progression model is supported by tissue culture and animal model studies, but it is difficult to reconcile with several well-established observations, principally among these are that up to 25% of early stage (Stage I/II), node-negative colorectal cancer (CRC) develop distant metastasis, and that circulating CRC cells are undetectable in peripheral blood samples of up to 50% of patients with confirmed metastasis, but more than 30% of patients with no detectable metastasis exhibit such cells. The mechanism responsible for this diverse behavior is unknown, and there are no effective means to identify patients with pending, or who are at high risk for, developing metastatic CRC.
Novel findings: Our previous studies of human breast and prostate cancer have shown that cancer invasion arises from the convergence of a tissue injury, the innate immune response to that injury, and the presence of tumor stem cells within tumor capsules at the site of the injury. Focal degeneration of a capsule due to age or disease attracts lymphocyte infiltration that degrades the degenerating capsules resulting in the formation of a focal disruption in the capsule, which selectively favors proliferating or “budding” of the underlying tumor stem cells. Our recent studies suggest that lymphocyte infiltration also triggers metastasis by disrupting the intercellular junctions and surface adhesion molecules within the proliferating cell buds causing their dissociation. Then, lymphocytes and tumor cells are conjoined through membrane fusion to form tumor-lymphocyte chimeras (TLCs) that allows the tumor stem cell to avail itself of the lymphocyte's natural ability to migrate and breach cell barriers in order to intravasate and to travel to distant organs. Our most recent studies of human CRC have detected nearly identical focal capsule disruptions, lymphocyte infiltration, budding cells, and the formation of TLCs. Our studies have further shown that age- and type-matched node-positive and -negative CRC have a significantly different morphological and immunohistochemical profile and that the majority of lymphatic ducts with disseminated cells are located within the mucosa adjacent to morphologically normal appearing epithelial structures that express a stem cell-related marker.
New hypothesis: Based on these findings and the growth patterns of budding cells revealed by double immunohistochemistry, we further hypothesize that metastatic spread is an early event of carcinogenesis and that budding cells overlying focal capsule disruptions represent invasion- and metastasis-initiating cells that follow one of four pathways to progress: (1) to undergo extensive in situ proliferation leading to the formation of tumor nests that subsequently invade the submucosa, (2) to migrate with associated lymphocytes functioning as “seeds” to grow in new sites, (3) to migrate and intravasate into pre-existing vascular structures by forming TLCs, or (4) to intravasate into vascular structures that are generated by the budding cells themselves. We also propose that only node-positive cases harbor stem cells with the potential for multi-lineage differentiation and unique surface markers that permit intravasation.
Keywords: Lymphocyte infiltration, tumor capsule, tumor invasion, tumor metastasis, stem cell.
Introduction
Worldwide, colorectal cancer (CRC) is one of the most common human malignancies; it ranks second in Europe and third in the United States [1-3]. In China and other developing countries, the incidence of CRC malignancy and mortality is rising and it is now the third most common cancer [4-8]. It is estimated that more than one million people worldwide are diagnosed with CRC annually and that more than 600,000 die from the disease, with projections that the annual incidence may reach more than two millions within the next two decades [9]. Colorectal carcinogenesis is believed to be a multi-stage process that originates with a localized adenoma, which linearly progresses to an intra-mucosal carcinoma, to an invasive lesion, and finally to metastatic cancer that accounts for over 90% of CRC-related deaths [10-14]. Although this linear model is supported by tissue culture and animal model studies [15-17], it is difficult to reconcile with several well-established facts. First, up to 25% of early stage (Stage I/II), node-negative CRC develop distant metastasis, while over 75% of these cases can be cured by surgery alone [18-21]. Second, circulating CRC tumor cells are undetectable in peripheral blood samples of up to 50% of patients with confirmed metastatic disease, but more than 30% of patients with no detectable metastatic disease exhibit such cells [22-24]. Third, cancer of unknown primary site (CUP) is one of the 10 most frequent cancers and is ranked as the 4th commonest cause of cancer deaths, but its primary tumor site remains elusive [25]. Taken together, the above facts argue that the linear model may not be the only pathway for colorectal carcinogenesis and progression.
Our previous studies of human breast, prostate, cervical, lung, and skin cancer have led us to propose a novel hypothesis that tumor invasion and metastasis arise from the convergence of a tissue injury, the innate immune response to that injury, and the presence of tumor stem cells within the tumor capsules at the site of the injury [26-35]. Epithelial cells are normally contained within capsules. Focal capsule degeneration due to age or disease attracts lymphocyte infiltration, which degrades the degenerating capsules resulting in the formation of a focal disruption in the capsule. If the epithelial cells overlying the disruption are terminally differentiated, no or minimal cell proliferation will occur and the capsule will be repaired. If the focal disruption occurs in a region of the capsule underlying progenitor and/or tumor stem cells, however, these cells are afforded the opportunity to proliferate leading to their “budding” from the disruption and subsequent growth out into the surrounding stroma [26-35]. Once proliferating epithelial buds are formed, their interaction with infiltrated lymphocytes gives rise to an early pathway for metastasis. Specifically, as lymphocytes infiltrate the tumor buds, they disrupt the intercellular junctions and surface adhesion molecules facilitating the dissociation of some proliferating cells from the bud. During this process, lymphocytes and tumor cells can form stable pairings that we refer to as tumor-lymphocyte chimeras (TLCs). Our confocal microscopic observations have shown that the interaction between lymphocytes and budding cells involves simple membrane fusion that does not trigger phagocytosis or tumor cell destruction [36,37]. This fusion may be mediated by the binding of lymphocyte function-associated antigen expressed on immune cells and integrins or other surface molecules on the tumor cells as demonstrated in cell culture studies [38-40]. This perilous coupling allows tumor stem cell to avail itself of the lymphocyte's natural ability to migrate and to breach cell barriers in order to intravasate and to travel to distant organs where the pair can extravasate, providing the potential for tumor stem cell to form the seed of a metastatic lesion.
There are three central tenants of our hypothesis. The first is that tumor stem cells are co-located with normal, benign, and malignant cells within capsules; however, they are the only cells that retain the potential for unlimited proliferation and multi-lineage differentiation in the adult stage [41-44]. The second is that tumor invasion or metastasis may occur at any stage of carcinogenesis if focal capsule disruption and lymphocyte infiltration occur at a site where the overlying epithelium contains a tumor stem cell. Further, lymphocyte infiltration occurs in response to tissue injury rather than the presence of malignancy. It is thus unlike the cytotoxic T lymphocyte infiltration that occurs after the tumor cells start to express tumor-associated antigens. The third is that the interaction of lymphocytes with tumor stem cells can lead to stable adhesions between the two creating a TLC that facilitates the dissemination of the tumor stem cell. Our hypothesis of tumor invasion has been recognized as more compatible with existing experimental evidence [45-47] than the traditional “protoelytic enzyme” theory [48]. Furthermore, our hypothesis of tumor metastasis is applicable to all epithelium-derived cancer and can reasonably explain all major events of metastasis, which includes dissemination from the primary site, intravasation, circulation, extravasation, migration, and colonization at new sites. Finally, our hypothesis postulates, for the first time, a morphologically defined precursor, the TLC, of metastatic cancer [36,37].
Similar to other epithelium-derived structures, the normal and pre-invasive colorectal epithelia are physically separated from small vascular structures in the mucosa by the basement membrane (BM), and from the larger vascular structures in the submucosa by the muscularis mucosae (MM) [49]. The disruption of the BM and MM is thus a pre-requisite for tumor invasion and metastasis [50-52]. A wide spectrum of histopathological and immunohistochemical features occurring at the primary CRC site have been assessed for their ability to correlate with the presence of distant metastasis and to serve as signatures for clinical prognosis [53-63]. Of these, tumor budding at the invasive front has been recognized as a strong predictor of lymph node involvement, distant metastasis, local recurrence, and poor disease-free survival [60-63]. However, a recent meta-analysis of 76 selected articles from 602 citations concluded that: (1) “no single histopathological feature of colorectal cancer reliably predicted lymph node metastases, and (2) several risk factors that correlate highly with nodal disease are not routine components of standard pathology reports” [64]. More importantly, tumor budding at the invasive front is primarily, if not solely, seen at the invasive stage of disease, which is often beyond the point where surgery alone is curative. Consequently, detection of tumor budding at the invasive front is unlikely to have significant value for early detection and intervention of CRC metastasis. Similarly, no other immunohistochemical feature can reliably predict the invasive or metastatic potential of a given CRC or identify the specific individuals with pending or at increased risk for metastatic CRC [64].
Our recent studies have tested the applicability of our hypotheses in CRC. Paraffin-embedded tissue sections from age- and type-matched CRC with (N=37) and without (N=55) positive lymph nodes were subjected to double immunohistochemistry to assess the potential correlation among focal capsule disruption, immune cell infiltration, and budding cells with the following markers: (a) leukocyte common antigen (LCA, clone: 2B11+PD7/26. Dako, Carpinteria, CA), which is expressed by all normal hematopoietic cells and their neoplastic transformations; (b) cytokeratin (CK) AE1/AE3 (clone: AE1/AE3, Dako, Carpinteria, CA), which is expressed in all epithelium-derived cells; (c) CK-19 (clone: clone: RCK108), (d) collagen IV (clone: CIV22, Dako, Carpinteria, CA), which is the main building block of the tumor capsule, and (e) D2-40 (clone: D2-40, Signet, Dedham, MA), which is a marker for lymphatic endothelium.
Lymphoid follicles, lymphocyte aggregates, and tumor-infiltrating lymphocytes were defined according to commonly accepted criteria [38]. A focal disruption in the BM or a fragmentation in the MM was defined as a physical gap, larger than the combined diameter of at least three epithelial cells, in these structures that was observed in at least three consecutive tissue sections. Budding tumor cells were defined as individual cells, or small cell clusters, observed to be physically separated from the tumor core in at least three consecutive tissue sections. To compare the frequencies of focal BM disruptions, MM fragmentations, and dissociated tumor cells in structures adjacent to and distant from, lymphoid follicles and large lymphocyte aggregates, the entire epithelium, lamina propria (LP), and MM of the pre-invasive tissue component was digitally photographed. The digital images were enlarged to 400X and reviewed under the screen of a standard computer. The mean frequencies of focal BM disruptions, MM fragmentations, and dissociated tumor cells between these tissue sites were statistically compared with the Pearson's Chi-square test. Statistical significance was defined as p<0.05. To assess the correlation of lymphocyte infiltration on tumor invasion, all junctions of pre-invasive and invasive lesions were identified and examined to determine whether these junctions are exclusively or preferentially associated with lymphoid follicles or infiltrating lymphocyte aggregates.
These studies have detected tumor budding at the invasive front in both node-positive and node-negative CRC. As shown in Figure 1, these structures with budding cells show distinct malignancy associated morphological changes, including the loss of the BM, lumen and polarity, increased cytoplamic-nuclear ratio, and multiple nucleoli. These structures with budding cells are predominately, if not solely, seen within the submucosa associated with
Tumor budding at the invasive front. A set of two adjacent human CRC tissue sections were immunostained for CK AE1/3 (red) plus collagen IV (brown; A-B) or leukocyte common antigen (LCA; brown; C-D). Circles identify tumor nests with budding cells. Thick and thin arrows identify budding tumor cells and associated lymphocytes, respectively. Note that tumor nests and budding cells show distinct malignancy-associated morphological changes, and that most budding cells are physically associated with lymphocytes. A & C: 150X. B & D: a higher magnification (400X) of A & C, respectively.
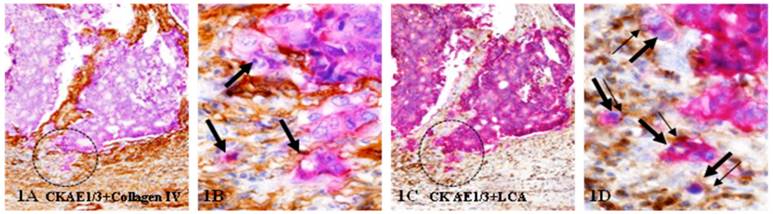
invasive lesions and infiltrated lymphocytes.
More importantly, our studies have detected nearly identical focal capsule disruptions, lymphocyte infiltration, budding cell clusters, and the formation of TLCs in normal colorectal epithelial structures both distant from, and adjacent to, malignant lesions [65-66]. Compared to their morphologically similar counterparts, node-positive CRC have the following unique alterations:
A: A significantly higher frequency of focal BM disruptions and MM fragmentations
The BM and MM distant from invasive lesion were continuous in ~90% of node-negative CRC (Fig 1A-B), but about one-third of node-positive cases harbored epithelial structures with focally disrupted BM overlying fragmented MM (1C-D). The 37-node positive cases harbored 269 epithelial structures with focally disrupted BM and 191 MM fragmentations, compared to 86 and 65, respectively, in the 55- node negative cases. The frequency of BM and MM disruptions was significantly higher (p<0.0001) in node-positive than in node-negative CRC (Fig. 2).
Physical status of BM and MM in node-negative and node-positive CRC. CRC tissue sections from node-negative (A-B) and node-positive (C-D) cases were immunostained for collagen IV (brown). Arrowheads identify MM. Arrows identify the BM. Circles identify epithelial structure with disrupted BM and budding cells. A&C: 150X; B&D:400X.
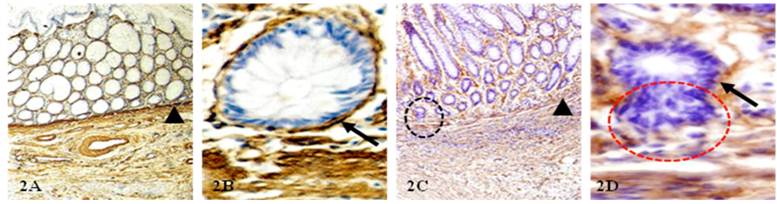
B: A significantly higher frequency of open lumen lymphatic ducts within the mucosae
In sections immunostained for D2-40 (lymphatic duct specific maker), the frequency of open lumen lymphatic ducts within the mucosa was significantly higher in node-positive than in node-negative cases (Table 1). This can be seen in Figure 3 where most open lumen lymphatic ducts are located within the submucosae in node-negative cases, but they are mainly seen within the mucosa of node-positive cases.
Open lumen lymphatic ducts within the mucosa of cases with and without positive lymph nodes.
| Type of cases | N= | Total number detected | Mean ± SD | P |
|---|---|---|---|---|
| With positive nodes | 37 | 361 | 9.76 ± 5.52 | < 0.0001 |
| Without positive nodes | 55 | 81 | 1.45 ± 1.61 |
The frequency and location of open lumen lymphatic ducts between node negative (A) and positive (B) cases. CRC tissue sections from node-negative (A) and node-positive (B) cases were immunostained for D2-40 (brown). Thick and thin arrows identify lymphatic ducts within the mucosa and submucosa, respectively. Asterisks identify MM. 200X.
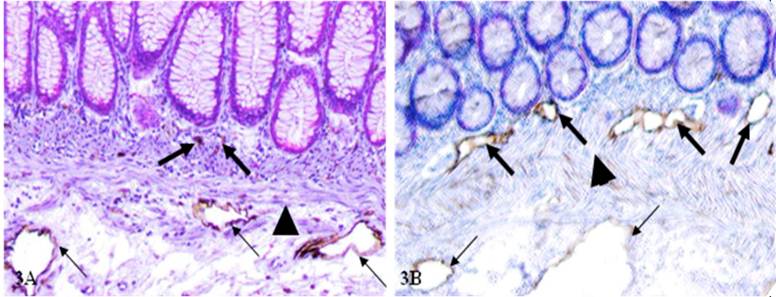
C: Open lumen lymphatic ducts with disseminated cells within the mucosa
Of 37 node-positive cases, 14 (37.8%) harbored multiple lymphatic ducts with disseminated cells within the mucosa (Fig 4), whereas only one (1.8%, with confirmed CRC metastasis to lung) in 55 node-negative cases. The frequency of open lumen lymphatic ducts with disseminated cells within the mucosa is significantly (p < 0.0001) in node-positive than in node-negative cases.
Open lumen lymphatic ducts with disseminated cells within mucosa. CRC tissue sections from node-negative (A-B) and node-positive (C-D) cases were double immunostained for CK-19 (red) and D2-40 (brown). Circles identify a lower magnification review of B and D. Thick and thin arrows identify budding cells and lymphatic ducts, respectively. Red arrows identify lymphatic ducts with disseminated cells that are only seen in the node-positive case. A&C: 80X. B&D: 400X.
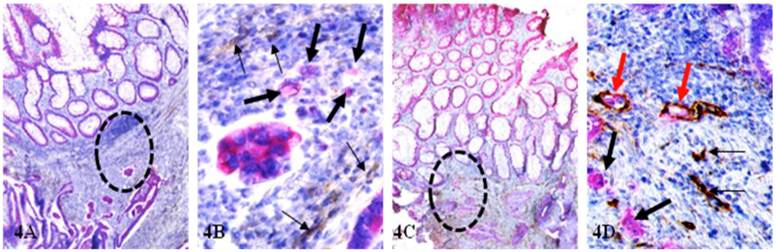
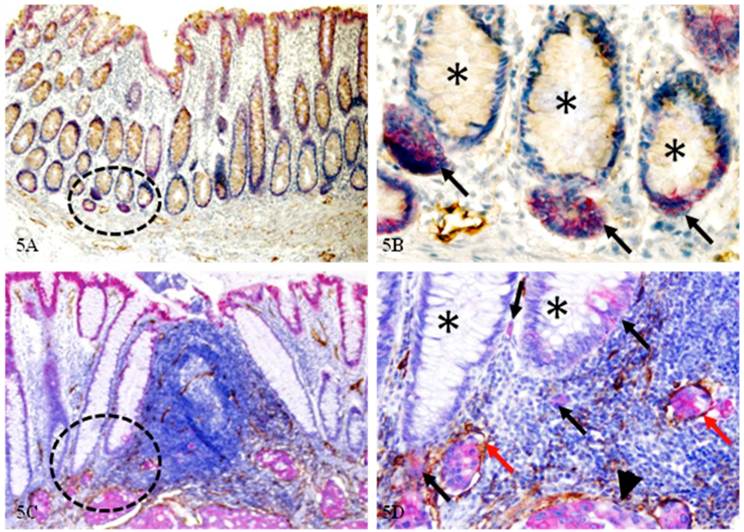
D: Significantly more epithelial structures with isolated cytokeratin (CK)-19-positive cell clusters
Of the 14 node-positive cases harboring lymphatic ducts with disseminated cells within the mucosa, all contained multiple normal epithelial structures with isolated CK-19-positive cell clusters. These CK-19 positive structures were interspersed with normal CK-19 negative epithelial structures, and were seen both distant from (Fig 5A-B) and adjacent to (Fig 5C-D) invasive lesions. As shown in Figure 5A-B, these CK-19 positive budding cell clusters distant from the invasive lesion are morphologically and immunohistochemically distinct from their adjacent CK-19-negative counterparts. As shown in Figure 5C-D, these CK-19 positive budding cells adjacent to the invasive lesions are morphologically and immunohistochemically similar to invasive cancer cells (arrowhead). Several budding cell clusters are located within lymphatic ducts (red arrows). Node-negative cases harbored significantly fewer and smaller such isolated CK-19-positive cell clusters, which are located distant from the MM.
Normal epithelial structures with isolated CK-19-positive cell clusters. Normal and CRC sections from node-positive cases were double immunostained for CK-19 (red) and D2-40 (brown). Circles identify CK-19-positive structures under low magnification. Asterisks identify epithelial structures with budding cells (black arrows). Red arrows identify open lumen lymphatic ducts with disseminated cells. Arrowhead identifies the invasive lesion. A & C: 100X. B & D: 300X.
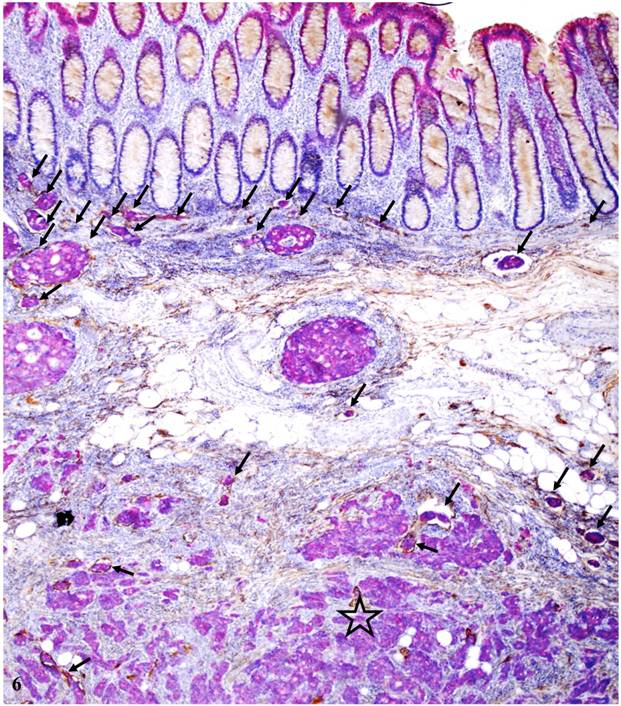
A total of 259 open lymphatic ducts with disseminated epithelial cells were found, of which 235 (90.7%) were in node-positive and 24 (9.3%) in node-negative cases (P < 0.0001); 211 (81%) were seen in the mucosa and submucosa, while 48 (19%) were seen in the muscularis propria and subserosa (p<0.001). As shown in Figure 6, a vast majority of open lymphatic ducts with disseminated tumor cells are located within the mucosa and submucosa, suggesting that metastatic spread may represent an early event in colorectal carcinogenesis and progression.
Distribution of open lymphatic ducts with disseminated cells at different locations within the colon wall. A CRC tissue section from a node-positive case was double immunostained for CK-19 (red) and D2-40 (brown). Arrows identify open lumen lymphatic ducts with disseminated tumor cells. Note that the vast majority of these ducts are located within the mucosa and submucosa with few located in the muscularis propria (star). 80X.
Our most recent studies detected nearly identical focal capsule disruptions, lymphocyte infiltration, budding cell clusters, and disseminated cells within lymphatic ducts in the mucosa in about 10% human colorectal adenoma (Jiang et al. Manuscripts in preparation.
Hypothesis
Based on the above observations, we hypothesize that metastatic spread of CRC cells is an early event in, not a late consequence of, colorectal carcinogenesis and progression, which can occur even in normal appearing epithelial structures through the following pathways:
1. Lymphocyte infiltration causes focal capsule disruptions facilitating stem cell proliferation
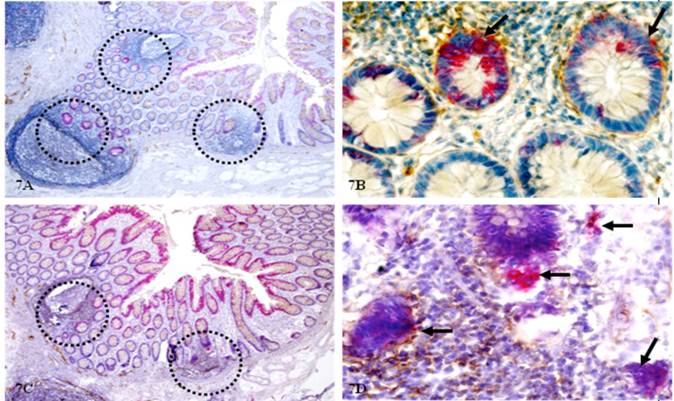
As CK-19 is a well-recognized stem cell marker and CK-19-positive cells are detected in peripheral blood of patients with metastatic cancer [67-75], our studies assessed the involvement of CK-19 and related signaling pathways in the initiation of CRC invasion and metastasis. As shown in Figure 7, some isolated epithelial clusters adjacent to lymphoid follicles exhibit strong CK-19 immunostaining within an otherwise negative background. These structures are morphologically indistinguishable from adjacent counterparts under low magnification in H&E stained sections, but under higher magnification, they differ markedly in size, shape, nuclear structures, and density, and with a higher proliferation index. Although similar CK-19 positive cell clusters are also seen in node-negative cases, they are generally located at the middle of a given crypt (as shown in Fig 7A-B) with no distinct morphological alterations. In contrast, CK-19 positive cell clusters in node-positive cases are generally baso-laterally located (as shown in Fig 7C-D) with malignant morphology and are often adjacent to open lumen lymphatic ducts.
Morphological and immunohistochemical alterations in epithelial structures near lymphatic follicles. Normal tissue sections distant from tumor of node(-) and (+) cases were double immunostained for CK-19 (red) and D2-40 (brown). Circles identify epithelial structures adjacent to lymphatic follicles. Arrows identify epithelial structures with CK-19 expression and signs of increased proliferation. A & B: 80X. C & D: Higher magnification (400X) of A and C, respectively.
2. Lymphocyte infiltration results in dissociation of some cells from the main epithelial structures
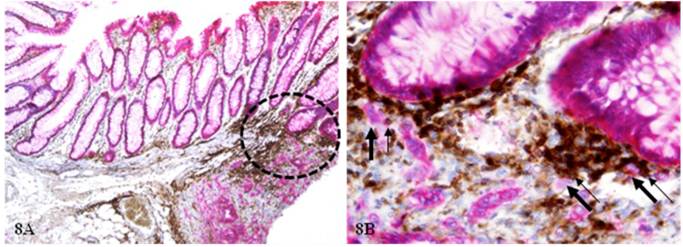
As shown in Figure 8, the normal epithelial structures adjacent to a small lymphocyte aggregate harbor multiple dissociated cells (thick arrows), which are all associated with lymphocytes (thin arrows). Dissociated cells are absent in the vicinity of epithelial structures distant from the lymphocyte aggregate.
Epithelial cell dissociation from morphologically normal appearing epithelial structures. A CRC tissue section from a node-positive case was double immunostained for CK AE1/3 (red) and leukocyte common antigen (LCA; brown). Circles identify epithelial structures adjacent to a small lymphatic aggregate. Thick arrows identify dissociated epithelial tumor cells and thin arrows identify associated lymphocytes. A: 100X. B: a higher magnification (400X) of A.
3. Dissociated cells form invasive or metastatic lesions through different pathways
(1). Dissociated cells undergo in situ proliferation resulting in distinct invasive cancer
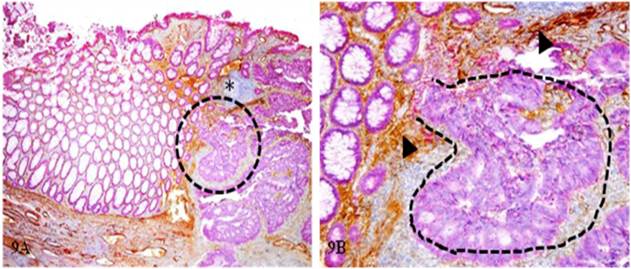
Some of the dissociated tumor stem cells can undergo extensive in situ proliferation near the focal disruption, forming tumor nests that gradually invade the submucosa. As shown in Figure 9, a tumor nest (curve lines) extends from the mucosa to the submucosa through a focal disruption in the MM.
In situ proliferation of budding cells into invasive cancer. A CRC tissue section from a node-negative case was double immunostained for CK AE1/3 (red) and collagen IV (brown). The circle identifies the low magnification of the structures in B. Arrowheads identify the MM. Asterisk identifies a lymphocyte aggregate. Curve lines identify a tumor nest extending from the mucosa into the submucosa through a focal disruption in the MM. A: 100X. B: 400X.
(2). Dissociated cells are pushed or dragged by associated lymphocytes to different locations
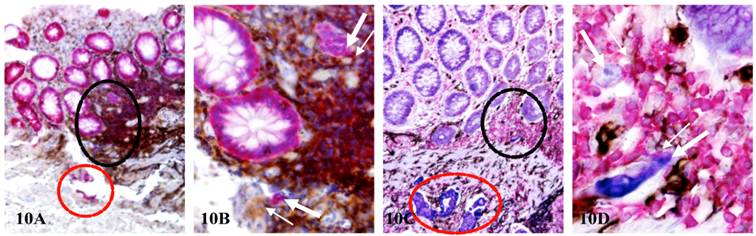
Lymphocytes and tumor cells can form stable pairing through membrane fusion to form TLCs. As lymphocytes have the natural capability to migrate, they can physically shuttle their associated tumor cells to different locations during migration. As shown in Figure 10, each of two node-positive cases harbor three isolated cell clusters(thick arrows and red circles) located in the mucosa, the MM, and the submucosa, respectively. All these cell clusters in different locations are physically associated with lymphocytes and also share very similar morphological and immunohistochemical features.
TLCs shuttle tumor cells to different histological locations. CRC tissue sections from two node-positive cases were double immunostained for CK AE1/3 (red) and LCA (brown; A-B) and triple immunostained for CK AE1/3 (blue), CD34 (brown) and LCA (red; C-D). Thick and thin arrows identify budding cells and associated lymphocytes located in the mucosa and MM, respectively. Black circles identify the low magnification views of structures in B and D. Red circles identify invasive cancer nests in the submucosa. A&C: 150X. B & D: a higher magnification (400X) of A&C, respectively.
(3). Dissociated cells enter pre-existing lymphatic ducts as TLCs
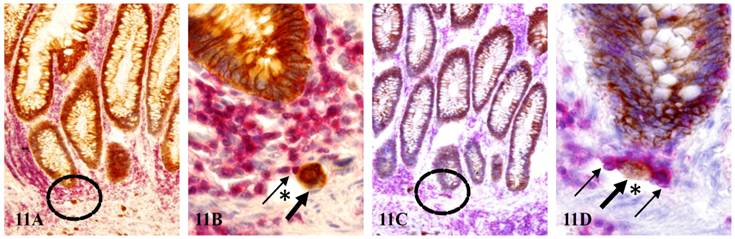
As shown in Figure 11, two dissociated epithelial cells (thick arrows) near morphologically normal appearing epithelial structures are physically conjoined with lymphocytes (thin arrows), which appear to physically drag these dissociated epithelial cells into two small lymphatic ducts (asterisks).
Tumor cells entering pre-existing lymphatic ducts through formation of TLCs. Normal tissue sections distant from tumor of node-positive cases were double immunostained for CK AE1/3 (brown) and LCA (red). Circles identify the low magnification views of B and D. Thick and thin arrows identify dissociated epithelial cells and associated lymphocytes, respectively. Asterisks identify lymphatic ducts. A & C: 150X. B & D: a higher magnification (500X) of A&C, respectively.
(4). Dissociated cells generate their own vascular structures facilitating distant migration
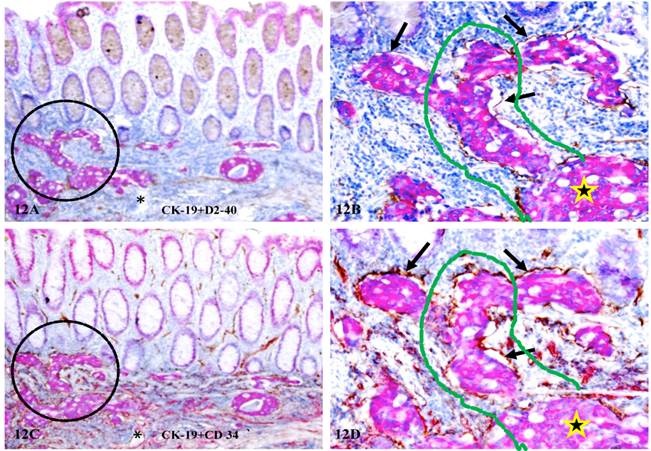
In node-positive cases, some of the budding cells from normal appearing structures are capable of forming their own vascular structures. As shown in Figure 12, these budding cells are located within tube-like structures that co-express both D2-40 (a lymphatic duct phenotypic marker) (A-B) and CD34 (a blood vessel phenotypic marker) (C-D). Budding cells in the mucosa appear to be in physical continuity with invasive cancer (star) within the submucosa through the vascular channel (curve lines). These tube-like structures are likely to represent newly formed vascular structures for the following reasons: (1). they are exclusively associated with dissociated CK-19 positive cell clusters, (2). the wall (the endothelial cell layer) of these structures is often incomplete with CK-19 positive cells physically connected with adjacent stromal cells or tumor cells within the stroma, and (3). the adjacent morphologically distinct lymphatic ducts or blood vessels without disseminated epithelial cells express only one of the two phenotypic markers.
Formation of vascular structures within mucosa by budding cells. A set of two adjacent human CRC tissue sections from a node-positive case were double immunostained for CK-19 (red) plus D2-40 (A-B) or CD34 (C-D) (brown), respectively. Circles identify low magnification views of the structures in B and D). Arrows identify the wall of the vascular structures. Stars identify invasive cancer nests in the submucosa. Curve lines identify the vascular channels spanning the mucosa, the MM, and the submucosa. Asterisks in A and C identify a tube-like structure, which harbors no disseminated epithelial cells and only expresses CD34. A & C: 150X. B & D: a higher magnification (400X) of A&C, respectively.
Together, we have proposed that budding cells from normal appearing epithelial structures are likely to represent a population of tumor stem cells. As only stem cells in the adult organs retain the potential for unlimited proliferation and multi-lineage differentiation, these budding cells are likely to be invasion and metastasis-initiating cells. The concept of budding cells presented in our current study differs fundamentally from the concept of “tumor budding in invasive front” in two main aspects: (1) “budding” cells represent a much earlier event that could potentially occur at the benign or even “normal” stage, whereas “tumor budding in invasive front” is a late stage event, occurring primarily, if not solely, at the invasive stage; and (2) “budding” cells are physically associated with normal epithelial structures, whereas “tumor budding in invasive front” are only associated with malignant structures.
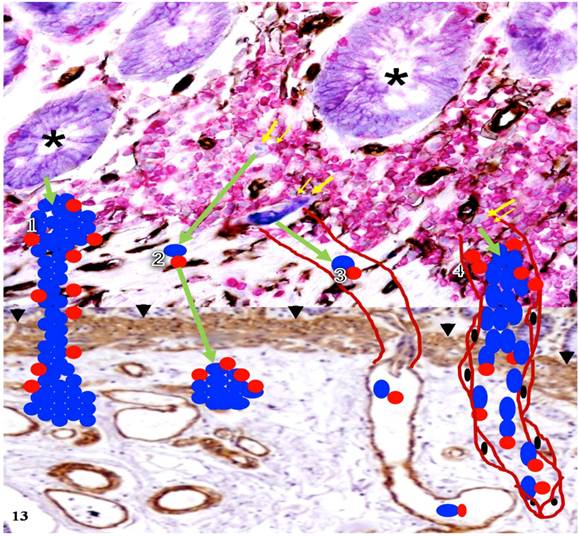
Our new hypothesis is further summarized in Figure 13 where the four pathways that tumor cells can follow after their dissociation from the tumor bud are depicted. These pathways are supported by the immunohistological findings presented in Figures 9-12, respectively.
Hypothesized pathways of dissociated cells from normal appearing epithelial structures. Human CRC tissue sections from a node-positive case were immunostained for tumor cells (purple), lymphocytes (red), blood vessels (black), and MM (brown). This figure is the combination of two figures. The up half of this figure demonstrates cell budding from normal appearing epithelial structures. The lower half elucidates the potential pathways in which dissociated epithelial cells progress to invasive or metastatic lesions. Asterisks identify normal appearing epithelial structures. Thick and thin yellow arrows identify dissociated epithelial cells and associated lymphocytes, respectively. Arrowheads identify the MM. Green arrows indicate the direction of the progression. Blue and red circles represent dissociated epithelial cells and associated lymphocytes. Curve lines represent pre-existing (3) and newly generated vascular structures (4), respectively.
Implications of our hypothesis
Our studies have consistently shown that, compared to its age- and type-matched counterpart, the primary tumor of node-positive CRC have a significantly higher frequency of the following alterations: (a) open-lumen lymphatic ducts within the mucosa, (b) open-lymphatic ducts with disseminated cells within the LP, and (c) normal epithelial structures with isolated CK-19-positive cell clusters that are morphologically or immunohistochemically similar to adjacent invasive cancer. It is not known why such or similar findings have not been previously reported. It is our speculation that the most likely reason might be that these alterations, especially the co-expression of D2-40 and CD34, can be seen only in double immunostained tissue sections, but double immunohistochemistry is barely used in the clinical laboratories.
The mechanism for the differential profile between node positive and node negative cases is unknown, but it is likely that the mucosal epithelial cells of node-negative CRC may have retained a significantly fewer number of stem cells than their node-positive counterparts, or their budding cells belong to a partially committed tumor progenitor population that either lacks the surface molecules to form TLCs for intravasation, or lacks the potential for multi-lineage differentiation. Thus, node-negative CRC may be able to utilize the first two pathways to progress to invasive lesions, but they are deficient to progress to metastatic lesions. Our speculation is consistent with a well-established fact that the relative proportion of the stem cell population varies significantly among different tumors of the same type or different tumor types, from 0.1% to 2% [76-82]. A recent mathematical model even shows that cancer stem cells “may comprise any possible proportion of the tumour, and that the higher the proportion the more aggressive the tumour is likely to be” [83]. Our speculation is also supported by the fact that the adult stem cell population progressively loses the breadth of their developmental potency during the aging process, which is significantly influenced by a great number of known and unknown factors [84-90]. As the stem cells retain the potential for unlimited proliferation, multi-lineage differentiation, and colonization in new sites [91-94], the residual stem cells of node-positive cases may be able to generate their own vasculatures such that tumor cells of these cases may utilize both newly formed and pre-existing vasculatures for dissemination to distant sites.
Although a vast majority of those isolated CK-19 positive colonic epithelial cell clusters seen in our current study appear to be morphologically normal, they are associated with a number of tumor invasion- and metastasis-associated alterations, including (1) focal disruptions of the BM, (2) focal fragmentations of the MM, (3) elevated proliferation, (4) budding from the parent structures, (5) direct physical continuity with invasive cancer nests, and (6) dissemination to lymphatic ducts or blood vessels. Thus, these CK-19 positive cell clusters may represent a previously undefined malignant entity. Consistent with our speculation are the facts: (1) breast clinging ductal carcinoma in situ (DCIS) is morphologically indistinguishable from normal breast epithelial structures, but it shares a very similar molecular and clinical profile with other types of DCIS [95], (2) the prostate tissues of a subset of aged men or normal-appearing prostate tissues adjacent to prostate cancer harbor a DNA phenotype that is identical to invasive and metastatic prostate cancer [96], and (3) genetic comparison of these microdissected cells and their adjacent and distant cancer cells has shown that they share a very similar molecular profile with their cancer counterparts, including a significantly elevated frequency of the loss of heterozygosity (LOH), expression of tumor invasion and metastasis-related genes, and aberrant expression of micro-RNAs 146a and 146b-5p [29-35]. These CK-19 positive cell clusters may also represent the direct “seeds” of invasive or metastatic CRC, judged from Figures 5 and Figures 8-12 of our current studies.
Thus, the metastatic potential of a given tumor is likely to be determined at the early stages of carcinogenesis. The presence of open lumen lymphatic ducts with disseminated cells in the mucosa may represent an earlier and more reliable predictor than tumor budding at the invasive front for node status. If confirmed, our findings and hypothesis may have the following scientific and clinical implications:
1. To assist early detection of patients with pending or at risk for invasive or metastatic CRC
As the disruption of the BM and MM is an absolute prerequisite for CRC invasion or metastasis, morphological and immunohistochemical assessment of the physical integrity of these two physical barriers in biopsy samples may significantly facilitate early detection of patients with pending, or at increased risk to develop, invasive or metastatic CRC. A previous study assessed the correlation between the physical integrity of the BM and the survival status in patients with CRC at different stages using immunohistochemistry for collagen IV and laminin (two primary components of the BM) [97]. The study revealed that a total loss or considerable discontinuity of the BM was adversely correlated with the survival status (p = 0.066 and p = 0.014 for collagen IV and laminin, respectively), whereas no association with the stage of disease was noticed [97].
2. To facilitate clinical differential diagnosis and the selection of the optimal treatment option
As open-lumen lymphatic ducts with disseminated tumor cells are exclusively seen in the mucosa of node-positive and confirmed cases with metastasis, immunohistochemical assessment of the biopsy samples before the operation may substantially benefit the decision making process for the selection of the operation extent and the treatment options.
3. To benefit the development of effective interventions to prevent CRC invasion and metastasis
As aberrant lymphocyte infiltration appears to be a trigger for both tumor invasion and metastasis, the development of specific approaches to manipulate lymphocyte infiltration or to disrupt the formation of tumor-lymphocyte chimeras may represent a more effective approach for CRC interventions and prevention. Our speculation is consistent with a recent report published in Lancet Oncology [98], which has revealed that regular use of Aspirin, a non-steroidal anti-inflammatory drug, reduces the long-term risk of CRC and other cancer and the risk of distant metastasis.
4. To facilitate the development of more effective therapeutic agents
As our previous studies of human breast and prostate tumors [26-37] have consistently shown that cell clusters overlying focally disrupted tumor capsules exhibit a substantially different molecular and immunohistochemical profile, compared to their adjacent counterparts distant from the disruption, the further assessment of the molecular and biochemical profiles of these budding cells may lead to the identification of more novel molecules associated with the early events of carcinogenesis. As the frequency and pattern of focal capsule disruptions and cell budding seen in our studies of CRC are almost identical to those seen in breast, prostate, lung, cervical, and skin tumors [26-37], the unique molecules expressed in these budding cells are very likely to represent the common trigger factors for the onset of invasion and metastasis of all epithelium-derived tumors. Consequently, the identification of the unique molecules expressed in these budding cells could potentially lead to the development of more effective therapeutic approaches for early detection, intervention, and prevention of cancer invasion and metastasis.
In summary, this is a hypothesis paper, based on our previously published experimental data of studies in human breast and prostate tumors [26-37] and recent unpublished findings in human CRC [65,66]. Our current study is the expansion of our previous studies with same concept and technical approaches. Thus, the experimental procedures were referred to previous publications, and the results were presented in an abstract style. As about 25% of early stage (stage I or II) node negative CRC and about 50% of CRC across all stages develop systemic and/or recurrent CRC, while none of the current concept of approaches could specifically identify these patients for early intervention, the primary goal of our paper is to present our novel concept in order to elicit more research interests in the field.
Acknowledgements
This study was supported in part by grants 200601060, 201108004 from Science and Technology Bureau of Nanjing, ZKX11004from Health Bureau of Nanjing, LZ11105 from Jiangsu Province Bureau of Traditional Chinese Medicine to Jiang Bin. This study was also supported in part by grants 2008-02 from the US Military Cancer Institute and Henry M. Jackson Foundation and 2006CB910505 from the Chinese Ministry of Science and Technology to Yan-gao Man and Xichen Zhang, by Merit Award ONCA-037-10F from the Veterans Health Administration awarded to Jeff Mason, and by RO1-DE12880 from NIDCR-NIH to Anahid Jewett.
Competing Interests
The authors have declared that no competing interest exists.
References
1. de Wijkerslooth TR, Bossuyt PM, Dekker E. Strategies in screening for colon carcinoma. Neth J Med. 2011;69(3):112-9
2. Cummings LC, Cooper GS. Colorectal cancer screening: update for 2011. Semin Oncol. 2011;38(4):483-9
3. Vu HT, Burke CA. Advances in colorectal cancer screening. Curr Gastroenterol Rep. 2009;11(5):406-12
4. Zhou MG, Wang XF, Hu JP. et al. Geographical distribution of cancer mortality in China, 2004-2005. Zhonghua Yu Fang Yi Xue Za Zhii. 2010;44(4):303-8
5. Lei T, Chen WQ, Zhang SW. et al. Prevalence trend of colorectal cancer in 10 cities and counties in China from 1988 to 2002. Zhonghua Zhong Liu Za Zhi. 2009;31(6):428-33
6. Mahmodlou R, Mohammadi P, Sepehrvand N. Colorectal cancer in northwestern Iran. ISRN Gastroenterol. 2012:968560
7. Shin A, Joo J, Bak J. et al. Site-specific risk factors for colorectal cancer in a Korean population. PLOS One. 2011;6(8):e23196
8. Moghimi-Dehkordi B, Safaee A. An overview of colorectal cancer survival rates and prognosis in Asia. World J Gastrointest Oncol. 2012;4(4):71-5
9. Karsa LV, Lignini TA, Patnick J, Lambert R, Sauvaget C. The dimensions of the CRC problem. Best Pract Res Clin Gastroenterol. 2010;24(4):381-96
10. Michor F, Iwasa Y, Rajagopalan H. et al. Linear model of colon cancer initiation. Cell Cycle. 2004;3(3):358-62
11. Aaltonen LA. The multistep process of colon carcinogenesis. Cytokines Mol Ther. 1996;2(2):111-4
12. Luebeck EG, Moolgavkar SH. Multistage carcinogenesis and the incidence of colorectal cancer. Proc Natl Acad Sci USA. 2002;99(23):15095-100
13. Barugel ME, Vargas C, Krygier Waltier G. Metastatic colorectal cancer: recent advances in its clinical management. Expert Rev Anticancer Ther. 2009;9(12):1829-47
14. Shimada H, Tanaka K, Endou I, Ichikawa Y. Treatment for colorectal liver metastases: a review. Langenbecks Arch Surg. 2009;394(6):973-83
15. Ernst M, Ramsay RG. Colorectal cancer mouse models: integrating inflammation and the stroma. J Gastroenterol Hepatol. 2012;27(1):39-50
16. Sussman DA, Santaolalla R, Strobel S. et al. Cancer in inflammatory bowel disease: lessons from animal models. Curr Opin Gastroenterol. 2012;28(4):327-33
17. Roper J, Hung KE. Priceless GEMMs: genetically engineered mouse models for colorectal cancer drug development. Trends Pharmacol Sci. 2012;33(8):449-55
18. Labianca R, Merelli B. Screening and diagnosis for colorectal cancer: present and future. Tumori. 2010;96(6):889-901
19. Chen SL, Steele SR, Eberhardt J. et al. Lymph node ratio as a quality and prognostic indicator in stage III colon cancer. Ann Surg. 2011;253(1):82-7
20. Glimelius B, Cavalli-Björkman N. Metastatic colorectal cancer: current treatment and future options for improved survival. Medical approach--present status. Scand J Gastroenterol. 2012;47(3):296-314
21. Mathis KL, Larson DW, Dozois EJ. et al. Outcomes following surgery without radiotherapy for rectal cancer. Br J Surg. 2012;99(1):137-43 doi: 10.1002/bjs.7739
22. Gerges N, Rak J, Jabado N. New technologies for the detection of circulating tumour cells. Br Med Bull. 2010;94:49-64
23. Gerges N, Jabado N. Biomarkers in cancer micrometastasis: where are we at? Bioanalysis. 2010;2:881-99
24. Mahmoud W, Sukhanova A, Oleinikov V. et al. Emerging applications of fluorescent nanocrystals quantum dots for micrometastases detection. Proteomics. 2010;10(4):700-16
25. Pentheroudakis G, Briasoulis E, Pavlidis N. Cancer of unknown primary site: missing primary or missing biology? Oncologist. 2007;12(4):418-25
26. Man YG, Sang QXA. The significance of focal myoepitehlial cell layer disruptions in breast tumor invasion: a paradigm shift from the “protease-centered” hypothesis. Exp Cell Res. 2004;301:103-18
27. Yousefi M, Mattu R, Gao C, Man YG. Mammary ducts with and without focal myoepithelial cell layer disruptions show a different frequency of white blood cell infiltration and growth pattern: Implications for tumor progression and invasion. AIMM. 2005;13:30-7
28. Man YG, Shen T, Zhao YG, Sang QX. Focal prostate basal cell layer disruptions and leukocyte infiltration are correlated events: A potential mechanism for basal cell layer disruptions and tumor invasion. Cancer Detect Prev. 2005;29:161-9
29. Man YG, Tai L, Barner R. et al. Cell clusters overlying focally disrupted mammary myoepithelial cell layers and adjacent cells within the same duct display different immunohistochemical and genetic features: implications for tumor progression and invasion. Breast Cancer Res. 2003;5:R231-41
30. Man YG, Zhang Y, Shen T. et al. et al. cDNA expression profiling identifies elevated expressions of tumor progression and invasion related genes in cell clusters of in situ breast tumors. Breast Cancer Res Treat. 2005;89:199-208
31. Man YG, Fu S, Liu AJ. et al. Aberrant expression of chromogranin A, mir-146a, and mir-146b-5p in prostate structures with focally disrupted basal cell layers: an early sign of invasion and hormone-refractory cancer? Cancer Genom & Proteom. 2011;8(5):235-44
32. Man YG. Focal degeneration of aged or injured myoepithelial cells and the resultant auto-immunoreactions are trigger factors for breast tumor invasion. Med Hypoth. 2007;69(6):1340-57
33. Man YG, Gardner WA. Focal degeneration of basal cells and the resultant auto-immunoreactions: a novel mechanism for prostate tumor progression and invasion. Med Hypoth. 2008;70:387-408
34. Man YG. Tumor cell budding from focally disrupted tumor capsules: a common pathway for all breast cancer subtype derived invasion? J Cancer. 2010;1:27-31
35. Man YG. A seemingly most effective target for early detection and intervention of prostate tumor Invasion. J Cancer. 2012;1:63-69
36. Man YG, Harley R, Mason J, Gardner WA. Contributions of leukocytes to tumor invasion and metastasis: the “piggy-back” hypothesis. Cancer Epidem. 2012;34(1):3-6
37. Man YG, Mason J, Harley R. et al. Leukocyte- facilitated tumor dissemination: findings from multiple types of human tumors. J Cell Biochem. 2011;112(4):1154-67
38. Neeson PJ, Thurlow PJ, Jamieson GP. Characterization of activated lymphocyte-tumor cell adhesion. J Leukoc Biol. 2000;67(6):847-55
39. Neeson PJ, Thurlow PJ, Jamieson GP, Bradley C. Lymphocyte-facilitated tumour cell adhesion to endothelial cells: the role of high affinity leucocyte integrins. Pathology. 2003;35(1):50-5
40. Bakker GJ, Eich C, Torreno-Pina JA. et al. Lateral mobility of individual integrin nanoclusters orchestrates the onset for leukocyte adhesion. Proc Natl Acad Sci USA. 2012;109(13):4869-74
41. Moinfar F, Man YG, Arnould L. et al. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: Implications for tumorigenesis. Cancer Res. 2000;60:2562-6
42. Vaiopoulos AG, Kostakis ID, Koutsilieris M, Papavassiliou AG. Colorectal cancer stem cells. Stem Cells. 2012;30(3):363-71
43. Biddle A, Mackenzie IC. Cancer stem cells and EMT in carcinoma. Cancer Metastasis Rev. 2012 epub
44. Borovski T, De Sousa E Melo F, Vermeulen L, Medema JP. Cancer stem cell niche: the place to be. Cancer Res. 2011;71(3):634-9
45. Schedin P. Pregnancy-associated breast cancer and metastasis. Nat Rev Cancer. 2006;6(4):281-91
46. Min Hu and Kornelia Polyak. Microenvironmental regulation of cancer development. Curr Opin Genet Dev. February. 2008;18(1):27-34
47. Puspa Raj Pandey PR, Saidou J, Watabe K. Role of myoepithelial cells in breast tumor progression. Front Biosci. 2011;15:226-36
48. Goldfarb RH, Liotta LA. Proteolytic enzymes in cancer invasion and metastasis. Semin Thromb Hemost. 1986;12(4):294-307
49. Noffsinger A, Fenoglio-Preiser CM, Maru D, Gilinsky N. Gastrointestinal Diseases. Washington DC: American Registry of Pathology and Armed Forces Institute of Pathology. 2007
50. Lazaris ACh, Tzoumani AN, Thimara I. et al. Immunohistochemical assessment of basement membrane components in colorectal cancer: prognostic implications. J Exp Clin Cancer Res. 2003;22(4):599-606
51. Ambiru S, Miyazaki M, Ito H. et al. Type IV collagenase activities in human colorectal cancers and its role in cancer invasion and metastasis. Nihon Shokakibyo Gakkai Zasshi. 1993;90(7):1555-61
52. Hagedorn EJ, Sherwood DR. Cell invasion through basement membrane: the anchor cell breaches the barrier. Curr Opin Cell Biol. 2011;23(5):589-96
53. Bayar S, Saxena R, Emir B, Salem RR. Venous invasion may predict lymph node metastasis in early rectal cancer. Eur J Surg Oncol. 2002;28(4):413-7
54. An JY, Baik YH, Choi MG. et al. Predictive factors for lymph node metastasis in early gastric cancer with submucosal invasion: analysis of a single institutional experience. Ann Surg. 2007;246(5):749-53
55. Rasheed S, Bowley DM, Aziz O, Tekkis PP, Sadat AE, Guenther T. et al. Can depth of tumour invasion predict lymph node positivity in patients undergoing resection for early rectal cancer? A comparative study between T1 and T2 cancers. Colorectal Dis. 2008;10(3):231-8
56. Yamauchi H, Togashi K, Kawamura YJ. et al. Pathological predictors for lymph node metastasis in T1 colorectal cancer. Surg Today. 2008;38(10):905-10
57. Tateishi Y, Nakanishi Y, Taniguchi H, Shimoda T, Umemura S. Pathological prognostic factors predicting lymph node metastasis in submucosal invasive (T1) colorectal carcinoma. Mod Pahtol. 2010;23(8):1068-72
58. Huh JW, Kim HR, Kim YJ. Lymphovascular or perineural invasion may predict lymph node metastasis in patients with T1 and T2 colorectal cancer. J Gastrointest Surg. 2010;14(7):1074-80
59. Zlobec I, Lugli A. Epithelial mesenchymal transition and tumor budding in aggressive colorectal cancer: tumor budding as oncotarget. Oncotarget. 2010;1(7):651-61
60. Nakamura T, Mitomi H, Kanazawa H. et al. Tumor budding as an index to identify high-risk patients with stage II colon cancer. Dis Colon Rectum. 2008;51(5):568-72
61. Ogawa T, Yoshida T, Tsuruta T. et al. Tumor budding is predictive of lymphatic involvement and lymph node metastases in submucosal invasive colorectal adenocarcinomas and in non-polypoid compared with polypoid growths. Scand J Gastroenterol. 2009;44(5):605-14
62. Wang LM, Kevans D, Mulcahy H. et al. Tumor budding is a strong and reproducible prognostic marker in T3N0 colorectal cancer. Am J Surg Pathol. 2009;33(1):134-41
63. Märkl B, Renk I, Oruzio DV. et al. Tumour budding, uPA and PAI-1 are associated with aggressive behaviour in colon cancer. J Surg Oncol. 2010;102(3):235-41
64. Glasgow SC, Bleier JI, Burgart LJ, Finne CO, Lowry AC. Meta-analysis of Histopathological Features of Primary Colorectal Cancers that Predict Lymph Node Metastases. J Gastrointest Surg. 2012;16(5):1019-28
65. Jiang B, Mason J, Jewett A, Cho WC, Man YG. Tube-like structures with co-expression of D2-40 and CD34: newly formed vasculatures? Int J Biol Sci. 2012;8(8):1206-16
66. Bin Jiang, Yan-gao Man, Anton Bilchik. et al. Differential lymphatic involvement in primary site of node positive and negative colorectal cancer. Manuscript in the process for submission.
67. Michel M, Török N, Godbout MJ, et al Keratin 19 as a biochemical marker of skin stem cells in vivo, in vitro. keratin 19 expressing cells are differentially localized in function of anatomic sites, and their number varies with donor age and culture stage. J Cell Sci. 1996;109( Pt 5):1017-28
68. Larouche D, Hayward C, Cuffley K, Germain L. Keratin 19 as a stem cell marker in vivo and in vitro. Methods Mol Biol. 2005;289:103-10
69. Naujok O, Francini F, Jörns A, Lenzen S. An efficient experimental strategy for mouse embryonic stem cell differentiation and separation of a cytokeratin-19-positive population of insulin-producing cells. Cell Prolif. 2008;41(4):607-24
70. Larouche D, Lavoie A, Paquet C, Simard-Bisson C, Germain L. Identification of epithelial stem cells in vivo and in vitro using keratin 19 and BrdU. Methods Mol Biol. 2010;585:383-400
71. Chang CC, Yang SH, Chien CC. et al. Clinical meaning of age-related expression of fecal cytokeratin 19 in colorectal malignancy. BMC Cancer. 2009;9:376
72. Yang SH, Huang CJ, Lee CL. et al. Fecal RNA detection of cytokeratin 19 and ribosomal protein L19 for colorectal cancer. Hepatogastroenterology. 2010;57(101):710-5
73. Sun GR, Dong XY, He QS, Qu H, Wang YM. Expression and clinical significance of CK19 and CK20 expressions in transverse mesocolon biopsies from patients with gastric carcinoma. Cell Biochem Biophys. 2012;62(2):361-4
74. Kim H, Choi GH, Na DC. et al. Human hepatocellular carcinomas with "Stemness"-related marker expression: keratin 19 expression and a poor prognosis. Hepatology. 2011;54(5):1707-17
75. Roy S, Majumdar AP. Signaling in colon cancer stem cells. J Mol Signal. 2012;7(1):11
76. Lapidot T, Sirard C, Vormoor J. et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645-8
77. Al-Hajj M, Wicha MS, Benito-Hernandez A. et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100(7):3983-8
78. Dick JE. Breast cancer stem cells revealed. Proc Natl Acad Sci USA. 2003;100(7):3547-9
79. Nakanishi Y, Seno H, Fukuoka A. et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat Genet. 2012;45(1):98-103
80. Yeung TM, Gandhi SC, Wilding JL, Muschel R, Bodmer WF. Cancer stem cells from colorectal cancer-derived cell lines. Proc Natl Acad Sci USA. 2010;107(8):3722-7
81. Ricardo S, Vieira AF, Gerhard R. et al. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. J Clin Patho. 2011;64:937-46
82. Wang B, Jacob ST. Role of cancer stem cells in hepatocarcinogenesis. Genome Med. 2011;3(2):11
83. Johnston MD, Maini PK, Jonathan Chapman S. et al. On the proportion of cancer stem cells in a tumour. J Theor Biol. 2010;266(4):708-11
84. Smith LG, Weissman IL, Heimfeld S. Clonal analysis of hematopoietic stem-cell differentiation in vivo. Proc.Natl.Acad.Sci.USA. 1991;88:2788-92
85. Spangrude GJ, Brooks DM, Tumas DB. Long-term repopulation of irradiated mice with limiting numbers of purified hematopoietic stem cells: in vivo expansion of stem cell phenotype but not function. Blood. 1995;85:1006-1016
86. Offner F, Kerre T, DeSmedt M, Plum J. Bone marrow CD34+ cells generate fewer T cells in vitro with increasing age and following chemotherapy. Bri J Haematology. 1999;104:801-8
87. Osawa M, Hanada K-i, Hamada H, Nakauchi H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 1996;273:242-5
88. Sudo K, Ema H, Morita Y, Nakauchi H. Age-associated characteristics of murine hematopoietic stem cells. J. Exp. Med. 2000;192:1273-80
89. Liang Y, Van Zant G. Genetic control of stem-cell properties and stem cells in aging. Curr Opin Hematol. 2003;10:195-202
90. Liang Y, Van Zant G. Aging stem cells, latexin, and longevity. Exp Cell Res. 2008;314(9):1962-72
91. Vaiopoulos AG, Kostakis ID, Koutsilieris M, Papavassiliou AG. Colorectal cancer stem cells. Stem Cells. 2012;30(3):363-71
92. Zeki SS, Graham TA, Wright NA. Stem cells and their implications for colorectal cancer. Nat Rev. Gastroenterol Hepatol. 2011;8(2):90-100
93. Biddle A, Mackenzie IC. Cancer stem cells and EMT in carcinoma. Cancer Metastasis Rev. 2012 [Epub ahead of print]
94. Borovski T, De Sousa E Melo F, Vermeulen L, Medema JP. Cancer stem cell niche: the place to be. Cancer Res. 2011;71(3):634-9
95. Moinfar F, Man YG, Bratthauer GL, Ratschek M, Tavassoli FA. Genetic abnormalities in mammary ductal intraepithelial neoplasia-flat type (Clinging ductal carcinoma in situ)-A simulator of normal mammary epithelium. Cancer. 2000;88:2072-81
96. Malins DC, Gilman NK, Green VM. et al. A cancer DNA phenotype in healthy prostates, conserved in tumors and adjacent normal cells, implies a relationship to carcinogenesis. Proc Natl Acad Sci USA. 2005;102:19093-6
97. Lazaris AC, Tzoumani AN, Thimara I. et al. Immunohistochemical assessment of basement membrane components in colorectal cancer: prognostic implications. J Exp Clin Cancer Res. 2003;22(4):599-606
98. Alga AM, Rothwell PM. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Onco. 2012 Epub
Author contact
![]() Corresponding author: Jiang Bin, MD, PhD. Associate Director, National Medical Centre of Colorectal DiseasesAssociate professor, Nanjing University of Chinese Medicine. The Third Affiliated Hospital, Nanjing University of Chinese Medicine, Nanjing, China. Phone: 18951755051. E-mail: jbfirsthcom.cn; or jbfirsth1com.
Corresponding author: Jiang Bin, MD, PhD. Associate Director, National Medical Centre of Colorectal DiseasesAssociate professor, Nanjing University of Chinese Medicine. The Third Affiliated Hospital, Nanjing University of Chinese Medicine, Nanjing, China. Phone: 18951755051. E-mail: jbfirsthcom.cn; or jbfirsth1com.