Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(3):279-288. doi:10.7150/ijbs.5996 This issue Cite
Research Paper
Interleukin-6 Receptor Blockade Selectively Reduces IL-21 Production by CD4 T Cells and IgG4 Autoantibodies in Rheumatoid Arthritis
Gustavo Carbone1, Augusta Wilson1, Sean A Diehl2, Janice Bunn3, Sheldon M. Cooper1, Mercedes Rincon1,2 ![]()
1. Department of Medicine, Division of Rheumatology1 and Division of Immunobiology2, University of Vermont, Burlington, VT 05405;
2. Department of Medicine, Division of Immunobiology, University of Vermont, Burlington, VT 05405;
3. Department of Mathematics and Statistics, University of Vermont, Burlington, VT05405, USA.
Received 2013-1-31; Accepted 2013-2-19; Published 2013-3-7
Abstract
Interleukin-6 (IL-6) levels are known to be increased in patients with rheumatoid arthritis (RA). Tocilizumab, a monoclonal antibody to the IL-6 receptor (IL-6R), reduces disease activity in RA, although its mechanisms of action remain unclear. Since IL-6 regulates cytokine production by CD4 T cells during activation, we investigated whether treatment with tocilizumab altered the phenotype and cytokine production by CD4 T cells in patients with rheumatoid arthritis. We show here that tocilizumab treatment does not change the production of cytokines by naïve CD4 T cells. However, tocilizumab treatment causes a selective decrease of IL-21 production by memory/activated CD4 T cells. Since IL-21 is known to promote plasma cell differentiation, we examined the effect of tocilizumab on the production of autoantibodies. We show that there is a decrease in the levels of IgG4 anti-CCP antibodies, but there is no effect on IgG1 anti-CCP antibodies. In addition, we show that IL-21 is a powerful inducer of IgG4 production by B cells. Thus, IL-6 contributes to the presence of IgG4-specific anti-CCP autoantibodies in RA patients, likely through its effect on IL-21 production by CD4 T cells, and IL-6R blockade down-regulates this pathway.
Keywords: Interleukin-6, IL-6, IL-21, CD4 T, rheumatoid arthritis, IgG4, auti-CCP, tocilizumab
Introduction
IL-6 is a cytokine produced by cells from the innate immune system (e.g. macrophages, dendritic cells, mast cells, neutrophils), B cells and, to a lesser extent, by some CD4 effector Th cells. In addition, in response to specific stimuli IL-6 is also secreted by a variety of non-leukocytes (e.g. endothelial cells, fibroblasts, epithelial cells, and some malignant cells) (1, 2, 3). IL-6 binds to its specific IL-6 receptor (IL-6R) which then associates with the signal transducer gp130 and triggers the JAK/STAT3 pathway as well as Ras/MAP kinase pathway. In addition to membrane bound IL-6R, a soluble form of IL-6R (sIL-6R) can be generated by cleavage or alternative splicing, bind to IL-6 and provide signals to cells lacking the receptor through a process call trans-signaling (4). Since elevated levels of IL-6 are found in a number of inflammatory conditions, IL-6 has been considered as a byproduct of an ongoing inflammation, rather than as a cytokine that drives the inflammatory response. As such, IL-6 is often used as a marker of inflammation. While TNFα and IL-1, the other two major proinflammatory cytokines, were soon after their identification used as targets for potential therapies, IL-6 was not thought to be an essential player in the pathogenesis of those inflammatory diseases.
During the last decade, however, a number of studies have shown that IL-6 is an important factor in determining the pattern of cytokine production by effector CD4 T cells (5). IL-6 inhibits Th1 differentiation and promotes Th2 differentiation through different mechanisms (6, 7). In addition, IL-6 in combination with TGFβ, but not IL-6 alone, promotes the generation of Th17 cells in mice (8, 9). IL-6 by itself is also sufficient and required for IL-21 production by naïve and memory CD4 T cells (10). The effect of IL-6 on IL-21 is independent of Th17 cell differentiation (11). The most well-characterized function of IL-21 in mouse and human is its role in promoting the differentiation of naïve and memory B cells into Ab-secreting plasma cells and the production of IgG (not IgM or IgE) (12-19). While IL-6 was identified as an inducer of B cell antibody production, recent studies have shown that the effect of IL-6 on Ab production is indirectly mediated by its effect on IL-21 production by CD4 T cells, both mouse and human (10, 20). IL-21 then promotes B cell differentiation and increases IgG production. In addition, IL-6 has been associated with the differentiation of T follicular helper (Tfh) cells, a cell population characterized by the production of IL-21 as effector cytokine (21, 22).
Increased levels of IL-6 are found both in serum as well as synovial tissues of patients with rheumatoid arthritis (RA) (2, 23), a chronic autoimmune inflammatory disease that targets the synovial membrane of multiple joints. RA is a complex disease in which a number of immune cells, including macrophages, B cells and CD4 T cells among others, contribute to the inflammatory cascade. Although the levels of IL-6 are elevated in RA, these levels do not necessary correlate with higher levels of TNFα, IL-1 or other proinflammatory cytokines, and they are not elevated in all RA patients. However, high levels of IL-6 were also shown to correlate with disease severity in RA (2), further suggesting that IL-6 contributes to RA pathogenesis. Studies using different mouse models of arthritis, including the collagen-induced arthritis (CIA) model and IL-6 deficient mice, were the first evidence that IL-6 contributes to arthritis onset and disease severity (24, 25). Several clinical trials have shown that tocilizumab, a humanized monoclonal Ab (mAb) against the IL-6 receptor (IL-6R), is effective in the treatment of RA (26-29). A recent clinical trial comparing tocilizumab with adalimumab (anti-TNF blocking antibody) has indicated that IL-6R blockade has superior efficacy than TNF blockade in RA patients as a monotherapy (EULAR 2012). Despite the growing evidence that supports a role of IL-6 in the pathogenesis of RA, it remains unclear how this cytokine specifically promotes inflammation and tissue damage.
We had a unique opportunity to explore the role of IL-6 in the pathogenesis of RA by performing a laboratory-based research study on patients with RA who were entered into an open-label Phase IIIb clinical study of tocilizumab. We obtained blood and serum samples from the patients at several fixed time points over 6 months, both prior to and after treatment with tocilizumab. The goal of the laboratory study was to determine if treatment with tocilizumab was associated with changes in levels of cytokines IL-21 and IL-17, immunoglobulins and composition of T cell subsets. The data show that blockade of the IL-6R in RA patients with tocilizumab is associated with reduced production of IL-21 by memory/activated CD4 T cells. Moreover, we found a selective reduction of IgG4 anti-CCP antibodies in this group. We also show that IL-21 is a major inducer of IgG4 production in purified human B cells, suggesting the presence of a pathway involving IL-6, IL-21 and IgG4 autoantibodies in RA.
Material and Methods
Study Population. Patients with RA who agreed to enter a Phase IIIb study of tocilizumab entitled, “An open-label, randomized study to evaluate the safety, tolerability and efficacy of tocilizumab (TCZ) monotherapy or TCZ in combination with non-biologic disease modifying antirheumatic drugs (DMARDs) in patients with active rheumatoid arthritis who have an inadequate response to current non-biologic or biologic DMARDs (ACT-STAR)” were offered the opportunity to enter a separate laboratory-based research study to investigate the mechanism of action of tocilizumab in RA. The laboratory study was entitled “IL-6 and IL-21 in rheumatoid arthritis.” Patients who agreed to enter the laboratory study signed a separate consent form that was approved by the Institutional Review Board of The University of Vermont.
Peripheral blood was obtained from 8 patients with RA who participated in the ACTSTAR protocol. Patients enrolled in the study were adult men and women with moderate to severe disease activity of RA, who were currently experiencing an inadequate clinical response or experiencing safety or tolerability related issues to stable doses of non-biological or biological DMARD therapy. Patients had to be ≥18 years of age, with moderate-to-severe RA of ≥6 months' duration, with a swollen joint count (SJC) of ≥4 and a tender joint count (TJC) of ≥4, Eligible patients receiving permitted DMARDs (methotrexate, hydroxychloroquine, sulfasalazine, azathioprine, and leflunomide) must have received stable doses for ≥7 weeks prior to study entry. Oral glucocorticoids (≤10 mg/day prednisone or equivalent) and nonsteroidal antiinflammatory drugs (NSAIDs)/cyclooxygenase 2 inhibitors were permitted but dose changes were not allowed. Patients were excluded if treated with rituximab within 6 months before screening. In patients who were receiving biologic drugs, these were discontinued prior to study entry, but traditional DMARDs were continued. After the initial screening visit, the 8 patients enrolled in the laboratory study were randomized in a 1:1 ratio to receive monthly infusions of either 4 mg/kg or 8 mg/kg tocilizumab over 6 months. There were 7 women and 1 man, with ages ranging from 32-75 years, and disease duration from 3-19 years. Two patients had never received a biologic drug, while the other 6 patients either had an inadequate response or intolerance to one or more biologic drug. All 8 patients were receiving one or more oral DMARD (methotrexate, sulfasalazine or hydroxychloroquine). Five patients were randomized to tocilizumab 8 mg /kg. Two of the three patients randomized to 4 mg/kg had the dose of tocilizumab increased to 8 mg/kg at week 8 because they had less than 20 % improvement in joint counts at that time point.
Whole heparinized blood and serum samples were obtained at baseline prior to the first infusion (0 month), and 1 month (prior to the second infusion), 3 months (prior to the fourth infusion), and 6 months (prior to the last infusion). Tender and swollen joint counts were obtained at the same time points. Heparin containing blood samples were used for the isolation of peripheral blood mononuclear cells, and a gelatin-containing tube was used to obtain serum. The levels of rheumatoid factor (RF), anti-cyclic citrullinated peptide (CCP), and anti-nuclear antibody (ANA) were determined at the same time points by the clinical laboratory of Fletcher Allen Health Care.
Cell purification and culture. Mononuclear cells from peripheral blood (PBMC) were isolated by Ficoll density-gradient centrifugation (Histopaque). CD4 T cells were purified by positive selection using the CD4 MACS kit (Miltenyi Biotec) as recommended by the manufacturer. CD4 T cells were then sorted into CD4 CD45 RO+ and CD4 CD45RA+ cells after releasing anti-CD4 Ab from the cells using the CD45RA MACS kit (containing the “Release” reagent) as recommended by the manufacturer. Purity of the cells in both populations (>95%) was determined by cell surface staining and FACS as described below.
CD4 CD45RA and CD4 CD45RO cells were activated (106 cells/ml in RPMI with 5% fetal bovine serum) with immobilized anti-CD3 antibody (Ab) (3 μg/ml) and soluble anti-CD28 Ab (BD Bioscience) (1 μg/ml) as previously described (30). Supernatants were collected after 24 h (CD4 CD45RO cells) or 48 h (CD4 CD45RA cells) and used for cytokine detection as described below.
B cells were obtained from peripheral blood of healthy volunteers by CD22 MACS microbead magnetic column selection (Miltenyi Biotec). Purity was > 97% and was determined by flow cytometry by staining for CD19 and CD20. We co-cultured B cells (2 - 2.5 × 105 cells/ml) on irradiated (50 Gray) CD40L-expressing L cell fibroblasts (5 × 104 cells/ml, > 96% surface CD154+) in 1 ml Iscove's Modified Eagle's Medium containing 8% fetal bovine serum and penicilin/streptomycin and with or without rhIL-4 (25 ng/ml, Peprotech) or rhIL-21 (10 ng/ml, Peprotech) in 24-well plates. After 6 days, we harvested supernatants and the levels of IgG4 in the supernatant were determined as described below.
Flow cytometry analysis. PBMC cells were stained with an anti-CD14 (monocytes), anti-CD19 (B cells) and anti-CD4, anti-CD45RA and anti-CD45RO Abs (Caltag). Stained samples were examined by flow cytometry using the LSRII flow cytometer (BD Bioscience).
Cytokine production and serum antibody analysis. The levels of IL-2, IL-17, IL-21, IL-4 and IFNγ in the culture supernatants were determined using the Human Cytokine/Chemokine Milliplex MAP Kits following the recommendations of the manufacturer (Millipore Co.) and using the fluorescence plate reader and analysis software (BioRad). For analysis of IL-21 gene expression, total RNA was extracted from 4 x 105 freshly isolated CD4 CD45RO cells using the RNAeasy kit (Qiagen) as recommended by the manufacturer. First-strand cDNA synthesis was performed as previously described (30). Quantitative RT-PCR was performed on cDNA using Assay on Demand probe/primer sets for IL-21 and HPRT (Applied Biosystems). Gene amplification was performed on an ABIPrism® 7700 instrument from Applied Biosystems. Expression of IL-21 was normalized to HPRT levels. Relative values were determined by the comparative Ct method. Patient 1 and 8 were not included in the analyses due to insufficient amount of RNA.
The levels of serum IgG1, IgG2, IgG3, IgG4, IgM and IgA were determined using the Human Immunoglobulin Isotype Milliplex MAP Kit following the recommendations by the manufacturer (Millipore). The levels of IgG4- and IgG1-specific anti-CCP Abs were determined using the QUANTA LiteTM CCP 3.1 ELISA kit (INOVA Diagnostic Inc.) containing a CCP-coated plate, but instead of using the horseperoxidase (HRP)-conjugated anti-human IgG/IgA Abs provided with the kit, we used a HRP-conjugated anti-human IgG4 (Invitrogen) or anti-human IgG1 (Invitrogen) Abs. Assay was performed as recommended by the manufacturer (INOVA Diagnostic Inc). Plates were read at 450 nm using an ELISA plate reader.
Statistical Analysis. Single-group repeated measures analysis of variance based on ranked data was used to determine differences over time of treatment in the various parameters examined. Repeated measures analysis of variance was also used to examine within-subject differences in fold reduction between IgG1 and IgG4, followed by tests of simple effects for examination of fold reduction over time of IgG1 and IgG4 separately. All analyses were performed using SAS 9.2.
Results
Tocilizumab treatment does not change the frequency of naïve and memory/activated CD4 T cell populations in the circulation of RA patients
There have been few longitudinal studies examining immunologic changes that may occur during treatment with tocilizumab. The goal of this study was to obtain evidence that could point to potential effects of tocilizumab on various immunologic parameters of a small group of patients followed longitudinally and sampled 4 times during 6 months of treatment. By clinical parameters, the patients responded to tocilizumab with a reduction of tender and swollen joints (Table I). By month 6 all patients achieved at least a 20% reduction in the number of tender and swollen joints, and 5 of 8 had a 70% or greater reduction. DAS28 scores also decreased, although the effect was progressive over time (Table I).
Disease severity is markedly attenuated by tocilizumab.
| 0 m | 1 m | 3 m | 6 m | p | |
|---|---|---|---|---|---|
| Tender Ja | 30.3 (12.9) | 13.8 (6.9) | 9.8 (5.3) | 7.8 (4.5) | <0.01 |
| Swollen Ja | 25.1 (9.0) | 10.1 (5.4) | 5.4 (4.0) | 4.0 (3.7) | <0.01 |
| DAS28b | 6.3 (1.3) | 4.6 (1.0) | 3.9 (1.0) | 3.4 (0.7) | <0.01 |
aNumber of tender and swollen joints prior to treatment (base line, 0 m) or during the treatment with tociluzumab (1, 3 and 6 m after initiation of the treatment). Mean (n=8) and standard deviation (in parenthesis) are shown. The significance of the difference in number over time was determined by single-group repeated measures analysis of variance based on ranked data. p value less than 0.05 was considered significant. bDAS28 score. Mean (n=8) and standard deviation are shown. The significance of the difference over time was determined by single-group repeated measures analysis of variance. p value less than 0.05 was considered significant.
We first examined the relative frequency of the immune cell populations in PBMC at the time points described in Methods by cell surface staining and flow cytometry. There was no significant change in the percentage of monocytes (as defined by CD14 expression), B cells (defined by the expression of CD19) or CD4 T cells in PBMC during the course of therapy with tocilizumab (Table II). In addition, within the CD4 T cell population, there were no statistically significant changes in the relative frequency of naïve (CD4 CD45RA) versus memory/activated (CD4 CD45RO) cells (Table II). Thus, blocking IL-6 signaling over 6 months does not appear to alter the frequencies of circulating monocytes, T or B cells in RA patients.
Blocking IL-6R does not affect the frequency of T cells, B cells or monocytesa.
| 0 m | 1 m | 3 m | 6 m | cp | |
|---|---|---|---|---|---|
| CD4 T cells | 38.6 (9.2) | 40.6 (12.1) | 42.4 (8.7) | 41.5 (6.5) | 0.56 |
| bCD45RA | 32.8 (10.9) | 31.3 (11.8) | 31.0 (12.2) | 42.6 (9.9) | 0.69 |
| bCD45RO | 47.5 (12.9) | 43.1 (11.2) | 43.4 (10.9) | 42.6 (10.7) | 0.10 |
| B cells | 6.1 (4.6) | 5.3 (3.1) | 5.0 (3.0) | 5.9 (4.3) | 0.27 |
| Monocytes | 15.7 (8.3) | 14.3 (8.7) | 13.3 (7.6) | 11.3 (8.8) | 0.76 |
aPercentage of the total CD4 T cells, B cells and monocytes in PBMC. Mean (n=8) and standard deviation (in parenthesis) are provided. bPercentage of CD45RA cells and CD45RO cells within the CD4 T cell population. Mean (n=8) and standard deviation (in parenthesis) are provided. cThe significance of the difference in percentage over time was determined by single-group repeated measures analysis of variance based on ranked data. p value less than 0.05 was considered significant.
Treatment with IL-6R blockade selectively reduces IL-21 production by CD4 RO T cells in RA patients
IL-6 is now recognized as a modulator of cytokine production by effector CD4 cells, and some of these cytokines are thought to be involved in the pathogenesis of RA (e.g. IL-17, IL-21). Thus, IL-6 is known to inhibit IFNγ production, promote Th2 differentiation, cooperate with TGFβ in CD4 Th17 differentiation, and induce by itself IL-21 production in both naïve and memory CD4 cells (5). The chronic presence of IL-6 in RA patients could therefore have an effect on the production of some or all of these cytokines. We therefore determined if blocking IL-6 signaling in vivo would alter cytokine production by CD4 cells in RA patients over time. Since cytokine levels are different in naïve and memory/activated CD4 cells, CD4 CD45RA and CD4 CD45RO cells were isolated from peripheral blood obtained from patients prior to or during the treatment with tocilizumab. CD4 CD45RA and CD4 CD45RO cells were activated in vitro with anti-CD3 and anti-CD28 Abs as we previously described (30). The levels of IL-2 (CD4 cell growth factor), IFNγ (Th1 cytokine), IL-4 (Th2 cytokine), IL-17 (Th17 cytokine) and IL-21 in culture supernatant were determined. There was no statistically significant change in the production of any of these cytokines by naïve CD4 CD45RA cells during the course of tocilizumab treatment (Table III).
Blocking IL-6R specifically reduces the production of IL-21 by CD4 CD45RO T cellsa.
| CD4 CD45RA | |||||
|---|---|---|---|---|---|
| 0 m | 1 m | 3 m | 6 m | p | |
| bIL-2 | 1.2 (1.2) | 0.8 (0.7) | 0.7 (0.5) | 0.9 (0.7) | 0.29 |
| bIFNγ | 2.5 (4) | 1.1 (1.8) | 1.1 (2.1) | 0.9 (1.1) | 0.80 |
| cIL-17 | 146 (116) | 128 (121) | 98 (95) | 155 (119) | 0.14 |
| cIL-4 | 66 (59) | 83 (108.3) | 70 (82.5) | 68 (60.4) | 0.34 |
| cIL-21 | 40 (55.3) | 23 (28.3) | 40 (74) | 15 (11) | 0.83 |
| CD4 CD45RO | |||||
| 0 m | 1 m | 3 m | 6 m | p | |
| bIL-2 | 3.5 (2.3) | 2.06 (1.3) | 2.2 (1.5) | 2.4 (1.9) | 0.02 |
| bIFNγ | 13.0 (11.5) | 7.7 (5.6) | 9.3 (6.9) | 10.2 (2.8) | 0.20 |
| cIL-17 | 477 (305) | 310 (193) | 321 (173) | 392 (231) | 0.16 |
| cIL-4 | 347 (298.4) | 373 (451.6) | 249 (140.4) | 309 (203.3) | 0.14 |
| cIL-21 | 50 (38.2) | 31 (33.8) | 36 (22.2) | 15.4 (8.5) | 0.004 |
aCD4 CD45RA and CD4 CD45RO T cells were isolated at the specified periods of time during the treatment with tocilizumab, activated with anti-CD3 and anti-CD28 Abs for 24 h (CD4 CD45RO cells) or 48 h (CD4 CD45 RA cells), and cytokine levels in the supernatant was determined by Multiplex analysis. Mean (n=8) and standard deviation (in parenthesis) are shown. The significance of the difference in cytokine production over time was determined by single-group repeated measures analysis of variance based on ranked data. p value less than 0.05 was considered significant. bcytokine values in ng/ml. ccytokine values in pg/ml.
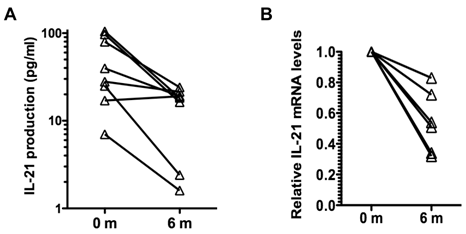
Analysis of cytokine production by memory CD4 CD45RO cells showed no effect of tocilizumab treatment in the production of IFNγ and IL-17, and a slight reduction in the production of IL-2 (Table III). However, there was a prominent reduction (over 3-fold) in the production of IL-21 by these cells during the course of tocilizumab treatment (Table III). Analysis of IL-21 production by CD4 CD45RO cells in individual patients showed a reduction of IL-21 production in seven out of the eight patients independent of the levels of IL-21 levels produced prior to the initiation of the treatment (Fig. 1A). We also examined the levels of IL-21 mRNA in CD4 CD45RO cells freshly isolated from the patients prior to and after tocilizumab treatment without further in vitro activation by quantitative real time RT-PCR. IL-21 mRNA levels in freshly isolated CD4 CD45RO cells at the end of the treatment were decreased compared with the initial levels in all examined patients (Fig. 1B). Considered together, these data suggest that IL-6 contributes to upregulation of IL-21 production in memory/activated CD4 T cells from RA patients, and that blocking IL-6 reverses this effect.
Tocilizumab treatment reduces production of IL-21 by CD4 CD45RO T cells in RA patients. (A) CD4 CD45RO cells were isolated from each patient at the indicated period of time and activated for 24 h with anti-CD3 and anti-CD28 Abs. IL-21 levels in the supernatant were determined by Multiplex analysis. (B) Relative IL-21 mRNA levels in freshly isolated CD4 CD45RO T cells from patients were determined by real time RT-PCR using HPRT as house keeping gene, using the delta delta CT analysis. IL-21 mRNA levels for each patients were relative to the levels at time 0 (prior to the first treatment). Levels of IL-21 mRNA at 6 months were statistically significantly reduced relative to the levels prior to the treatment (p=0.03) using Wilcoxon signed rank test.
Reduction of Serum IgG4 levels with tocilizumab treatment
IL-21 production plays a major role in antibody (Ab) production by B cells (12-17). It promotes the production of IgG, with no effect on IgM production and a negative effect on IgE. We have shown that the effect of IL-6 on B cell antibody production is an indirect effect that is mediated by IL-21 produced by CD4 cells (10). The presence of autoantibodies is a feature of RA, although they are not necessarily present in all patients. We examined whether the blockade of IL-6R with tocilizumab could have an effect on the total serum levels of known autoantibodies. Five patients were positive for RF, five were positive for anti-CCP Abs, and four were ANA positive. We found no significant change in the serum titers of these autoantibodies during tocilizumab treatment using the same time points described above (data not shown).
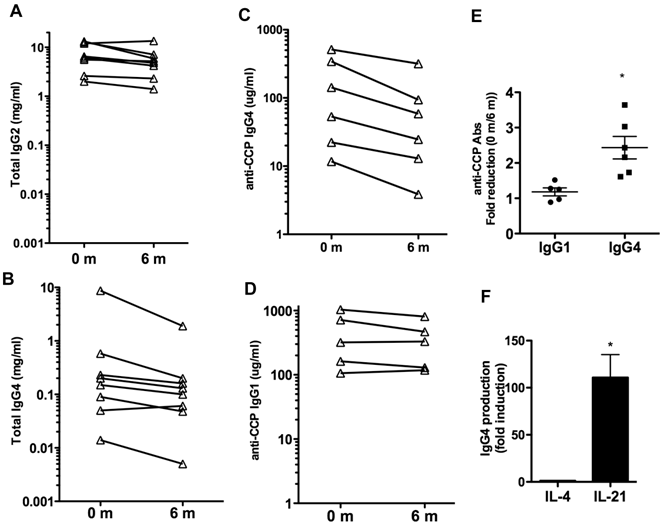
We also examined whether IL-6R blockade could affect the total levels of IgG or other Ig isotypes in serum. Total levels of IgG1, IgG2, IgG3, IgG4, IgM and IgA in serum were measured. The levels of IgG1, IgG3, IgM and IgA did not change over the 6 months of treatment (Table IV). A slight reduction of IgG2 levels, marginally significant (p= 0.06) was observed (Table IV and Fig. 2A). However, over the six months there was a prominent 4-fold reduction in the levels of IgG4 (p=0.06) (Table IV). The levels of IgG4 were markedly decreased in all but one patient (Fig. 2B). Thus, IL-6 blockade was associated with decreased production of certain IgG isotypes, predominantly IgG4.
Blocking IL-6R causes a selective decrease in the levels of IgG4a.
| 0 m | 1 m | 3 m | 6 m | p | |
|---|---|---|---|---|---|
| IgA | 1.1 (0.5) | 1.0 (0.5) | 1.0 (0.5) | 0.9 (0.5) | 0.35 |
| IgG1 | 4 (1.55) | 3.5 (1.1) | 3.5 (1.3) | 3.1 (1.1) | 0.38 |
| IgG2 | 7.6 (4.5) | 6.8 (3.9) | 6.1 (3.4) | 5.5 (3.7) | 0.06 |
| IgG3 | 0.4 (.3) | 0.3 (0.2) | 0.34 (0.3) | 0.36 (0.4) | 0.34 |
| IgG4 | 1.3 (3.0) | 0.9 (2.2) | 0.4 (0.8) | 0.3 (0.6) | 0.06 |
| IgM | 2.0 (2.2) | 1.8 (1.8) | 1.8 (1.8) | 1.7 (1.6) | 0.30 |
aLevels of total IgA, IgG1, IgG2, IgG3, IgG4 and IgM in serum at the specified periods of time during the treatment. Mean (n=8) in mg/ml and standard deviation (in parenthesis) are shown for each isotype. The significance of the difference in the levels of Ig over time was determined by one-group repeated measures analysis of variance based on ranked data. p value less than 0.05 was considered significant.
Effect of tocilizumab on IgG isotypes and IgG4 autoantibodies. (A) and (B) Serum levels of total IgG2 (A) and IgG4 (B) prior to (0 months) and 6 months after the treatment with tocilizumab in the eight patients. (C) IgG4-specific anti-CCP Ab levels in serum. Six patients (Patients #1, 2, 4, 5, 6 and 7) had detectable levels of IgG4-anti-CCP Abs in serum prior to the initiation of tocilizumab treatment (0 months). (D) IgG1-specific anti-CCP Ab levels in serum. Five patients (Patients #1, 2, 4, 6 and 7) showed detectable levels of IgG1-anti-CCP Abs prior to the treatment. (E) Fold reduction in the serum levels of IgG1-specific anti-CCP Abs and IgG4-specific anti-CCP Abs between 0 months (prior to the treatment) and 6 months after the initiation of the treatment. Results of the repeated measures analysis of variance suggest that the fold reduction in IgG1 differs from that observed in IgG4 (p=0.011 for the interaction effect). Follow-up tests of simple effects shows a statistically significant (p =0.011) fold-reduction in IgG4 anti-CCP Abs (denoted by *) while there was no reduction in IgG1 anti-CCP Abs levels (p=0.185). Note that a fold-reduction equal to 1 between 0 months and 6 months means no effect on IgG levels with treatment. (F) Purified B cells from healthy volunteers (n=4) were activated in vitro with CD40L-expressing cells in the presence of medium, IL-4 or IL-21. The levels of IgG4 in the supernatants were determined after 6 days. Fold induction for each subject in the levels of IgG4 produced by B cells activated with IL-4 or IL-21 relative to the levels by B cells activated with just medium is shown. The statistically significant difference between fold-induction obtained with IL-21 relative to IL-4 Abs was determined by paired t test analysis, p =0.0206.
Tocilizumab treatment reduces the levels of IgG4-specific anti-CCP antibodies
Although IgG1 is the predominant isotype among anti-CCP and other autoantibodies, recent studies have shown that IgG4 is the second most frequent isotype found in RA patients (31, 32). We therefore examined the levels of IgG4- and IgG1-specific anti-CCP Abs at baseline and after 6 months of therapy to determine if there was an isotype specific effect. Six patients had detectable IgG4-specific anti-CCP Abs, and five patients had detectable IgG1-specific anti-CCP Abs. Analysis in individual patients showed a pronounced reduction (2-3 fold) in the serum levels of IgG4-specific anti-CCP Abs in all patients (p= 0.011) (Fig. 2C), but no statistically significant reduction in the levels of IgG1-anti-CCP Abs (p= 0.185) (Fig. 2D). Results of the repeated measures analysis of variance for the difference between the levels of IgG4 and IgG1 specific anti-CCP Abs indicated that the fold reduction in IgG4 levels with the treatment differs from that observed in IgG1 (p=0.011) (Fig. 2E).
The presence of elevated levels of IgG4 has been considered to be an indicator of a Th2 immune response in human (33) but there is no clear Th2 response in RA. Since IL-21 is an important plasma cell differentiation factor, we compared the ability of IL-21 relative to IL-4 to enhance IgG4 production by B cells. B cells were isolated from the peripheral blood of healthy volunteers and activated in vitro with CD40L-expressing cells in the presence of medium alone, IL-4 or IL-21. IgG4 levels in the supernatant were determined 6 days later. IL-21 strongly induced B cell production of IgG4 when compared to the production in the presence of medium or IL-4 (Fig. 2F). This supports the hypothesis that IL-21 may be responsible for the selective production of IgG4 in RA and other non-Th2 type autoimmune diseases.
Discussion
Elevated IL-6 levels have long been associated with RA disease activity and progression. The success of tocilizumab in the treatment of RA established that IL-6 also had a role in promoting disease, although the mechanisms by which IL-6 promotes RA pathogenesis are poorly understood. The goal of this pilot study was to see if we could find changes in cytokine profiles over time that could be correlated with IL-6 suppression with tocilizumab. Since IL-6 regulates cytokine production by naïve and activated/memory CD4 T cells, it was important to separate and examine both naïve and activated/memory CD4 T cells since the autoreactive cells would be expected to be in the latter (CD45RO) population.
Despite the evidence that IL-6 promotes Th2 and inhibits Th1 differentiation in vitro, our data suggest that IL-6R blockade over 6 months does not significantly affect IL-4 and IFNγ production by CD4 CD45RO cells. We were also unable to demonstrate a change in IL-17 production by CD4 CD45RO cells during treatment with tocilizumab. A recent study reported a reduction in the frequency of IL-17 producing CD4 T cells after 3 months of tocilizumab treatment (34). There are significant methodological differences between the two studies that could account for this apparent discrepancy. While our study was performed on purified CD4 CD45RO cells, the study by Samson et al. (34) examined total CD4 T cells. In addition, we measured the actual production of IL-17 in the supernatant after stimulation with anti-CD3/CD28 Abs, while the study by Samson et al. examined the frequency of IL-17 cells (0.5-1.5% of CD4 T cells) by intracellular staining upon stimulation with PMA and ionomycin. Thus, it is possible that although the frequency of IL-17 producing cells is reduced with tocilizumab treatment, the overall amount of cytokine produced by these cells is not changed. IL-6-independent pathways that promote the development of Th17 cells have been reported (35). We also cannot rule out a potential effect of tocilizumab on the production of IL-17 by other T cell subsets such as γδ T cells (36) or the recently identified innate lymphoid cells (37).
The results from our study however indicate that IL-6 blockade significantly reduced the production of IL-21 by CD4 CD45RO cells. In seven out of the eight patients there was a decrease in the production of IL-21 by these cells with a more prominent effect in those patients with the highest production of IL-21 prior to the initiation of treatment. In the last few years, IL-21 is emerging as another potential target for RA based on studies showing that blockade of IL-21 signaling with an IL-21R.Fc fusion protein interferes with the development of CIA in mice and adjuvant-induced arthritis in rats (38). In addition, IL-21R knockout mice are more resistant to the development of arthritis using the K/BxN mouse model (39). IL-6 is sufficient and necessary to induce IL-21 by both naïve and memory CD4 T cells (10). Our data provides evidence that in patients with RA, treatment with tocilizumab is associated with decreased production of IL-21 by activated/memory CD4 T cells. This suggests that one of the effects of IL-6 blockade may due to reduction in levels of IL-21. There is an ongoing Phase I clinical trial with an anti-IL-21 blocking antibody in healthy and RA subjects (ClinicalTrials.gov).
IL-21 is a major factor in the differentiation of human B cells into Ab-producing plasma cells (12-19). It is known that IL-6 also regulates Ab production. However, using mouse models, we have previously shown that IL-6 promotes the in vivo and in vitro production of IgG by B cells indirectly by inducing IL-21 production by CD4 cells, and IL-21 acting directly on B cells (10). Similar results have been obtained in human (20). The results from our study provide evidence for a potential association between IL-6 blockade and a reduction in IgG4 levels. Studies have shown that while IgG1 is the most abundant autoantibody isotype in RA, IgG4 is the second most abundant isotype for both anti-CCP Abs and anti-citrullinated fibrin autoantibodies (31, 32). IgG4 is also the predominant RF isotype together with IgG1 (40). Although we did not find a decrease in IgG1-anti-CCP antibodies, our data indicate an association between the blockade of IL-6R and a reduction in IgG4-specific anti-CCP Abs. Treatment with TNF blocking agents in RA patients has also been reported to preferentially reduce IgG4 isotype anti-citrullinated fibrinogen Abs (41). Although total IgG4 levels also decreased with the treatment, Spearman correlation coefficient statistical analysis showed no correlation between the reduction of IgG4-specific anti-CCP Abs and the reduction in the levels of total IgG4 (rs= -0.09, p= 0.81). Thus, the reduced levels of IgG4-specific anti-CCP Abs do not appear to be simply the consequence of having less total IgG4. The effect of tocilizumab on IgG4-specific anti-CCP Abs is likely mediated through its effect on IL-21 since we show that IL-21 is far more potent in promoting IgG4 production in human B cells from healthy volunteers than IL-4. A recent study has reported an increased frequency of Tfh cells (characterized by their production of IL-21) and IL-21 expression in CD4 T cells in RA patients compared with healthy subjects (42). Interestingly, this study also reports a correlation between Tfh cell frequency and the levels of anti-CCP Abs.
Although the relevance of autoantibodies in the pathogenesis of RA has been questioned for long time, anti-citrullinated protein antibodies (ACPA) are emerging as potential players in the development of RA (43, 44). ACPA have been found in individuals before disease onset and the presence of ACPA was accepted as a criteria for RA diagnosis at the 2010 ACR/EULAR meeting. Some recent studies have shown that ACPA are independent of disease phenotype, but are associated with disease activity (45-47). No large studies have examined the isotype specificity of ACPA and other autoantibodies in RA.
Increased levels of IgG4 isotype are found among anti-neutrophil cytoplasm autoantibodies in Wegener's granulomatosis (48) and anti-dsDNA antibodies in lupus (49). Moreover, a number of inflammatory disorders that are characterized by elevated serum IgG4 levels (IgG4-related systemic diseases) have emerged (50, 51). The role of IgG4 in the pathology of these syndromes and other autoimmune diseases remains unclear. IgG4 does not fix complement and has no strong affinity for FcγR. However, studies have shown that IgG4 is associated with the presence of natural bispecific antibodies against CCP in RA (52, 53). We can speculate that bispecific Abs may be more pathogenic due to crosslinking of different molecules.
In summary, this pilot study of a small cohort of patients analyzed longitudinally over several months provides evidences that IL-6 blockade decreases IL-21 production by activated/memory CD4 T cells as well as serum levels of IgG4-specific autoantibodies. Larger clinical studies in patients treated with tocilizumab over time will be needed to further support these preliminary observations. Our small sample size does not provide sufficient power to detect correlations between laboratory parameters and response to tocilizumab. However, a recent study (ESPOIR cohort) in early rheumatoid arthritis has shown that IL-6 and IL-21 are associated with markers of B cell activation, presence of autoantibodies and disease progression (54), supporting the results from our study.
Acknowledgements
We would like to thank Shirley Frimpong and Dr. Tina Thornton for assistance with RNA isolation, Timothy Hunter and Mary Lou Shane (DNA Sequencing Facility, University of Vermont, Burlington, VT) for assistance with real time RT-PCR analysis, Colette Charland for help with flow cytometry analysis (Flow Cytometry Facility, University of Vermont, Burlington, VT). The study was supported by Hoffman-La Roche Inc.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rincon M. Interleukin-6: from an inflammatory marker to a target for inflammatory diseases. Trends Immunol. 2012;33:571-7
2. Tanaka T, Kishimoto T. Targeting interleukin-6: all the way to treat autoimmune and inflammatory diseases. Int J Biol Sci. 2012;8:1227-36
3. Rincon M. Special issue on interleukin-6 (IL-6). Int J Biol Sci. 2012;8:1225-6
4. Rose-John S. IL-6 Trans-Signaling via the Soluble IL-6 Receptor: Importance for the Pro-Inflammatory Activities of IL-6. Int J Biol Sci. 2012;8:1237-47
5. Dienz O, Rincon M. The effects of IL-6 on CD4 T cell responses. Clin Immunol. 2009;130:27-33
6. Diehl S, Anguita J, Hoffmeyer A, Zapton T, Ihle JN, Fikrig E. et al. Inhibition of Th1 differentiation by IL-6 is mediated by SOCS1. Immunity. 2000;13:805-15
7. Diehl S, Chow CW, Weiss L, Palmetshofer A, Twardzik T, Rounds L. et al. Induction of NFATc2 expression by interleukin 6 promotes T helper type 2 differentiation. J Exp Med. 2002;196:39-49
8. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M. et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235-8
9. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179-89
10. Dienz O, Eaton SM, Bond JP, Neveu W, Moquin D, Noubade R. et al. The induction of antibody production by IL-6 is indirectly mediated by IL-21 produced by CD4+ T cells. J Exp Med. 2009;206:69-78
11. Suto A, Kashiwakuma D, Kagami S, Hirose K, Watanabe N, Yokote K. et al. Development and characterization of IL-21-producing CD4+ T cells. J Exp Med. 2008;205:1369-79
12. Ozaki K, Spolski R, Ettinger R, Kim HP, Wang G, Qi CF. et al. Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of Blimp-1 and Bcl-6. J Immunol. 2004;173:5361-71
13. Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, Sher A. et al. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298:1630-4
14. Suto A, Nakajima H, Hirose K, Suzuki K, Kagami S, Seto Y. et al. Interleukin 21 prevents antigen-induced IgE production by inhibiting germ line C(epsilon) transcription of IL-4-stimulated B cells. Blood. 2002;100:4565-73
15. Shang XZ, Ma KY, Radewonuk J, Li J, Song XY, Griswold DE. et al. IgE isotype switch and IgE production are enhanced in IL-21-deficient but not IFN-gamma-deficient mice in a Th2-biased response. Cell Immunol. 2006;241:66-74
16. Ettinger R, Sims GP, Fairhurst AM, Robbins R, da Silva YS, Spolski R. et al. IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J Immunol. 2005;175:7867-79
17. Ettinger R, Sims GP, Robbins R, Withers D, Fischer RT, Grammer AC. et al. IL-21 and BAFF/BLyS synergize in stimulating plasma cell differentiation from a unique population of human splenic memory B cells. J Immunol. 2007;178:2872-82
18. Kuchen S, Robbins R, Sims GP, Sheng C, Phillips TM, Lipsky PE. et al. Essential role of IL-21 in B cell activation, expansion, and plasma cell generation during CD4+ T cell-B cell collaboration. J Immunol. 2007;179:5886-96
19. Diehl SA, Schmidlin H, Nagasawa M, van Haren SD, Kwakkenbos MJ, Yasuda E. et al. STAT3-mediated up-regulation of BLIMP1 Is coordinated with BCL6 down-regulation to control human plasma cell differentiation. J Immunol. 2008;180:4805-15
20. Diehl SA, Schmidlin H, Nagasawa M, Blom B, Spits H. IL-6 Triggers IL-21 production by human CD4(+) T cells to drive STAT3-dependent plasma cell differentiation in B cells. Immunol Cell Biol. 2012;90:802-11
21. Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L. et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. 2008;29:138-49
22. Eto D, Lao C, DiToro D, Barnett B, Escobar TC, Kageyama R. et al. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T cell (Tfh) differentiation. PLoS One. 2011;6:e17739
23. Houssiau FA, Devogelaer JP, Van Damme J, de Deuxchaisnes CN, Van Snick J. Interleukin-6 in synovial fluid and serum of patients with rheumatoid arthritis and other inflammatory arthritides. Arthritis Rheum. 1988;31:784-8
24. Sasai M, Saeki Y, Ohshima S, Nishioka K, Mima T, Tanaka T. et al. Delayed onset and reduced severity of collagen-induced arthritis in interleukin-6-deficient mice. Arthritis Rheum. 1999;42:1635-43
25. Alonzi T, Fattori E, Lazzaro D, Costa P, Probert L, Kollias G. et al. Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med. 1998;187:461-8
26. Emery P, Keystone E, Tony HP, Cantagrel A, van Vollenhoven R, Sanchez A. et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis. 2008;67:1516-23
27. Genovese MC, McKay JD, Nasonov EL, Mysler EF, da Silva NA, Alecock E. et al. Interleukin-6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease-modifying antirheumatic drugs: the tocilizumab in combination with traditional disease-modifying antirheumatic drug therapy study. Arthritis Rheum. 2008;58:2968-80
28. Kremer JM, Blanco R, Brzosko M, Burgos-Vargas R, Halland AM, Vernon E. et al. Tocilizumab inhibits structural joint damage in rheumatoid arthritis patients with inadequate responses to methotrexate: results from the double-blind treatment phase of a randomized placebo-controlled trial of tocilizumab safety and prevention of structural joint damage at one year. Arthritis Rheum. 2011;63:609-21
29. Dayer JM, Choy E. Therapeutic targets in rheumatoid arthritis: the interleukin-6 receptor. Rheumatology (Oxford). 2010;49:15-24
30. Briso EM, Dienz O, Rincon M. Cutting edge: soluble IL-6R is produced by IL-6R ectodomain shedding in activated CD4 T cells. J Immunol. 2008;180:7102-6
31. Engelmann R, Brandt J, Eggert M, Karberg K, Krause A, Neeck G. et al. IgG1 and IgG4 are the predominant subclasses among auto-antibodies against two citrullinated antigens in RA. Rheumatology (Oxford). 2008;47:1489-92
32. Chapuy-Regaud S, Nogueira L, Clavel C, Sebbag M, Vincent C, Serre G. IgG subclass distribution of the rheumatoid arthritis-specific autoantibodies to citrullinated fibrin. Clin Exp Immunol. 2005;139:542-50
33. Punnonen J, Aversa G, Cocks BG, McKenzie AN, Menon S, Zurawski G. et al. Interleukin 13 induces interleukin 4-independent IgG4 and IgE synthesis and CD23 expression by human B cells. Proc Natl Acad Sci U S A. 1993;90:3730-4
34. Samson M, Audia S, Janikashvili N, Ciudad M, Trad M, Fraszczak J. et al. Inhibition of IL-6 function corrects Th17/Treg imbalance in rheumatoid arthritis patients. Arthritis Rheum. 2012Aug;64(8):2499-503
35. Kimura A, Naka T, Kishimoto T. IL-6-dependent and -independent pathways in the development of interleukin 17-producing T helper cells. Proc Natl Acad Sci U S A. 2007;104:12099-104
36. Silva-Santos B. gammadelta cells making IL-17. Blood. 2011;118:3-5
37. Hirota K, Ahlfors H, Duarte JH, Stockinger B. Regulation and function of innate and adaptive interleukin-17-producing cells. EMBO Rep. 2012;13:113-20
38. Young DA, Hegen M, Ma HL, Whitters MJ, Albert LM, Lowe L. et al. Blockade of the interleukin-21/interleukin-21 receptor pathway ameliorates disease in animal models of rheumatoid arthritis. Arthritis Rheum. 2007;56:1152-63
39. Jang E, Cho SH, Park H, Paik DJ, Kim JM, Youn J. A positive feedback loop of IL-21 signaling provoked by homeostatic CD4+CD25- T cell expansion is essential for the development of arthritis in autoimmune K/BxN mice. J Immunol. 2009;182:4649-56
40. Cohen PL, Cheek RL, Hadler JA, Yount WJ, Eisenberg RA. The subclass distribution of human IgG rheumatoid factor. J Immunol. 1987;139:1466-71
41. Bos WH, Bartelds GM, Vis M, van der Horst AR, Wolbink GJ, van de Stadt RJ. et al. Preferential decrease in IgG4 anti-citrullinated protein antibodies during treatment with tumour necrosis factor blocking agents in patients with rheumatoid arthritis. Ann Rheum Dis. 2009;68:558-63
42. Ma J, Zhu C, Ma B, Tian J, Baidoo SE, Mao C. et al. Increased frequency of circulating follicular helper T cells in patients with rheumatoid arthritis. Clin Dev Immunol. 2012;2012:827480
43. Willemze A, Trouw LA, Toes RE, Huizinga TW. The influence of ACPA status and characteristics on the course of RA. Nat Rev Rheumatol. 2012;8:144-52
44. Meyer O. Anti-citrullinated peptide/protein antibodies and structural prognosis of rheumatoid arthritis: quantity versus quality. J Rheumatol. 2012;39:675-6
45. Shiozawa K, Kawasaki Y, Yamane T, Yoshihara R, Tanaka Y, Uto K. et al. Anticitrullinated protein antibody, but not its titer, is a predictor of radiographic progression and disease activity in rheumatoid arthritis. J Rheumatol. 2012;39:694-700
46. Suwannalai P, van de Stadt LA, Radner H, Steiner G, El-Gabalawy HS, Zijde CM. et al. Avidity maturation of anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheum. 2012;64:1323-8
47. Sokolove J, Bromberg R, Deane KD, Lahey LJ, Derber LA, Chandra PE. et al. Autoantibody epitope spreading in the pre-clinical phase predicts progression to rheumatoid arthritis. PLoS One. 2012;7:e35296
48. Holland M, Hewins P, Goodall M, Adu D, Jefferis R, Savage CO. Anti-neutrophil cytoplasm antibody IgG subclasses in Wegener's granulomatosis: a possible pathogenic role for the IgG4 subclass. Clin Exp Immunol. 2004;138:183-92
49. Blanco F, Kalsi J, Ravirajan CT, Speight P, Bradwell AR, Isenberg DA. IgG subclasses in systemic lupus erythematosus and other autoimmune rheumatic diseases. Lupus. 1992;1:391-9
50. Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366:539-51
51. Khosroshahi A, Stone JH. IgG4-related systemic disease: the age of discovery. Curr Opin Rheumatol. 2011;23:72-3
52. Wang W, Li J. Identification of natural bispecific antibodies against cyclic citrullinated peptide and immunoglobulin G in rheumatoid arthritis. PLoS One. 2011;6:e16527
53. van der Neut Kolfschoten M, Schuurman J, Losen M, Bleeker WK, Martinez-Martinez P, Vermeulen E. et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science. 2007;317:1554-7
54. Gottenberg JE, Dayer JM, Lukas C, Ducot B, Chiocchia G, Cantagrel A. et al. Serum IL-6 and IL-21 are associated with markers of B cell activation and structural progression in early rheumatoid arthritis: results from the ESPOIR cohort. Ann Rheum Dis. 2012;71:1243-8
Author contact
![]() Corresponding author: Mercedes Rincon, Ph.D. Professor, Department of Medicine/Immunobiology, Given C331, University of Vermont, 89 Beaumont Ave. Burlington, VT 05405. Phone: 802-656.0937 Fax: 802-656-3854 mrinconedu.
Corresponding author: Mercedes Rincon, Ph.D. Professor, Department of Medicine/Immunobiology, Given C331, University of Vermont, 89 Beaumont Ave. Burlington, VT 05405. Phone: 802-656.0937 Fax: 802-656-3854 mrinconedu.