Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Methods and Materials
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(5):444-454. doi:10.7150/ijbs.6507 This issue Cite
Research Paper
Nanos3 Gene Targeting in Medaka ES Cells
Guijun Guan1,2, Yan Yan2, Tiansheng Chen2, Meisheng Yi3, Hong Ni2, Kiyoshi Naruse1, Yoshitaka Nagahama4, ![]() , Yunhan Hong2,
, Yunhan Hong2, ![]()
1. Department of Bioresource, National Institute for Basic Biology, Okazaki, Aichi 444-8585, Japan;
2. Department of Biological Sciences, National University of Singapore, 14 Science Drive 4, Singapore 117543;
3. School of Marine Sciences, Sun Yat-Sen University, Guangzhou, 501275, China;
4. Institution for Collaborative Relations, Matsuyama 790-8577, Ehime University, Japan.
Received 2013-4-18; Accepted 2013-5-8; Published 2013-5-9
Abstract
Gene targeting (GT) by homologous recombination offers the best precision for genome editing in mice. nanos3 is a highly conserved gene and encodes a zinc-finger RNA binding protein essential for germ stem cell maintenance in Drosophila, zebrafish and mouse. Here we report nanos3 GT in embryonic stem (ES) cells of the fish medaka as a lower vertebrate model organism. A vector was designed for GT via homologous recombination on the basis of positive-negative selection (PNS). The ES cell line MES1 after gene transfer and PNS produced 56 colonies that were expanded into ES cell sublines. Nine sublines were GT-positive by PCR genotyping, 4 of which were homologous recombinants as revealed by Southern blot. We show that one of the 4, A15, contains a precisely targeted nanos3 allele without any random events, demonstrating the GT feasibility in medaka ES cells. Importantly, A15 retained all features of undifferentiated ES cells, including stable self-renewal, an undifferentiated phenotype, pluripotency gene expression and differentiation during chimeric embryogenesis. These results provide first evidence that the GT procedure and genuine GT on a chromosomal locus such as nanos3 do not compromise pluripotency in ES cells of a lower vertebrate.
Keywords: ES, gene targeting, homologous recombination, nanos3, pluripotency.
Introduction
Gene targeting (GT) is a powerful tool for precise genome editing via homologous recombination (HR) to study genes' functions (1). In this approach, a precise alteration is introduced into the genome of ES cells at a particular locus, and targeted ES cells are introduced into early developing embryos for germline chimera formation. Crossing of germline chimeras leads to animals that are heterozygous or homozygous for the targeted locus. Although GT is possible also in human ES cells (2), the full technology, ranging from GT in ES cells to production of whole animals from targeted ES cells, has so far been limited to rodents (1).
Efforts have been made towards GT in lower vertebrates, in particular in small laboratory fish models zebrafish and medaka, in which ES or ES-like cell lines are available. In zebrafish, a successful GT has been reported in ES-like cells (3, 4). In medaka, there are stable lines of diploid ES cells (5), haploid ES cells (6, 7) and adult germ stem cells (8), and procedures for chimera formation (9) and GT including gene transfer (8, 10-12) and positive-negative selection (PNS) (13). However, reproducible and efficient bona fide GT in cell culture has so far been a major challenge in fish (14), raising a question as to whether lower vertebrates are comparable to mouse for precise genome editing by HR-mediated GT.
Site-specific genome editing can also be achieved by using engineered sequence-specific endonucleases such as zinc finger nucleases (15, 16) and transcription activator-like effector nucleases (17). Both approaches usually introduce minor additions or deletions in an unpredictable manner. Because of precision for gene disruption and correction via gene replacement, HR-based GT continues to be the approach of choice for experimental settings where accuracy is required. Therefore, the HR-based GT in ES cells followed by germline transmission still represents the approach of choice to engineer a genome with the best precision (1).
The Drosophila nanos gene is required for abdominal patterning and primordial germ cells formation and migration (18), and germ stem cell maintenance (19). nanos is present in diverse animal species and its role in germ cell development appears to be highly conserved (20-24). In medaka, four nanos genes have been identified, which are nanos1a and 1b, nanos2 and nanos3 (25).
Here we report a success in HR-mediated GT in medaka ES cells by using the nanos3 as a gene model. We show that medaka ES cells retain the genetic stability and developmental pluripotency after genuine GT and long-term drug selection, providing convincing evidence for the feasibility of HR-mediated precise genome editing in a lower vertebrate.
Methods and Materials
Fish and embryos
Work with fish was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Advisory Committee for Laboratory Animal Research in Singapore and approved by this committee (Permit Number: 27/09). Medaka strains af and orange were kept at 26~28°C with a 14-h light/10-h darkness daily cycle, embryos were maintained at 28°C and staged as described (9).
Plasmids
Plasmid pGTnanos3 was constructed from pSTneo and pNEB-STk as outline in Supplementary Material: Fig. S1, which expresses the neo gene for resistance to neomycin or G418, and the human herpes simplex thymidine kinase (tk) gene for sensitivity to gancyclovir (Gc) (10). Briefly, a 2.3-kb 5' arm and a 5.2-kb 3' arm of nanos3 sequence were PCR-amplified from MES1-derived genomic DNA by using primers primFW3 plus primRV11 and primFW6 plus primRV7, respectively (Supplementary Material: Table S1), and sub-cloned into pGEM-T Easy (Promega). The CMVgfp from pEGFP-N1 (Clontech) was fused in frame to neo by insertion between XbaI and XhoI in pSTneo, leading to pCMVgfp:neo, to which the 5' HR arms was inserted ahead of cassette CMVgfp:neo, resulting in pNos3gfp:neo. The 3' HR arm was inserted upstream of the STk in pNEB-STk to produce p3nosSTk. The 4.3-kb fragment covering the 5' HR arm plus cassette CMVgfp:neo was released from pNos3gfp:neo and combined with the 3' HR arm at the NotI-NheI sites in p3nosSTk, ultimately resulting in the GT vector pGTnanos3, in which gfp:neo serves a positive selectable marker to confer resistance to G418. To facilitate further cloning, multiple cloning sites within pEGFP-N1 were removed by digestion with NheI and SmaBI. Plasmid DNA was prepared by using the Maxi and Midi prep kits (Qiagen, Germany). Vectors pCVpf and pCVpr used for cell labeling were as described (12).
Sequencing
Genomic PCR products were cloned into pGEM-T (Promega) and sequenced on the ABI3100 automatic sequencer (Applied Biosystems). Sequence analyses were performed by using the Vector NTI package (Invitrogen).
Cell culture and transfection
MES1 is an ES cell line derived from mid-blastula embryos of medaka (5), which was maintained in medium ESM4 on gelatin-coated culture dishes at 28°C under feeder-free culture conditions. Alkaline phosphatase staining was performed as described (5, 26). pGTnanos3 was linearized with SacII and transfected into MES1 cells by using the Gene Juice reagent as described (8, 10, 11). For the PNS procedure, G418 (Gibco; 500 µg/mL) and Gc (Cymevan, Syntex Arzneimittel GmbH, Switzerland; 5 µM) were added to culture medium. Stable transgenic cells were maintained in the presence of two respective drugs at half concentrations.
Cell colony isolation
After 2~3 days of culture in 6-well plates, cells transfected with pGTnanos3 were seeded at 106 cells/10-cm dishes. Cells were grown in the absence or presence of G418 and/or Gc for up to 28 days to allow for colony formation. Dishes with ≤ 200 colonies were washed twice with PBS and submerged in 12 mL of PBS. Under a stereo microscope in a cell culture hood, only distinct colonies consisting of undifferentiated ES cells and lacking any overlapping with others were sucked in 40 µL of PBS by using a p200 Eppendorf tip and transferred to 96-well plates, in which 200 µL of drug-containing ESM4 medium was pre-added per well. Cells were pipetted for several times for dissociation. A monolayer formed during 14~21 days of culture. Colonies were split into three 96-well plates, which were used for PCR genotyping (plate A), Southern analysis (plate B) and master plate (plate C) for propagation into stable clones.
Genomic DNA extraction
DNA samples from 96-well plates were isolated by using the micro-extraction procedure (27) with minor modifications. Briefly, the DNA was extracted by adding cell lysis buffer (10 mM Tris-HCl, pH7.5, 10 mM EDTA, 10 mM NaCl, 0.5% sarcosyl and 500 µg/mL proteinase K), followed by incubation at 55°C for 12-16 h and precipitation by NaCl and cold ethanol (1.5 µL of 5 M NaCl to 100 µL of absolute ethanol). After the 70% ethanol wash, DNA was dissolved and restored in 50 µL of water at 4ºC until use.
Genotyping
PCR screening was conducted on pooled cell populations or single PNS-resistant colonies by using nested PCR primers that flank the GT unique junction regions (Supplementary Material: Table S1). PCR was run in a 25-µL volume containing 50 ng of genomic DNA for 35 cycles of 96°C for 30 s, 60°C for 30 s and 72°C for 3 min. PCR products were separated on 1% agarose gels and validated by sequencing.
Southern blot analyses
Genomic DNA was similarly extracted as described above but to a larger scale and digested with appropriate restriction enzyme(s) at 5 U/µg DNA. Digests (5~10 µg) were separated on a 0.8% agarose gel in 1 X TBE buffer, and transferred to a Hybond N+ nylon membrane (GE health, USA) overnight in 10 × SSC buffer. The membrane was washed once in 2 × SSC, submerged in 5 mL of DIG-Easy solution (Roche) in a hybridization bag containing 100 µg/mL of sonicated salmon sperm DNA fragments (≤ 2 kb in size), and pre-hybridized at 42°C. The membrane was then hybridized to probes (3'-external and gfp:neo vector specific; for detail see Supplementary Material: Table S1) labeled by using the DIG-DNA-labeling Kit (Roche) at 42°C overnight. After washing twice under low stringency with 2 × SSC-0.1% SDS at 65°C for 10 min each, and twice under high stringency with 0.1 × SSC-0.1% SDS at 65°C for 10 min each, the membranes were incubated with an anti-DIG-AP antibody (Roche; 1:5000 dilution) overnight at 4°C and subjected to chemiluminescent detection by using the CDP-Star detection reagent (GE lifesciences). Signals were captured by the LAS-3000 Imaging system (Fujifilm).
RT-PCR analysis
Total RNA was isolated by using the Trizol Reagent (Invitrogen). Synthesis of cDNA templates was primed with oligo (dT)18 by using M-MLV transcriptase (Invitrogen). The cDNA reaction was diluted with water to 10 ng/µL. RT-PCR was run as previously described (6, 8). PCR primers are listed in Supplementary Material: Table S1.
Cytogenetic analysis
Chromosomes from cell cultures were prepared as described (6). Briefly, cells at ~80% confluence were incubated with colchicines (1 μg/ml) for 2~4 hours, harvested from the plate with 0.5% trypsin, subjected to hypotonic treatment with 40 mM KCl, and fixed in freshly prepared Carnoy's fixative (methanol : acetic acid at 3:1 vol/vol) with three changes. Cell suspension after the final fixation was dropped onto cold wet slides, which were air-dried at room temperature and stained with 5% Giemsa solution.
Induced cell differentiation
Induced cell differentiation MES1 cells (RFP labeled) and nanos3-targeted cell line A15 (GFP labeled) were subjected to EB formation in suspension culture in the presence of all-trans retinoic acid (RA; 5 µM) for induced differentiation (5, 7). Cell differentiation was monitored by phenotype and studied by RT-PCR analyses of expression of pluripotency and lineage-specific genes (6, 28).
Chimera formation
A15 and MES1 cells were transplanted into dechorionated blastula embryos of strain af and the resultant chimeric embryos were monitored regularly as described (9, 11, 29).
Microscopy
Observation and photography on Leica MZFIII stereo microscope, Zeiss Axiovertinvert and Axiovert upright microscopes were as described (6, 9, 30).
Results
Vector design and construction
The pGTnanos3 vector was designed to target the medaka nanos3 via HR on the basis of PNS (Fig. 1), which was constructed in multiple steps (Supplementary Material: Fig. S1A). This vector is 14.5 kb and has two homology arms (a 2.3-kb 5'-arms and a 5.2-kb 3'-arm) that flank cassette gfp:neo expressing a fusion between gfp and neo, while STk was positioned at the end of 3'-arm (Supplementary Material: Fig. S1B). Upon an HR event, the gfp:neo would be co-integrated, while the STk would get lost, resulting in homologous recombinants that are resistant to G418 and gancyclovir (Gc) owing to the expression of neo and the absence of tk. Upon random integration (RI) events, both gfp:neo and STk would be co-integrated, leading to the formation of colonies that are resistant to G418 but sensitive to Gc due to the expression of both neo and tk. PNS by G418 and Gc double selection enriches for the HR event via eliminating cells arisen from any RI events.
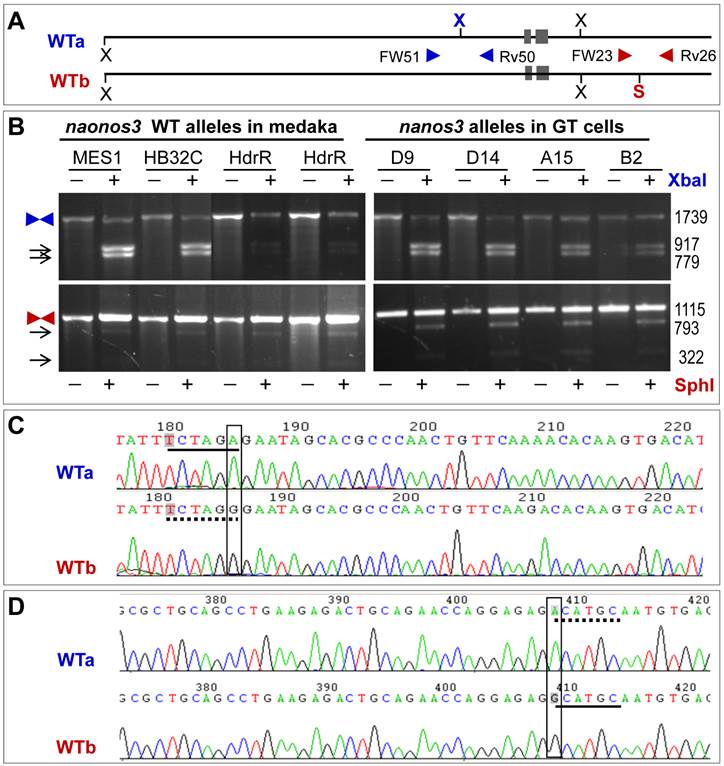
Schematic nanos3 gene targeting in medaka ES cells. arrowhead, positions and extension directions of PCR primers for genotyping; cross, HR region: gfp:neo, cassette that expresses the fusion between GFP and Neo; STk, the cassette expresses HSP-tk; S, SphI.

Medaka genome sequencing has been conducted (31) and nanos3 sequence from strain HdrR is available (http://www.ensembl.org/index.html). MES1 is from strain HB32C. Isogenic DNA fragments were PCR-amplified from MES1 for vector construction. PCR-based genotyping was designed to rapidly screen for the putative GT event using a combination of an external primer (GN1) in the immediate upstream region and an internal primer (gfpR) within the gfp:neo, which allows for the amplification of a 3.2kb fragment specific to the targeted locus (Fig. 1 and Supplementary Material: Fig. S2).
Analysis of the nanos3 locus in MES1
After HR with introduced pGTnanos3, one of the two wildtype (WT) alleles will become a targeted allele, and the use of an appropriate probe on Southern blots of genomic digests will detect the disappearance of a WT allele and the simultaneous appearance of a new GT allele. We observed an allelic polymorphism in the nanos3 locus in MES1, which allows an unambiguous detection of both WT and GT alleles on Southern blots of genomic digests upon hybridization with different probes.
Allelic polymorphism results from the presence or absence of two restriction sites (Fig. 2A). One is the XbaI site in the region upstream of the nanos3 coding sequence, which makes the nanos3 locus into two WT alleles referred to as WTa and WTb. This site is present in WTa but absent in WTb (Fig. 2A). The other is SphI site in the region downstream of the nanos3 coding sequence, which is absent in WTa but present in WTb (Fig. 2A). In short, WTa has the XbaI site but lacks the SphI site, whereas WTb lacks the XbaI site but possesses the SphI site. A PCR analysis revealed that this polymorphism existed in MES1 and strain HB32C from which MES1 was derived, as well as strain HdrR (Fig. 2B). Sequencing of the cloned PCR products validated polymorphisms at the sites concerned: WTa has TCTAGA (XbaI site) and ACATGC, whereas WTb has TCTAGG and GCATGC (SphI site) (Fig. 2C and D). Detailed sequence analyses confirmed this observation and revealed more polymorphic positions (Supplementary Material: Fig. S3 and S4).
Allelic polymorphism of the nanos3 locus in medaka. (A) Schematic structure of WTa and WTb alleles. N, NheI; S, SphI; X, XbaI. Polymorphic sites are highlighted in bold color. Arrowheads depict primers for PCR analyses. (B) PCR analyses. PCR was run using primers indicated. PCR products were not digested (-) or digested (+) with XbaI or SphI and separated on agarose gels. Medaka strains (HB32C and HdrR), MES1 and GT sublines are indicated. Arrows depict digested products. Sizes in bp are given to the right. (C) Sequences of WTa and WTb spanning the polymorphic XbaI site, which is located in the region upstream of the 5' homology arm. (D) Sequences of WTa and WTb spanning the polymorphic SphI site, which is located in the region downstream of the 3' homology arm. broken underline, absence of the site; solid underline, presence of the site.
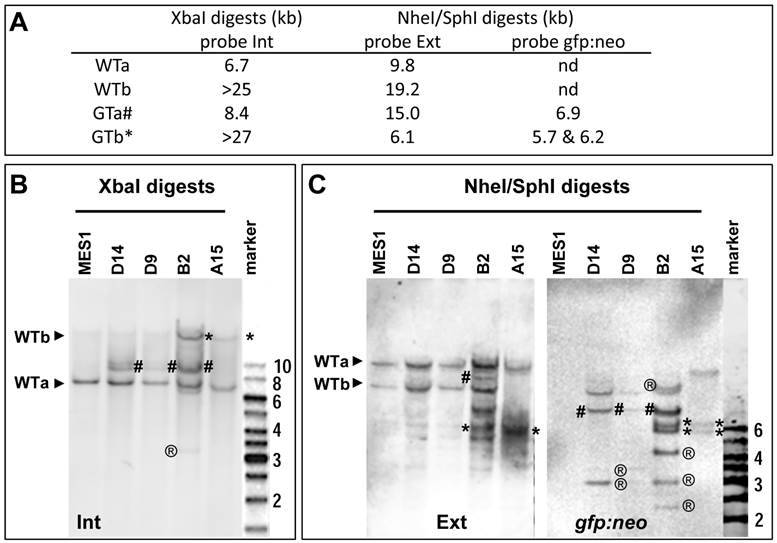
Upon HR, the two WT alleles will converted to two GT alleles, namely GTa and GTb, respectively. Three probes were prepared for the analysis of WT and GT alleles, which are the 5'-internal probe Int, transgene probe gfp:neo and 3'-external probe Ext (Fig. 3A and Supplementary Material: Fig. S5). These probes are expected to produce complicated band patterns on Southern blots of XbaI digests or NheI-SphI double digests, which are unique to either WTa or WTb and their GT counterparts (Fig. 3B and C).
Analyses of nanos3 gene targeting. (A) Predicted number and size of WT and GT alleles of genomic digests as detected by three probes. For detailed information on the positions of probes and sites for XbaI and SphI see Figure 2 and Figure 3. (B) Southern blot of XbaI digests probed with Int. (C) Southern blot of NheI/SphI digests probed with Ext (left panel) followed by reprobing with gfp:neo (right panel). WT alleles (WTa and WTb) are depicted by arrowheads. GT alleles are highlighted by hash (GTa) or asterisks (GTb). ®, putative RI bands. Parental MES1 is negative for probe gfp:neo.
Efficiency of nanos3 gene targeting
MES1 cells after transfection with pGTnanos3 were seeded at a low density for clonal growth in the presence of both G418 and Gc for PNS. Colonies formed after 2~3 weeks of clonal growth. Like the parental MES1 cells (5), the colonies from pGTnanos3-transfected MES1 cells fell into 3 major types according to their morphology. Type-1 colonies (~65%) appeared to represent ES cell colonies due to their morphology and in particular, their remarkable potential of differentiation and self-renewal to expand to ES cell populations. They had a smooth appearance and were very tightly compacted, with their cells being uniform in morphology. Half of type-1 colonies consisted exclusively of undifferentiated ES-like cells, all exhibiting a uniform and strong AP staining. The remainder had ES-like cells as well as varying numbers of differentiated cells with variable morphologies. Type-II colonies (30%) contained variable numbers of AP-positive and negative cells. They did not show the typical morphology of ES cells but more of differentiated cells. Type-III colonies (5%) showed varying appearance and little AP staining at all. Hence, pGTnanos3-transfection and the PNS procedure do not compromise the ability of subclonal culture and the relative efficiency of type-I pure ES cell colony formation.
Only distinct type-I pure ES colonies without overlapping with others were individually picked up in 96-well plates and expanded for further analyses. From five independent experiments, we analyzed 56 PNS-resistant colonies (Supplementary Material: Table S2). Of these, 9 produced the GT-specific PCR product (Supplementary Material: Fig. S2A). Sequencing validated the predicted junction sequence of the targeted nanos3 locus (Supplementary Material: Fig. S2B). Taken together, genuine GT has indeed taken place in MES1 cells at a PCR-detectable efficiency.
Accuracy of nanos3 gene targeting
Four putative GT colonies were successfully expanded into stable sublines, which were analyzed in more detail by Southern blot hybridization. As illustrated in Fig. 3A and Supplementary Material: Fig. S5, WTa and WTb are detected as 6.7-kb and ~25-kb bands by probe Int on the Southern blot of XbaI digests, whereas their targeted alleles, namely GTa and GTb, should form 8.4-kb and ~27-kb bands, respectively. On a Southern blot of SphI-NheI double-digests hybridized with external probe Ext, WTa and WTb should be detected as 19.2-kb and 9.8-kb bands, respectively. During HR, WTa and WTb will become two targeted alleles GTa and GTb, which acquire a co-integrated a SphI site within cassette gfp:neo. Consequently, GTa will appear as a 15-kb band, whereas GTb will appear as a 6.1-kb band on a Southern blot of SphI-NheI double-digests. When the same blot is analyzed by probe gfp:neo, no signals were obtained in parental MES1 cells for the absence of gfp:neo, whereas GTa and GTb should be detectable due to the presence of gfp:neo.
Southern blot analyses of the 4 sublines indeed generated a complicated band pattern that conforms to the prediction. Specifically, on the Southern blot of XbaI digests, probe Int detected not only 6.7-kb (WTa) and ~25-kb (WTb) bands but also an 8.4-kb (GTa) band in B2, D9 and D14, and a similar ~27-kb (GTb) band in B2 and A15, with the ~25-kb WTb band being indistinguishable in position from the ~27-kb GTb band (Fig. 3B). Moreover, probe Ext detected the 19.2-kb and 9.8-kb bands for WTa and WTb alleles in parental MES1 cells (Fig. 3C), as well as B2, D9, D14, indicating the presence of intact WTa and WTb in those sublines. Of the four sublines from pGTnanos3-transfectants, B2 had allele GTa, as the 15-kb band was detected in B2 (Fig. 3C). The 6.1-kb band was seen in B2 and A15 (Fig. 3C), suggesting that WTb was targeted to GTb. Only in A15, WTb was completely absent, indicating a successful GT on WTb by a replacement of GTb in all cells of this clone (Fig. 3C). This was further verified by reprobing the same membrane with gfp:neo, the transgenic probe capable of detecting transgenic genomes arisen from either RI or HR. Fragments of 5.7 kb and 6.2 kb were detected in B2 and A15, coincident with the result of GTb in B2 and A15 detected by Ext probe. The 6.9-kb fragment for GTa was detected in D14, D9 and B2 with probe gfp:neo (Fig. 3C). The intensity of this fragment in D9 and D14 was insufficient for clear detection with probe Ext. In addition, multiple bands with varying sizes were observed (e.g. band of 3-kb present in all three sublines, bands of 4-kb and 2.5-kb in B2, or 3.5-kb band in D9 and D14), indicating they were a mixture of cells with and without GT, and/or RI. The ectopic GT is a byproduct event which has been reported in mice (32). In contrast, only the expected 5.7-kb and 6.2-kb bands were observed in A15 (Fig. 3C). The band patterns retained unchanged in A15 and B2 when analyzed after seven additional passages (Supplementary Material: Fig. S6). Taken together, A15 consists of a pure population of ES cells that contains a precisely targeted nanos3 allele without any random events, whereas the other sublines appear to be mixtures of cells containing GTa or GTb alleles. Therefore, GT can occur at either of nanos3 WT alleles. These results offer first evidence for bona fide GT in medaka ES cells.
Genetic stability
To evaluate the genetic stability in nanos3-targeted ES cells, we performed a cytogenetic analysis in MES1 and A15 (Supplementary Material: Figure S7). Specifically, 62% of MES1 cells had the normal diploid number of chromosomes (2n = 48), with the remainder frequently having fewer chromosomes (Supplementary Material: Figure S7A), due in part to chromosome loss during metaphase preparation (5). Similarly, 49% of A15 cells had 48 chromosomes (Supplementary Material: Figure S7B) and the remainder had fewer chromosomes (Supplementary Material: Figure S7C and D). There was no significant difference in percentage of cells having a normal diploid chromosome number between MES1 and A15, suggesting that the GT procedure and disruption of one nanos3 allele have little adverse effect on genetic stability of ES cells in culture.
Retention of pluripotency in vitro
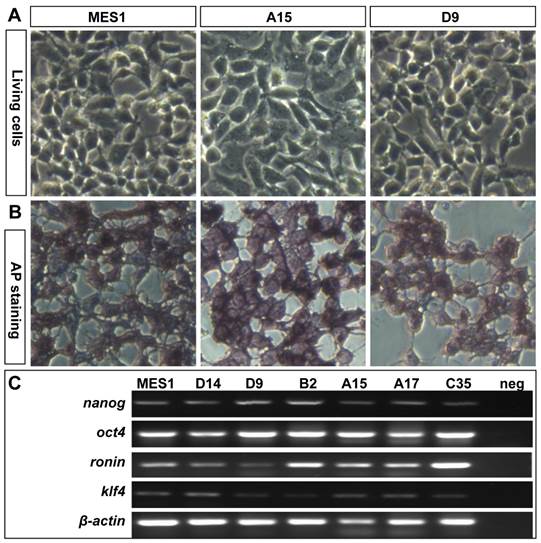
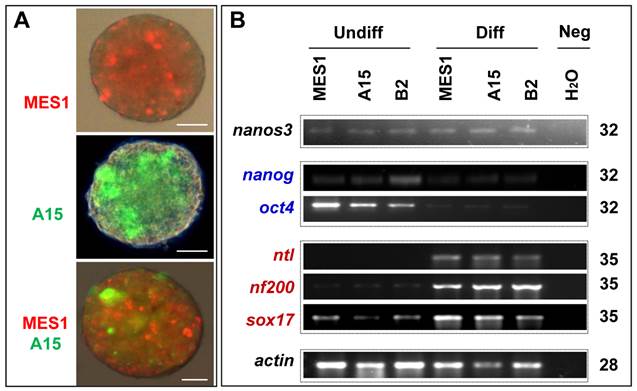
All the four clones resembled MES1 in stable growth, an ES-cell phenotype (such as a small size and round shape), and high alkaline phosphatase activity (Fig. 4A and B) an ES phenotype that is not different from that of MES1 (Figure 4C). ES cells in suspension culture are capable of forming a spherical structure called embryoid body (EB), in which cell differentiation is induced or promoted (5, 6). This ability was examined with clone A15. A15 was labeled with GFP via transfection with pCVpf and mixed with RFP-labeled MES1 for EB formation in suspension culture. A15 indeed retained this ability (Fig. 5A). Several molecular markers have been reported to be associated with an undifferentiated state of medaka ES cells in culture (6, 28). These markers were used to analyze the cellular phenotype at the molecular level. When maintained for undifferentiated growth, nanos3-targeted cells resembled the parental MES1 line in gene expression profile (Fig. 5B). Specifically, pluripotency genes nanog and oct4 were detectable at a similar level in MES1, A15 and B2, whereas differentiation markers were essentially absent (ntl) or barely detectable (nf200 and sox17). More importantly, upon EB formation in the presence of retinoic acid (RA; 5 µM) as a standard differentiation inducer, the loss of pluripotency genes' expression accompanied upregulation or activation of differentiation markers' expression in both MES1 cells and nanos3-targeted cells (Fig. 5B). Nevertheless, a low level of nanos3 RNA expression was detected before and after EB-induced differentiation in MES1 and nanos3-targeted clones (Fig. 5B). Thus, the GT procedure and disruption of one nanos3 allele do not compromise pluripotency at the molecular level, and nanos3 expression appears to be independent of pluripotency in vitro.
Retention of pluripotency in vitro. (A) Phenotype of growing ES cells of parental MES1 and nanos3-targeted clones. (B) AP staining of parental MES1 and nanos3-targeted clones. (C) Expression of pluripotency genes in parental MES1 and nanos3-targeted clones (numerals on lanes). neg, negative control without cDNA template.
Differentiation in vitro. (A) EB formation. MES1, parental ES cell line; A15, nanos3-targeted clone derived from MES1. MES1 and A15 were genetically labeled by RFP and GFP, respectively, and mixed at a 1:1 ratio and seeded onto cell culture Petri dishes. Shown is an EB at day 18 post suspension culture. Scale bars, 200 µm. (B) Gene expression profile of MES1 line and its nanos3-targeted clones (A15 and B2). ES cells were maintained in adherent culture for undifferentiated (Undiff) growth or for induced differentiation (Diff) by EB formation in suspension culture for 10 days. Numbers of PCR cycles are indicated to the right. Genes chosen are markers for pluripotency (oct4 and nanog) and differentiated lineages (nf200, ectoderm; ntl, mesoderm; sox17, endoderm). β-actin served as a loading control. Neg, negative control by using H2O as a template. Notably, the nanos3 RNA was barely detectable in all samples.
Retention of pluripotency in vivo
In order to elucidate whether targeted medaka ES cells retained pluripotency in vivo, we performed chimeric formation, a standard assay of pluripotency in developing embryos (29, 30). To this end, GFP-labeled A15 cells and RFP-labeled MES1 cells were co-transplanted into blastula hosts, and chimeric embryos were monitored. A15 was found to resemble MES1 in wide distribution, because GFP signals overlapped the RFP signal in various embryonic compartments (Fig. 6A-C) and organ systems, including the brain, eyes, spinal cord, heart and surface of the head and trunk (Fig. 6D-I). Convincingly, A15 is not different from MES1 in vivo, demonstrating the pluripotency retention after long-term drug selection and genuine GT at one nanos3 allele.
Retention of pluripotency in vivo. Clone A15 (green) and MES1 cells (red) were co-transplanted into blastula embryo hosts and photographed at stages indicated. (A-C) stage 21, showing wide distribution of A15 and MES1 cells throughout the entire embryo and yolk sac. (D-I) Stage 28, showing distribution of A15 and MES1 cells in various organ systems. br, brain; ey, eye; hd, head; ht, heart; od, oil droplet; sc, spinal cord; tk, trunk. Scale bars, 100 µm.

Discussion
In this study, we have explored HR-mediated GT in medaka ES cells using the nanos3 gene as a model. We reveal that GT can precisely occur at ~2% efficiency in MES1. This efficiency is within the range 0.1~42.6% reported in mouse ES cells, where the HR efficiency varies considerably depending on loci (33). Whether there is a relationship between the GT efficiency and genetic loci also in fish remains unknown. Our work provides a starting point to address this important issue in a lower vertebrate. The precision of GT at the medaka nanos3 locus also points to the high fidelity for the HR process in the genome of a lower vertebrate.
We have provided three lines of evidence that convincingly demonstrate a success in genuine GT. First, a PCR-based approach reproducibly detects GT-specific junction sequences in transfected cells, multiple colonies and their expanded clones, suggesting that medaka ES cells have a relatively high cellular HR activity, which is in accordance with our previous report (10). Second, we defined allelic polymorphism at the nanos3 locus in MES1 cells, which allows for unambiguous identification of gene replacement by the disappearance of either allele and the formation of a new allele from GT. This is exactly the case with clone A15, where the loss of one of wildtype alleles, WTb, accompanies the acquisition of a targeted allele, GTb. Finally, GT may occur free of any random events, as it has been observed again in clone A15.
Our observation that ablation of one nanos3 allele in medaka ES cells does not compromise the pluripotency conforms to our previous observation on pluripotency retention in MES1 after long-term gene transfer and drug selection (34), as targeted disruption of one nanos3 allele allows for ES cells to stably self-renew in an undifferentiated state and to differentiate under proper conditions in vitro and in vivo. It has been well documented that gene expression dosage plays a critical role in early cell fate decision in vivo and in vitro. For example, aberrant Oct4 expression leads to mouse ES cell differentiation in vitro (35), p73 shows haploinsufficiency in mouse models of aging and Alzheimer's disease (36), and oskar dosage determines the number of Drosophila germ cells in vivo (37). nanos3 is expressed in germ stem cells of mouse (21) and medaka (25), pointing to an essential role in germ cells and/or stem cells. Two observations made in this study point to the haplosufficiency of nanos3 for pluripotency in vitro: One is that nanos3 is expressed in medaka ES cells, and the other is that loss of a single nanos3 allele does not affect the ES cell phenotype. Alternatively, nanos3 may be dispensable for pluripotency but indispensable for the germline. In consistency with this possibility is our observation that nanos3 RNA expression exhibits a little difference before and after ES differentiation. Further in support of this notion is the report that embryos without nanos3 can develop into adult mice (21) and zebrafish (22). Future work is needed to pin point the role of nanos3 in vivo.
Together with successful GT in zebrafish ES-like cells (3, 4) and on the p53 locus of medaka ES cells (38), our data clearly underscores the feasibility for precise genome editing by PNS-assisted GT procedures in lower vertebrates such as medaka.
Site-specific gene alterations can also be achieved by using engineered sequence-specific endonucleases such as zinc finger nucleases (15) and transcription activator-like effector nucleases (17). Both approaches usually introduce minor additions or deletions in an unpredictable manner. In addition, these approaches may also produce off-target alterations that are difficult to predict and detect. Because of precision and freedom for gene disruption and correction via gene replacement, HR-based GT continues to be the approach of choice for experimental settings where precision is required. Although random integration and ectopic GT may occur frequently, these unwanted events can easily be detected by Southern analysis, as we have shown in this study. Therefore, the HR-based GT in ES cells followed by germline transmission still represents the approach of choice to engineer a genome with the best precision (1).
One of the ultimate goals in GT experiments is to produce whole animal, which is routine in rodents (1, 39, 40). Germline transmission of long-term cell cultures has not yet been available in lower vertebrate organisms. A single study has reported germline transmission of a short-term zebrafish embryo culture, which was maintained for only weeks at a temperature lower than 20°C before chimera formation (41). Germ cells are pre-formed by maternal factors in zebrafish (42) and perhaps also in medaka (43), which raises a question as to whether fish ES cells after long-term culture, would be capable of germline chimera formation. In deserves to note that first haploid ES cells have been obtained in medaka, which are capable of germline transmission by semi-cloning (6). It will be interesting to determine whether GT in these haploid ES cells in combination with semi-cloning will allow for the production of knockout animals. The procedures and the success in bona fide GT reported in the present study will offer information and tools for GT in haploid medaka ES cells.
Abbreviations
ES, embryonic stem; GT, gene targeting; HR, homologous recombination; PNS, positive-negative selection; RI, random integration.
Acknowledgements
We thank Jiaorong Deng for breeding fish, Veron Wong and Choy Mei Foong for laboratory management. This work was supported by the National Research Foundation Singapore (NRF-CRP7-2010-03) and also supported in part by a Research Fellowship (no. 0773) of the Japan Society for Promotion of Science for Young Scientists.
Authors' contributions
G.G., Y.Y., T.C. and M.Y. performed the research; G.G. analyzed the data; Y.H. and Y.N. designed the research; G.G. and Y.H. wrote the paper.
Supplementary Material
Fig.S1-S7 and Table S1-S2.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Capecchi MR. Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat Rev Genet. 2005;6:507-12
2. Zwaka TP, Thomson JA. Homologous recombination in human embryonic stem cells. Nat Biotechnol. 2003;21:319-21
3. Fan L, Alestrom A, Alestrom P, Collodi P. Development of cell cultures with competency for contributing to the zebrafish germ line. Crit Rev Eukaryot Gene Expr. 2004;14:43-51
4. Fan L, Moon J, Crodian J, Collodi P. Homologous recombination in zebrafish ES cells. Transgenic Res. 2006;15:21-30
5. Hong Y, Winkler C, Schartl M. Pluripotency and differentiation of embryonic stem cell lines from the medakafish (Oryzias latipes). Mech Dev. 1996;60:33-44
6. Yi M, Hong N, Hong Y. Generation of medaka fish haploid embryonic stem cells. Science. 2009;326:430-3
7. Yi M, Hong N, Hong Y. Derivation and characterization of haploid embryonic stem cell cultures in medaka fish. Nat Protoc. 2010;5:1418-30
8. Hong Y, Liu T, Zhao H, Xu H, Wang W, Liu R, Chen T, Deng J, Gui J. Establishment of a normal medakafish spermatogonial cell line capable of sperm production in vitro. Proc Natl Acad Sci U S A. 2004;101:8011-6
9. Hong N, Li M, Zeng Z, Yi M, Deng J, Gui J, Winkler C, Schartl M, Hong Y. Accessibility of host cell lineages to medaka stem cells depends on genetic background and irradiation of recipient embryos. Cell Mol Life Sci. 2010;67:1189-202
10. Hong Y, Chen S, Gui J, Schartl M. Retention of the developmental pluripotency in medaka embryonic stem cells after gene transfer and long-term drug selection for gene targeting in fish. Transgenic Res. 2004;13:41-50
11. Yan Y, Du J, Chen T, Yi M, Li M, Wang S, Li CM, Hong Y. Establishment of medakafish as a model for stem cell-based gene therapy: efficient gene delivery and potential chromosomal integration by baculoviral vectors. Exp Cell Res. 2009;315:2322-31
12. Zhao H, Hong N, Lu W, Zeng H, Song J, Hong Y. Fusion gene vectors allowing for simultaneous drug selection, cell labeling, and reporter assay in vitro and in vivo. Anal Chem. 2012;84:987-93
13. Chen S, Hong Y, Schartl M. Development of a positive-negative selection procedure for gene targeting in fish cells. aquaculture. 2002;214:67-79
14. Hong N, Li Z, Hong Y. Fish stem cell cultures. Int J Biol Sci. 2011;7:392-402
15. Collin J, Lako M. Concise Review: Putting a Finger on Stem Cell Biology: Zinc Finger Nuclease-Driven Targeted Genetic Editing in Human Pluripotent Stem Cells. Stem Cells. 2011;29:1021-33
16. Chen J, Zhang X, Wang T, Li Z, Guan G, Hong Y. Efficient detection, quantification and enrichment of subtle allelic alterations. DNA Res. 2012;19:423-33
17. Huang P, Xiao A, Zhou M, Zhu Z, Lin S, Zhang B. Heritable gene targeting in zebrafish using customized TALENs. Nat Biotechnol. 2011;29:699-700
18. Kobayashi S, Yamada M, Asaoka M, Kitamura T. Essential role of the posterior morphogen nanos for germline development in Drosophila. Nature. 1996;380:708-11
19. Lehmann R, Nusslein-Volhard C. The maternal gene nanos has a central role in posterior pattern formation of the Drosophila embryo. Development. 1991;112:679-91
20. Lolicato F, Marino R, Paronetto MP, Pellegrini M, Dolci S, Geremia R, Grimaldi P. Potential role of Nanos3 in maintaining the undifferentiated spermatogonia population. Dev Biol. 2008;313:725-38
21. Tsuda M, Sasaoka Y, Kiso M, Abe K, Haraguchi S, Kobayashi S, Saga Y. Conserved role of nanos proteins in germ cell development. Science. 2003;301:1239-41
22. Draper BW, McCallum CM, Moens CB. nanos1 is required to maintain oocyte production in adult zebrafish. Dev Biol. 2007;305:589-98
23. Julaton VT, Reijo Pera RA. NANOS3 function in human germ cell development. Hum Mol Genet. 2011;20:2238-50
24. Koprunner M, Thisse C, Thisse B, Raz E. A zebrafish nanos-related gene is essential for the development of primordial germ cells. Genes Dev. 2001;15:2877-85
25. Aoki Y, Nakamura S, Ishikawa Y, Tanaka M. Expression and syntenic analyses of four nanos genes in medaka. Zoolog Sci. 2009;26:112-8
26. Hong Y, Schartl M. Isolation and differentiation of medaka embryonic stem cells. Methods Mol Biol. 2006;329:3-16
27. Ramirez-Solis R, Rivera-Perez J, Wallace JD, Wims M, Zheng H, Bradley A. Genomic DNA microextraction: a method to screen numerous samples. Anal Biochem. 1992;201:331-5
28. Wang D, Manali D, Wang T, Bhat N, Hong N, Li Z, Wang L, Yan Y, Liu R, Hong Y. Identification of pluripotency genes in the fish medaka. Int J Biol Sci. 2011;7:440-51
29. Hong Y, Winkler C, Schartl M. Production of medakafish chimeras from a stable embryonic stem cell line. Proc Natl Acad Sci U S A. 1998;95:3679-84
30. Hong N, Chen S, Ge R, Song J, Yi M, Hong Y. Interordinal Chimera Formation Between Medaka and Zebrafish for Analyzing Stem Cell Differentiation. Stem Cells Dev. 2012
31. Naruse K, Hori H, Shimizu N, Kohara Y, Takeda H. Medaka genomics: a bridge between mutant phenotype and gene function. Mechanisms of development. 2004;121:619-28
32. Dellaire G, Lemieux N, Belmaaza A, Chartrand P. Ectopic gene targeting exhibits a bimodal distribution of integration in murine cells, indicating that both intra- and interchromosomal sites are accessible to the targeting vector. Mol Cell Biol. 1997;17:5571-80
33. Laible G, Alonso-Gonzalez L. Gene targeting from laboratory to livestock: current status and emerging concepts. Biotechnol J. 2009;4:1278-92
34. Hong Y, Winkler C, Liu T, Chai G, Schartl M. Activation of the mouse Oct4 promoter in medaka embryonic stem cells and its use for ablation of spontaneous differentiation. Mech Dev. 2004;121:933-43
35. Niwa H, Miyazaki J, Smith AG. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet. 2000;24:372-6
36. Cancino GI, Miller FD, Kaplan DR. p73 haploinsufficiency causes tau hyperphosphorylation and tau kinase dysregulation in mouse models of aging and Alzheimer's disease. Neurobiol Aging. 2012
37. Ephrussi A, Lehmann R. Induction of germ cell formation by oskar. Nature. 1992;358:387-92
38. Yan Y, Hong N, Chen T, Li M, Wang T, Guan G, Qiao Y, Chen S, Schartl M, Li CM, Hong Y. p53 Gene Targeting by Homologous Recombination in Fish ES Cells. Plos One. 2013;8:e59400
39. Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215-21
40. Tong C, Li P, Wu NL, Yan Y, Ying QL. Production of p53 gene knockout rats by homologous recombination in embryonic stem cells. Nature. 2010;467:211-3
41. Lin Z, Yu H, Gao S, Cheng J, Wang L. Development of the fragment constant method for estimating the partition coefficients of nonionic organic mixtures. Arch Environ Contam Toxicol. 2001;41:255-60
42. Raz E. Primordial germ-cell development: the zebrafish perspective. Nature reviews Genetics. 2003;4:690-700
43. Herpin A, Rohr S, Riedel D, Kluever N, Raz E, Schartl M. Specification of primordial germ cells in medaka (Oryzias latipes). BMC Dev Biol. 2007;7:3
Author contact
![]() Corresponding author: Prof. Yunhan Hong, dbshyhedu.sg. Department of Biological Sciences, National University of Singapore. 14 Science Drive 4, Singapore 117543; Fax: +65 6779 2486; Tel: +65 6516 2915 Or Prof. Yoshitaka Nagahama, E-mail: nagahama.yoshitaka.mhac.jp. Institution for Collaborative Relations, Ehime University, 3 Bunkyo-cho, Matsuyama 790-8577. Tel: 81-89-927-8513; Fax: 81-89-927-8820
Corresponding author: Prof. Yunhan Hong, dbshyhedu.sg. Department of Biological Sciences, National University of Singapore. 14 Science Drive 4, Singapore 117543; Fax: +65 6779 2486; Tel: +65 6516 2915 Or Prof. Yoshitaka Nagahama, E-mail: nagahama.yoshitaka.mhac.jp. Institution for Collaborative Relations, Ehime University, 3 Bunkyo-cho, Matsuyama 790-8577. Tel: 81-89-927-8513; Fax: 81-89-927-8820