Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(9):934-946. doi:10.7150/ijbs.7529 This issue Cite
Research Paper
SIRT1 Deacetylates FOXA2 and Is Critical for Pdx1 Transcription and β-Cell Formation
Rui-Hong Wang1, Xiaoling Xu1, Hyun-Seok Kim1, Zhen Xiao2, Chu-Xia Deng1 ![]()
1. Genetics of Development and Disease Branch, 10/9N105, National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland MD 20892, USA;
2. Laboratory of Proteomics and Analytical Technologies, Advanced Technology Program, SAIC-Frederick, Inc., NCI-Frederick, Frederick, Maryland 21702, USA.
Received 2013-8-29; Accepted 2013-9-19; Published 2013-9-21
Abstract
Pancreas duodenum homeobox 1 (PDX1) is essential for pancreas development and β-cell formation; however more studies are needed to clearly illustrate the precise mechanism regarding spatiotemporal regulation of Pdx1 expression during β-cell formation and development. Here, we demonstrate that SIRT1, FOXA2 and a number of proteins form a protein complex on the promoter of the Pdx1 gene. SIRT1 and PDX1 are expressed in the same set of cells during β-cell differentiation and maturation. Pancreas-specific disruption of SIRT1 diminished PDX1 expression and impaired islet development. Consequently, SIRT1 mutant mice develop progressive hyperglycemia, glucose intolerance, and insulin insufficiency, which directly correlate with the extent of SIRT1 deletion. We further show that SIRT1 interacts with and deacetylates FOXA2 on the promoter of the Pdx1gene, and positively regulates its transcription. These results uncover an essential role of SIRT1 in β-cell formation by maintaining expression of PDX1 and its downstream genes, and identify pancreas-specific SIRT1 mutant mice as a relevant model for studying insulin insufficiency.
Keywords: SIRT1, FOXA2, PDX1, insulin.
Introduction
Diabetes is a severe metabolic disease worldwide that can be caused by many factors. In human, inactivation of pancreas duodenum homeobox 1 (PDX1) causes early-onset type II diabetes mellitus [50, 51]. Experiments in mice revealed that PDX1 regulates the transcriptional network of pancreatic endocrine progenitor cells and that PDX1 deficiency blocks pancreas formation, suggesting that PDX1 is essential for pancreas development and β-cell formation [26, 27, 41, 42]. Thus, it is important to understand how PDX1 is regulated. Previous studies indicated that PDX1 is highly expressed throughout the entire pancreatic epithelium in the early stages of embryogenesis; however, its expression becomes more restricted to insulin-producing β-cells at later stages of pancreas development [41, 52].
The PDX1 promoter contains several nuclease hypersensitive sequences (HSS) that are regulated by transcriptional factors [16, 61]. This enhancer region, known as area I (bp -2761 to -2457), area II (bp -2153 to -1953, also referred as PH2 domain), area III (bp -1879 to -1600) [16], and area IV [17], is bound by transcription factors, including HNF1a, Foxa2, Foxa1, NHF6, Pax6, MafA and Ptf1 to direct Pdx1 expression in pancreatic progenitors/or in adult β-cells [2, 14, 25, 31, 37, 46, 47, 53]. Area II (PH2) is only present in the mammalian PDX1 gene promoter [16, 17]. Recently, GATA4 and GATA6 binding sites have been discovered in area III, which are critical for pancreas organogenesis [5, 65].
Forkhead transcription factor FOXA2 (formally HNF3β) binds to area II through a consensus FOXA binding site and is required for PDX1 expression [53]. Pancreas-specific disruption of FOXA2 diminishes PDX1 transcription and impairs pancreas formation in mice [32]. These data establish an important role for FOXA2 in regulating PDX1 expression and pancreas development. Several proteins, including PAX6, FOXA1, and FOXO1, have been reported to affect expression of PDX1 through their interactions with FOXA2 [14, 28, 45]. However, the underlying mechanism regarding their relationship with FOXA2 in regulation of PDX1 expression deserves further investigation.
Mammalian SIRT1, a homolog of yeast Sir2, is a founding member of a family of 7 members (SIRT1-7) of NAD+ dependent type III histone and protein deacetylases [15, 19-21, 23, 30, 36, 39, 48]. SIRT1 is indispensable for mammalian development as majority of SIRT1 mutant mice die during embryonic development or early postnatal stages [7, 38, 58]. Tissue specific knockout using a Cre-loxP mediated approach to overcome the early lethality revealed important roles of SIRT1 in calorie restriction, energy expenditure, lipid metabolism, and mammary lobuloalveolar development, as well as neuronal differentiation and neurodegeneration [6, 9, 12, 35, 43, 55, 56]. A study of β cell-specific SIRT1-overexpressing transgenic mice showed that increased dosage of SIRT1 in β-cells improves glucose tolerance and enhances insulin secretion in response to glucose [40]. Other investigations also demonstrated that chemicals that activate SIRT1 activity play protective roles on islet insulin secretion [22, 67]. Recently, a study identified an inherited heterozygous mutation in human SIRT1 that leads to an autoimmune-mediated β cell destruction, and diabetes [3]. However, a role for SIRT1 in β cell development and differentiation remains elusive due to the early lethality of SIRT1 null mice.
In this study, we performed a proteomic analysis and found that SIRT1, FOXA2 and a number of other proteins form a protein complex on the promoter of PDX1 in cultured cells. We further demonstrated that SIRT1 deacetylates FOXA2 and plays a critical role in regulating PDX1 expression. Finally, using the Cre-loxp conditional knockout approach, we showed that the absence of SIRT1 in the pancreas results in impaired β cells formation followed by progressively developed insulin insufficiency.
Materials and Methods
Generation of pancreatic-specific Sirt1 deletion animals
Mice that carry 3 loxP sites (a ploxPneo in intron 4 and the third loxP in intron 6) were crossed with EIIa-Cre [29] to delete the ploxPneo using a protocol as described [63]. The resulting mice carrying the floxed exon 5 and exon 6 of the Sirt1 gene were crossed with Pdx-cre transgenic mice to obtain the Sirt1flox5-6;Pdx-cre strain. Afterwards, males and females of Sirt1flox5-6;Pdx-cre were intercrossed to generate Sirt1PKO mice and controls. These mice are in a mixed genetic background of 129/FVB/Black Swiss and are genotyped with the following primers:
Floxed Sirt1 gene: Forward- 5' CTT CCT TGC CAC AGT CAC TC 3'; Reverse- 5' CAT CTA AAC TTT GTT GGC TGC 3'. Pdx-Cre: Forward- 5' CCT GTT TTG CAC GTT CAC CG 3'; Reverse- 5' ATG CTT CTG TCC GTT TGC CG 3'. All experiments were approved by the Animal Care and Use Committee of the National Institute of Diabetes, Digestive and Kidney Diseases (ACUC, NIDDK).
Transfection
All the transfections were carried out with LipofectamineTM 2000 (Invitrogen). pUse-Sirt1 is from Upsate. pCruz-SIRT1 and pCruz-Sirt1(HY) were used previously [60]. shRNA against mouse SIRT1(pBSU6-Sirt1) was constructed as described previously [58]. GFP-FOXA2 expression vector was previously described [49].
Oligo pull down and mass spectrometry
The biotin-labeled wild-type (WT: [Biotin 5] ACCAGAAGTGCTAAGCAAACATCCTGGGGTG) or mutant (MT: [Biotin-5] ACCAGAAGTGCTAAGCCCCTATCCTGGGGTG) oligo of FOXA2 binding site on Pdx1 promoter was used for biotin pull down assay with the nuclear lysates from 20 million GFP-FOXA2 transfeced 293HEK cells. The biotin-bound streptavidin beads were extensively washed with 500 mM KCl and loaded onto 4-12% Tris-SDS PAGE. The gel was later stained with Silver Staining (SilerQuestTM Staining Kit, Invitrogen). The bands were excised, distained, and digested with trypsin. The tryptic peptides were analyzed by nanoflow reversed-phase liquid chromatography (nanoRPLC) mass spectrometry using a linear ion-trap (LIT) mass spectrometer (LTQ, ThermoElectron, San Jose, CA). The LTQ was operated in a data-dependent mode in which the seven most abundant peptide molecular ions in every MS scan were sequentially selected for fragmentation and acquisition of MS2 spectra. Tandem mass spectra were searched against the human protein database using SEQUEST software (ThermoFinnigan, San Jose, CA). A similar in-gel digestion approach was also used to analyze the FOX2A gel band following in vitro deacetylation by SIRT1. Acetylation of lysine (+42.0106 Da) and oxidation of methionine (+15.9949 Da) residues were included as dynamic modification when the MS2 spectrum was searched against the human FOXA2A protein sequence. The SEQUEST filtering criteria for initial identification of peptides were cross-correlation (Xcorr) scores of 1.9 for [M + H]1+, 2.2 for [M + 2H]2+, 3.1 for [M + 3H]3+ ions; and a minimum delta correlation (ΔCn) of 0.08. The MS2 spectra of eligible peptides were manually inspected to confirm the identification of correct peptide sequence and modifications.
To validate the Mass Spectrometry data, pulldown with either WT or MT biotinylated oligos was performed and Western blot analysis was carried out with antibodies against individual protein. The following antibodies were used: PARP1 (Millipore), KU70 (Santa Cruz), KU80 (Abcam), and GFP (MP).
Acetylation of FOXA2 in vivo
Twenty million 293T cells were co-transfected with myc-p300 and GFP-FOXA2 or one of the flag-FOXA2 (wildtype, K226R, K275R or K226R/K275R). The transfectants were then treated with either TSA (5μM) or NIM (20mM) or both for 8 hours before harvest. The cellular lysates were immunoprecipitated with GFP antibody, Ac-K antibody, or flag antibody respectively. Generally, 2-3 mg nuclear lysates were used. For flag-tagged protein, after being immunoprecipitated with flag antibody (Sigma, M2), the beads were eluted with 3Xflag peptide (Sigma) and eluates were loaded onto 10% Tris-glycine SDS PAGE for Western blot with Ac-K antibody. When testing SIRT1 function on FOXA2 acetylation, myc-p300 and GFP-FOXA2 plasmids were co-transfected into 293 HEK cells, together with either GFP or SIRT1 or SIRT1 (HY).
In Vitro deacetylation assay
Flag-tagged SIRT1 or flag-tagged SIRT1(HY) was transfected into 293T cells. The flag-tagged FOXA2 was co-transfected with myc-p300. The cells were lysed in buffer (20mM Hepes, pH7.9, 180mM KCl, 0.2mM EGTA, 1.5mM MgCl2, 20% glycerol, 0.1% NP40) with 0.5uM TSA and 10mM Nicotinamide (NIM). The flag-tagged proteins were then immunoprecipitated with anti-Flag M2 agarose beads (Sigma) and eluted with 3X flag peptide. Afterwards, the purified proteins were suspended with 20ul deacetylation buffer (20mM Hepes, pH7.9, 67mM KCl, 5mM MgCl2, 0.2mM EDTA) with or without NAD+ (Sigma), incubated at 30 °C for 2hr, and then reactions were stopped by adding 2X loading buffer. After electrophoresis, the membrane was immunoblotted by anti-Ac-K antibody, followed with M2 antibody; then the gels were stained with Coomassie blue and processed for mass spectrometry analysis.
Site directed mutagenesis
To mutate the lysine residues that are acetylated in FOXA2, flag-tagged murine FOXA2 plasmid in a modified pCMV vector was used as a parental template. The PCR method (PCR SuperMix high fidelity, Invitrogen) was applied and mutation was confirmed with sequencing. Mutagenesis was carried out by BioInnovatise Inc. (Rockville, MD 20852). The primers are as follows: Foxa2F1: ggtggtggatccatgctgggagccgtgaagatgg; Foxa2R1: GGTGGTAAGCTT TTA GGA TGA GTT CAT AAT AGG; K226F1: cccgctcgccagacaggcctggcaagggctcc; K226R1: GGA GCC CTT GCC AGG CC T GTC TGG CGA GCG GG; K275F1: TGCGGC CAGTAGCGGA GGCA GGAG GA CCGCTCCTGG GTCCCAG; K275R1: CTG GGA CCC AGG AGC GGT CCT CCT GCC TCC GCT ACT GGC CGC A. “Red color” indicates mutant nucleotides.
Glucose measurement
Mice were maintained under fed condition with regular chow. At different ages, glucose was measured with ContourR glucose meter (Bayer) and ContourR blood glucose test strips (Bayer).
Insulin measurement
Serum was used for insulin measurements with either a Rat Insulin ELISA Kit (Crystal Chem, Inc.) or an RA method performed by the Metabolic Core Facility of NIDDK.
Islet isolation
Mice at different ages were euthanized with CO2. Collagenase Type V from Sigma was perfused into pancreas. The entire pancreas was collected, and digested at 37°C. Islets were purified by Ficoll gradient (Ficoll-paqueTM Plus, GE Healthcare), and hand-picked using a microscope.
qRT-PCR
Total RNA was isolated with STAT-60TM (Tel-Test, Inc.) from islets or MIN6 cell. cDNA was synthesized with Cells-to-cDNA TMII (Ambion, Inc.). Primers used are shown in the Supplementary (Additional file 1).
qChIP
MIN6 cells were cross-linked with 1% formalin for 15 min and ChIP was performed with either SIRT1 or FOXA2 antibody as described previously [59]. Primers used to amplify mouse Pdx1 promoter are: mPdx1 pF1: 5'ttcgggatctggattgagtc3', mPdx1 pR1: 5'cccaagaaagcaaaccatgt3', mPdx1-pF4: 5' ttctttttgcaaagcacagc3', mPdx1-pR4: 5' caccccaggatgtttgctta3', mPdx1-pF6: 5' aggcttacagcgctgagttc3', and mPdx1-pR6: 5'ctccttttcctccccagttc3'
Western blot and immunoprecipitation
Western blot was carried out with Licor (Lincoln, Nebraska) using antibodies against SIRT1 (Upstate), FOXA2 (Santa Cruz), acetyl-lysine (Chemicon and Abcam). Immunoprecipitation was carried out as described previously [59].
Luciferase activity
Pdx-PH2/pGL3B was obtained from Addgene. The plasmid was transfected into MIN6 cells or beta-TC6 cells either alone or together with SIRT1, FOXA2, FOXA2 mutant alone or both. The luciferase activity was assessed with the Dual-LuciferaseR Reporter Assay System (Promega).
Pathological analysis of pancreas
The entire pancreas was collected, flattened, and fixed in 10% formalin. The fixed pancreas was embedded with paraffin. Five μm sections were cut with MICROM (HM 340E). The sections were stained with H&E, or processed for immunohistochemistry or immunofluorescence. The antibodies used are: anti-Sir2a (Millipore), anti-glucogon and anti-insulin (Sigma), anti-proinsulin (R&D system), anti-somastatin (MP), anti-pancreatic polypeptide (ICN), anti-Glut2 (Chemicon), anti-Pdx-1 (a gift from Christopher Wright, Vanderbilt University), anti-F4/80 (Serotec), anti-MPO (Biocare Medical), anti-NF-κB (Santa Cruz), anti-CD3 (Dako), anti-IL6 (Becton Dickenson), anti-TNFα (R&D), anti-IL1β (R&D). The immunohistochemistry was performed with HistomouseTM SP (Invitrogen). The secondary antibodies for immunofluorescence staining were from Invitrogen and Sigma.
Statistical analysis
Student's t-test is used for statistical analysis. ±SD is reflected in corresponding panels as bars.
Results
A protein complex in the promoter of Pdx1 gene revealed by proteomics
To identify potential factors involved in regulating Pdx1 expression through the interaction with FOXA2, the consensus FOXA2 binding site in the region of PH2 domain of Pdx1 promoter was utilized for a biotin pull-down assay. A GFP-FOXA2 or FLAG-FOXA2 construct was expressed in either HEK 293 cell line that was originally derived from human embryonic kidney or Min6 cell line, a pancreatic beta cell line derived from a transgenic mouse expressing the large T-antigen [24]. The nuclear lysates from the transfectants were used for a pulldown analysis with biotinylated wild-type and mutant oligos for the consensus FOXA2 binding site in the PH2 domain (Fig. 1A). Wild-type oligo precipitated several distinct protein bands, while mutant oligo did not pull down any of these proteins (Fig. 1B). Analysis of the differential bands by mass spectrometry identified several proteins, including PARP1, HLTF1, KU70, KU86, SSBP, and SIRT1 (Fig. 1B). Individual oligo pull-downs and Western blots by antibodies against PARP1, KU70, KU86 and SIRT1 confirmed their interaction with the wild-type oligo in both 293 and Min6 cells (Fig. 1C-D). Once the consensus FOXA2 binding site was mutated, SIRT1, PARP1, KU70, KU86 and FOXA2 disappeared together, indicating that the binding of SIRT1, PARP1, KU70, and KU86 to Pdx1 promoter is mediated by FOXA2 recognition sequence.
A protein complex formed in the promoter of Pdx1 revealed by oligo pulldown and proteomics. (A) Wild-type and mutant oligo sequences that were used for biotin pulldown. In wild-type oligo, AAAC is the consensus site for FOXA2 binding. In mutant oligo, AAAC is mutated to CCCT. (B) A silver stained picture from GFP-FOXA2 pulldown sample is shown. (C-D) Biotin pulldown validation was performed in 293T (C) and Min 6 (D) cells using nuclear extracts that over expressing FLAG-FOXA2 to confirm the identity. (E-F) Reciprocal immunoprecipitation to show SIRT1 and FOXA2 interact with each other. Input: 10% of total protein. (E) 293T cells with overexpression of Flag-FOXA2. (F) Min6 cells with endogenous protein lysates. (G) FOXA2 is acetylated by p300. 293T cells were transfected with FLAG-FOXA2 and p300, then the lysates were immunoprecipated with anti-FLAG antibody, western blotted with either FOXA2 antibody or Ac-K antibody. (H) Treatment of NIM or SIRT1 mutant (SIRT1(HY)) can enhance the acetylation level on FOXA2. (I) Wild-type SIRT1deacetylates FOXA2 in vitro. Purified FLAG-FOXA2 and SIRT1 or SIRT1(HY) were added together into a test tube with or without NAD+. (J) Mutation of K226 and K275 completely abolished the acetylation on FOXA2 in Min 6 cells.
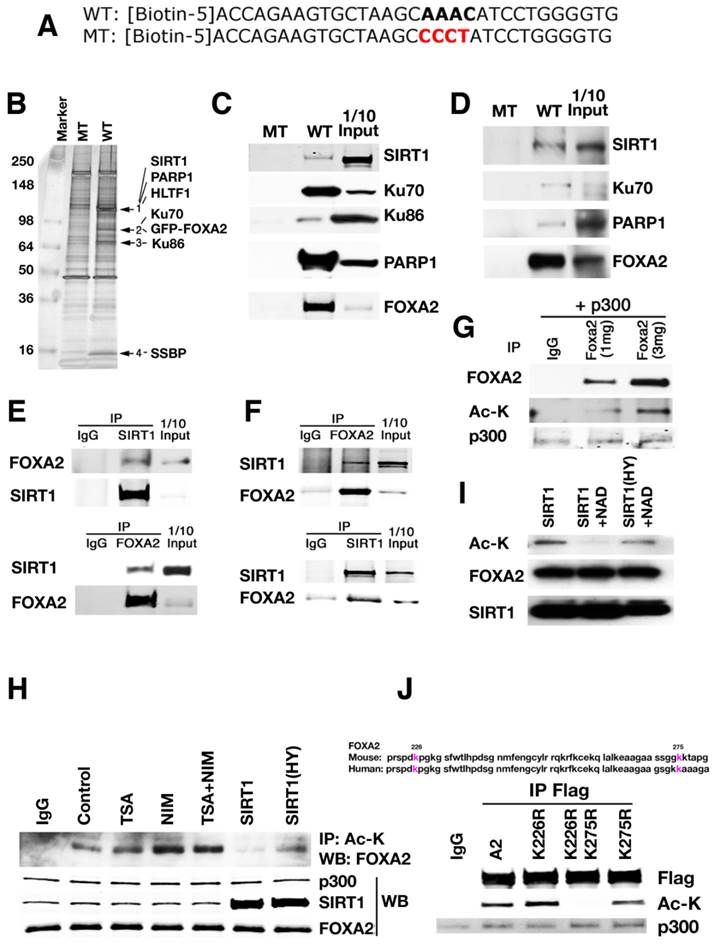
SIRT1 can deacetylate FOXA2
Two of the proteins identified as part of the complex, PARP1 [44], and KU70 [10] were reported to interact with SIRT1 as substrates. A third protein, KU86, could be acetylated as well although the corresponding acetyltransferase was not identified [8]. Therefore, we chose to study SIRT1's relationship with FOXA2 and PDX1 expression.
Using reciprocal immunoprecipitation, we confirmed the interaction between SIRT1 and FOXA2 in both 293T cells (Fig. 1E) and Min6 cells (Fig. 1F), suggesting FOXA2 could be one of the SIRT1's substrates. Next, we tested whether FOXA2 is acetylated and whether SIRT1 could deacetylate FOXA2. 293T cells were transfected with p300 and FLAG-FOXA2. Then FLAG-FOXA2 was pulled down with anti-FLAG M2 antibody and western blotted with anti-acetyl lysine antibody. The reverse way was also performed with the same lysates. As shown in figure 1G, FOXA2 was acetylated in the presence of p300 and the level of acetylation is correlated with the amount of FOXA2. The acetylation of FOXA2 was enhanced in the presence of nicotinomide (NIM) (Fig. 1H), indicating that class III HDACs are involved. Wild-type SIRT1, but not a deacetylase deficient mutant form of SIRT1, was able to deacetylate FOXA2 (Fig. 1H). A similar acetylation pattern was observed when purified FLAG-FOXA2 and SIRT1were added together into a test tube in the presence or absence of NAD+ (Fig. 1I). Only in the presence of NAD+, was wild-type SIRT1 capable of taking off the acetyl group from FOXA2. The deacetylase deficient mutant of SIRT1 (SIRT1-HY) was unable to catalyze the removal of acetyl group from FOXA2 (Fig. 1H-I). Mass spectrometry (MS) analysis identified two potential acetylation sites within FOXA2 (i.e., K226 and K275). K226 resides in FOXA2 DBD domain, and K275 is at the junction between Wing 2 and NLS (Nuclear localization sequence). K226 and K275 are conserved in both human and mouse FOXA2 (Fig. 1J). Site mutagenesis experiments were performed to mutate these two lysine residues (K) to arginine residues (R). Three FOXA2 mutants were generated (FOXA2K226R, FOXA2K275R and FOXA2K226R/K275R=DM). In Min6 cells, mutation of both sites to arginine residues completely abolished acetylation of FOXA2 by p300 (Fig. 1J). Mutating each lysine residue individually does not have an obvious effect on the acetylation status of FOXA2 (Fig. 1J), confirming the identification of only two p300-dependent acetylation sites within FOXA2 by MS under our experimental condition.
SIRT1 positively regulates Pdx1 level together with FOXA2
Min6 cell line was applied to study the relationship between SIRT1 and FOXA2 further. Acute knockdown of Sirt1 resulted in significant decrease of Pdx1 and a number of its downstream genes, including insulin, glucokinase (Gk) and Glut2 (Fig. 2A). Of note, overexpression of SIRT1 in these cells resulted in only a slight up-regulation of Pdx1 (Additional file 1: Supplementary Fig. 1A, B), suggesting that the amount of SIRT1 is not a rate-limiting factor for the expression of Pdx1 in these Min6 cells.
Regulation of Pdx1 transcription by SIRT1 and FOXA2. (A) shRNA-mediated knockdown of Sirt1 in Min6 cells reduces expression of genes that are involved in pancreas development and insulin production. *p<0.01. (B) Both SIRT1 and FOXA2 bind to the PH2 domain of the Pdx1 promoter revealed by qChIP. Arrows show the positions of primers used in the assay. (C) Wild-type SIRT1 (T1) and FOXA2 (A2) synergistically elevate the luciferase activity driven by the PH2 domain of the Pdx1 promoter. Insert shows expression of SIRT1 and FOXA2. (D) Knockdown of Sirt1 reduces the activity of FOXA2 on Pdx1 promoter. Insert shows level of SIRT1 knockdown. (E) Mutation of two lysine residues in FOXA2 abolished the synergistic effect of FOXA2 and SIRT1 on Pdx1 promoter, but improve the FOXA2 transcriptional activity. All the transfection contained the same amount of DNA, GFP plasmid was used to make up the total DNA amount. (F) Mutation of two lysine residues in FOXA2 enhances the binding ability of FOXA2 to Pdx1 promoter assessed by ChIP.
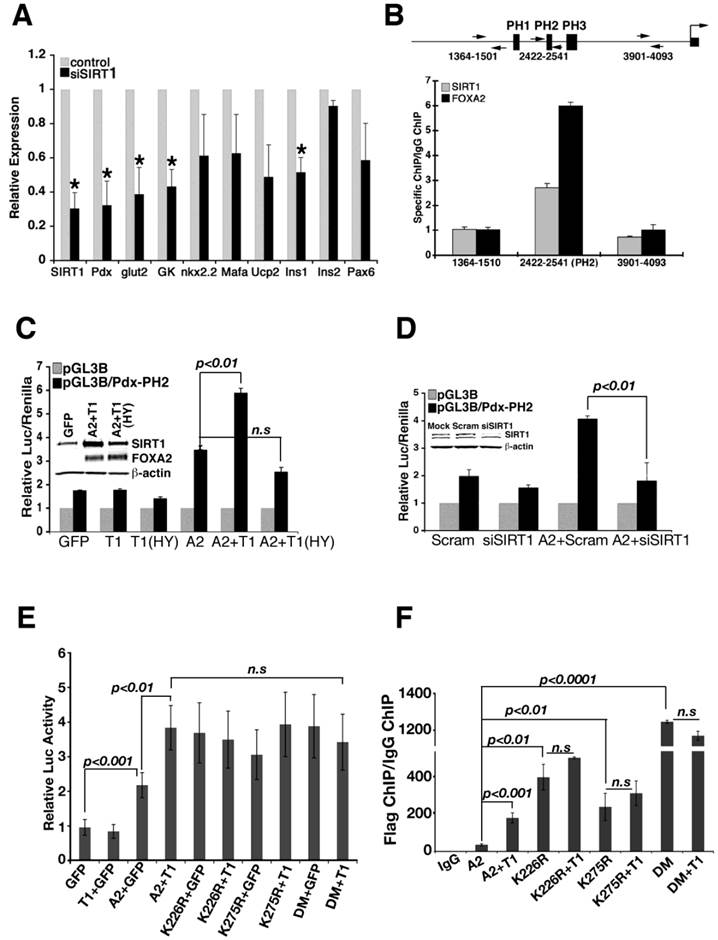
To investigate whether SIRT1 binds to Pdx1 promoter, we performed a chromatin immunoprecipitation (ChIP) assay and found that SIRT1 occupies the Pdx1 promoter near FOXA2 binding site (PH2) (Fig. 2B) [16]. Using a luciferase reporter for Pdx1 transcription, we found that expression of FOXA2 and wild type SIRT1, but not SIRT1-HY, synergistically induced Pdx1 reporter activity (Fig. 2C), suggesting that deacetylase activity of SIRT1 is required for this action. siRNA-mediated knockdown of Sirt1 also reduced induction of Pdx1 promoter activity by FOXA2 (Fig. 2D), indicating that SIRT1 is required for FOXA2 to further elevate Pdx1 promoter.
Next, we studied whether K226 and K275 in FOXA2 play a role in SIRT1 induction of Pdx1 expression. Expression of FOXA2 K226 and/or K275 mutants in min6 cells were able to induce the Pdx-luc activity close to the level where SIRT1 and wild type FOXA2 combination could get (Fig. 2E). Of note, expression of SIRT1 does not further increase Pdx-luc activity on the top of FOXA2 mutations (Fig. 2E). Since arginine mutation mimics deacetylated form of lysine, these data is consistent with a role of SIRT1 deacetylating FOXA2 on these sites to increase its transcriptional activity on Pdx1 promoter.
With these data, we rationalized that mutation of lysine 226 and 275 to arginine enhances FOXA2 binding to Pdx1 promoter. To test this hypothesis, FOXA2 or its different mutants were co-transfected with SIRT1 into min6 cells, and ChIP experiments were performed with these transfectants (Fig. 2F). In the presence of SIRT1, FOXA2 binds to Pdx1 promoter stronger compared with FOXA2 alone (p<0.001). K226R or K275R single mutation enhanced the binding of FOXA2 to Pdx1 promoter (p<0.01). When both K226 and K275 were mutated to arginine (FOXA2 DM), FOXA2 DM achieved the highest binding to Pdx1 promoter. The binding synergy between SIRT1 and FOXA2 diminished when lysine to arginine mutants were applied. This evidence underscores the importance of SIRT1 and lysine sites K226/K275 in maintaining transcriptional activity of FOXA2 on Pdx1 promoter.
Pancreatic deficiency of SIRT1 results in progressive development of insulin insufficiency
To assess the potential impact of SIRT1 deficiency on β-cell formation and pancreas development, a pancreas-specific disruption of SIRT1 using a Cre-LoxP mediated approach was performed (Additional file 1: Supplementary Fig. 2A). The Sirt1 floxed mice were crossed with transgenic mice carrying a Cre gene under the control of a Pdx1 promoter (Pdx1-Cre) [18] to achieve pancreas-specific knockout of SIRT1 (Sirt1PKO) (Supplementary Fig. 2B-C).
The Sirt1PKO mice appeared normal during early postnatal developmental stages. At 14 days of age, all Sirt1PKO mice examined had a normal range of glucose. However starting from 3 weeks (P21), when the mice were weaned, 6 out of 41 (15%) of Sirt1PKO mice displayed a significant increase in glucose (Fig. 3A). Over time, the number of mice with hyperglycemia significantly increased. By 20 weeks of age, 60% of Sirt1PKO mice were hyperglycemic. Many of these Sirt1PKO mice died at later stage. This contributes to the high mortality rate of Sirt1PKO mice over 16 weeks of age (Fig. 3B).
Sirt1PKO mice display hyperglycemia and glucose intolerance. (A) Percentage of Sirt1PKO mice that contain high blood glucose level (>250 mg/ml) during development. Number of mice is show on the top of each bar. (B) Survival curve of Sirt1PKO mice during 4 months after birth. (C) Summary of blood glucose levels in animals at different ages. Twenty-three Sirt1PKO animals and twelve control mice were followed for their glucose level during period of P14 to 4 months. (D) Islet number and glucose level of P21 animals. Each group contains 4-6 animals. * p<0.001.
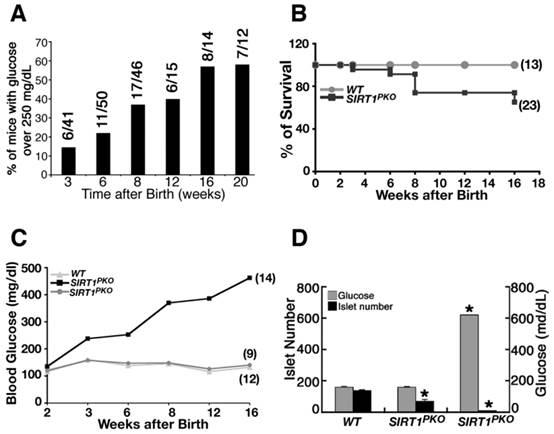
Dynamic changes in blood glucose levels of a cohort of 23 Sirt1PKO mice were followed up to 4 months of age. Of these mice, 5 at 3 weeks, 7 at 6 weeks, and 14 at 16 weeks displayed hyperglycemia (blood glucose levels >200 mg/dL) (Fig. 3C, Additional file 1: Supplementary Table 1A). In contrast, the blood glucose level remains almost steady in the wild-type control group (Fig. 3C, Supplementary Table 1B).
To understand the cellular and molecular basis for the observed hyperglycemia, we analyzed Sirt1PKO mice, irrespective of whether or not they were hyperglycemic. Analysis of mice at P21 indicated that hyperglycemic Sirt1PKO animals had an average of 8 islets, with the blood glucose concentration reaching over 600 mg/dL. The average number of islets of normaglycemic Sirt1PKO was 68, about half the number found in wild-type controls (Fig. 3D).
We suspected that phenotypic variation might be due to incomplete Cre-mediated recombination, which is always the case based on our experience on Cre-loxP mediated disruption of many other genes in various organs/tissues [34, 57, 62, 64]. Indeed, Western blot against SIRT1 from different genotypes of pancreases demonstrated that hyperglycemic Sirt1PKO mice contain much less SIRT1 compared with normaglycemic Sirt1PKO mice (Additional file 1: Supplementary Fig. 2C). Thus, the severity of SIRT1 mutant phenotype directly correlates with the extent of SIRT1 deletion, highlighting a critical role of SIRT1 in regulating and maintaining normal development of islets. Therefore, our further study focused on the hyperglycemic Sirt1PKO mice.
Pancreatic specific deletion of SIRT1 impairs islet formation
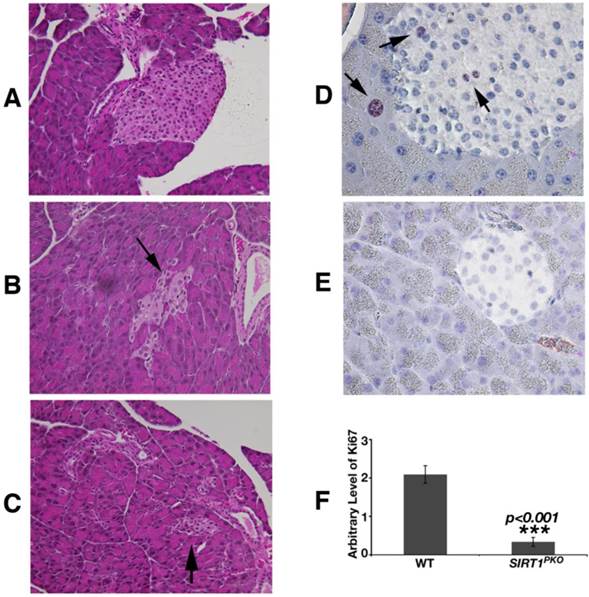
Histological analysis revealed that Sirt1PKO mice had reduced size and irregular shaped islets (Fig. 4A-C). Studying pancreas at P21, apoptosis (TUNEL assay) was not detected in the islets of either Sirt1PKO mice or wild-type mice. The proliferation status was investigated with Ki67 antibody staining. As shown in figure 4D-F, in wild-type pancreas, Ki67 positive cells can be observed in both acinar and islet, but SIRT1 deficient pancreases contain 6-fold reduction of Ki67 positive cells.
Sirt1PKO mice display abnormal islet development. (A-C) H&E staining of pancreas sections is shown from wild-type (A) and Sirt1PKO (B-C) mice. Sirt1PKO animals exhibit various abnormal morphology of the islet. Arrows indicate islets. (D-F) Ki67 staining of pancreas at P21. The pancreas from Sirt1PKO mice has 6 folds reduction of Ki67 positive cells. Arrows indicate the Ki67 positive cells.
It has been shown that over-expression of SIRT1 protects β-cells against cytokine toxicity by suppressing NF-kB signals [33]. Therefore, we examined whether loss of SIRT1 could trigger an autoimmune response that destroys islets. Monitoring immunohistological markers, such as IL-1β, TNFα, IL-6, CD3, F4/80, MPO, NF-kB, did not detect obvious signs of inflammation in the mutant islets (Additional file 1: Supplementary Fig. 3). These data ruled out the possibility that the impaired formation of islets in Sirt1PKO mice is caused by an autoimmune response triggered by SIRT1 deficiency.
SIRT1 deficiency in pancreas diminished expression of PDX1 and impaired β-cell formation
PDX1 is a master regulator for islet formation and expression of insulin. Due to the positive relationship between SIRT1 and PDX1 displayed above (Figs. 1, 2), a gene expression analysis was conducted to ascertain the molecular mechanism underlying the phenotype. Realtime RT-PCR analysis revealed that islets from hyperglycemic Sirt1PKO mice had about 5% of Sirt1 mRNA, markedly reduced transcription levels of Pdx1 and a number of Pdx1 downstream genes such as Glut2 and insulin (Fig. 5A). We followed insulin levels in Sirt1PKO mice for 6 months. Hyperglycemic Sirt1PKO mice consistently exhibited very low levels of insulin (Fig. 5B).
Deletion of SIRT1 in pancreas impaired β-cell formation. (A) Gene expression pattern in islets with different genotypes. Each group contains islets from 4 individual animals. * p<0.01. (B) Serum insulin level from animals. *p<0.001. (C) Immunofluorescent staining of the islet. (D-E) Reciprocal immunoprecipitation with whole pancreases lysates shows that SIRT1 and FOXA2 interact with each other in vivo. (F) FOXA2 is acetylated in vivo. The ratio of acetylated FOXA2 (top lane) vs total FOXA2 (second lane) is presented in number at the bottom. (G) FOXA2 binds to PH2 domain of PDX1 promoter in vivo. ChIP assay was performed with a FOXA2 antibody. For experiments in D-G, pancreases from 9 wild-type mice or 13 Sirt1PKO mice at P14 were pooled together to get enough samples for each experiment.
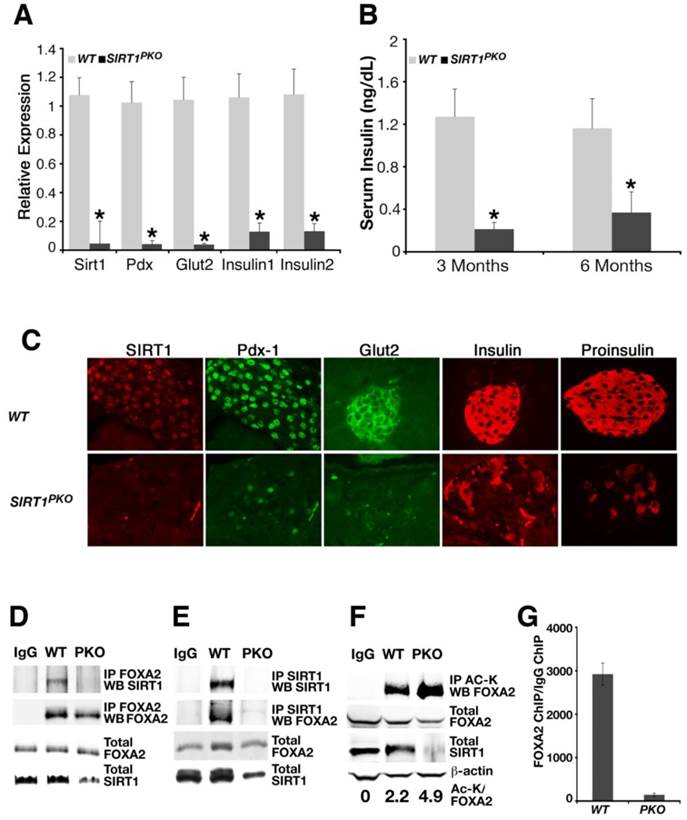
Next, we performed immuno-fluorescent staining to examine the impact of SIRT1 deletion on different cell types in pancreatic islets. Consistent with the real-time PCR data (Fig. 5A), a dramatic reduction of SIRT1 protein in the islet of hyperglycemic Sirt1PKO mice was observed compared with wild- type control mice (Fig. 5C). A graded reduction of cells that are positive for PDX1, GLUT2, insulin, and proinsulin was also observed in the islet of these mutant mice (Fig. 5C), suggesting the specific reduction of β-cells.
Notably, α cells (positive for glucagon) became randomly distributed in the mutant islets (Additional file 1: Supplementary Fig. 4A). They also seemed relatively more abundant in mutant islets compared to wild type islets. However, when serum glucagons was measured, no difference was detected between wild-type and Sirt1PKO mice. Conversely, there was no obvious increase of δ cells (positive for somatostatin) (Supplementary Fig. 4B) or PP cells (positive for pancreatic polypeptide) (Supplementary Fig. 4C).
Our in vitro studies illustrated that SIRT1 positively regulates Pdx1 expression through interacting with and deacetylating FOXA2 (Figs. 1 and 2). Using the Sirt1PKO pancreas as a negative control, we now ask whether SIRT1 and FOXA2 interact with each other in vivo. Whole pancreas lysates from wild-type or Sirt1PKO mice at P14 were used to perform a reciprocal CO-IP between SIRT1 and FOXA2. Indeed, SIRT1 and FOXA2 interact with each other in wild type mice. The interactions between these two proteins were not detected in the mutant samples (Fig. 5D-E). We next examined whether FOXA2 is acetylated in vivo at this stage. Whole pancreatic lysates were precipitated with acetyl-lysine antibody and Western blotted with FOXA2 antibody, and ratio of acetyl lysine vs total FOXA2 was quantified. The results indicated that FOXA2 is acetylated and Sirt1PKO samples contain higher level of acetylation on FOXA2 compared with controls (Fig. 5F). Finally, we investigated whether FOXA2 binds to PDX1 promoter in vivo and whether loss of SIRT1 affects its binding by ChIP assay. Clearly, FOXA2 binds to PH2 domain of PDX1 promoter in wild type pancreas, and deletion of Sirt1 greatly reduced this binding (Fig. 5G). Thus, with Sirt1PKO mice model, we demonstrated that SIRT1 and FOXA2 indeed interact with each other in vivo during pancreas development. SIRT1 deacetylates FOXA2 and enhances FOXA2 transcriptional activity on Pdx1 promoter either directly or by affecting FOXA2's ability to recruit other transcriptional activators/repressors, and this process is critical for pancreatic β-cell formation.
Discussion
SIRT1 deacetylates many proteins that are implicated in multiple important biological processes, including DNA damage repair, cell cycle checkpoints, metabolic regulation, animal aging, and cancer [11, 13, 20, 21, 23, 30, 36, 48, 55, 56, 66]. Here, our results demonstrate that SIRT1 positively regulates pancreatic β cell formation and differentiation by interacting with FOXA2 and synergistically regulates PDX1 transcription. Since mutation of K226 and K275 to arginines (which mimic the deacetylated form) in FOXA2 abolished the synergistic induction of FOXA2 and SIRT1 on PDX1-Luc reporter, enhanced the binding of FOXA2 to endogenous PDX1 promoter, and increased FOXA2 transcriptional activity on Pdx1-Luc vector in min6 cells, we conclude that SIRT1 activates FOXA2 through deacetylation on K226 and K275. We further showed loss of SIRT1 impairs expression of PDX1 and blocks β-cell formation, leading to severe hyperglycemia.
A recent study showed that FOXA2 acetylation on K259 promotes FOXA2 transcriptional activity to regulate hepatic lipid oxidation in ob/ob and db/db mice [54]. In our study, SIRT1 deacetylates FOXA2-K226 and K275 in both human HEK-293 cell line and mouse pancreas beta cell derived Min6 line, whereas K259 was not detected. Since acetylation on K259 only takes place when fasting or glucogon treatment was applied, and SIRT1 displays the highest activity in fasting condition [54], it is possible that K259Ac is not a direct SIRT1 target under normal physiological condition. It is also plausible that SIRT1 deacetylates different residues of FOXA2 in different tissue context. Nonetheless, these data picture a complex mechanism in finely tuning FOXA2 transcriptional activity during development, differentiation and metabolism. This issue deserves further investigation in order to fully understand disease progress in liver and pancreas.
Of note, it has been previously shown that overexpression of SIRT1 in β cells in transgenic mice enhances insulin secretion in response to glucose through inhibiting transcription of the Ucp gene [40]. Other study also showed that a transgenic mouse strain with moderate overexpression of SIRT1 exhibits normal insulin sensitivity but exhibits decreased food intake and locomotor activity, resulting in diminished energy expenditure [1]. In cultured β cell lines, SIRT1 represses transcription of the Ucp2 gene by binding directly to the Ucp2 promoter [4]. Here we demonstrate for the first time that SIRT1 is critical in β cell differentiation and maturation, since SIRT1 deficiency inhibits β-cell formation. We further manifested that SIRT1 deacetylates FOXA2 and forms a protein complex with FOXA2 on the promoter of Pdx1 and serves as a potent regulator for expression of Pdx1 during the early stages of pancreas development and islet formation. Of note, our pulldown using the consensus FOXA2 binding site in the PH2 domain revealed a protein complex including SIRT1, FOXA2, PARP1, HLTF1, KU70, KU86, and SSBP on the Pdx1 promoter, it is likely that these proteins form a complex in the promoter of Pdx1. The spatial relationship of each component in the complex might be an interesting topic to be addressed in future. Furthermore, SIRT1 is also a histone deacetylase, it is possible that SIRT1 might affect this protein complex formation through epigenetic modification on histones on the promoter of Pdx promoter. All these issues deserve further studies. Nonetheless, because PDX1 acts as a master factors in the pancreas and regulates expression of many genes that are essential for insulin production and secretion [26, 27, 41], we conclude that the impact of SIRT1 on β-cell formation is primarily through its regulation of Pdx1 expression (Fig. 6).
An integrated model displays a critical role of SIRT1 in maintaining PDX1 expression and β-cell formation. (A) SIRT1 interacts with, and deacetylates FOXA2 on the promoter of PDX1 and serves as a potent regulator for expression of PDX1 and its downstream genes during the early stages of pancreatic development and β-cell formation. Concurrently, expression of SIRT1 is also subjected to spatial and temporal regulation. (B) Targeted disruption of SIRT1 increases the acetylated form of FOXA2, impairs PDX1 expression, and blocks β-cell formation, leading to hyperglycemia and insulin insufficiency.
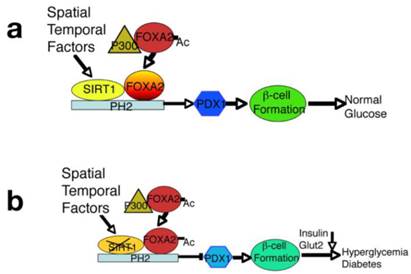
There is no doubt that Sirt1 transcription is subjected to spatial and temporal regulation. Our previous study showed that the tumor suppressor BRCA1 could positively regulate Sirt1 expression during tumorigenesis [60]. Given the important roles of SIRT1 in numerous biological processes [11, 13, 21, 23, 30, 36, 48], the underlying mechanism for SIRT1 regulation should be an important focus for future investigation. The regulation and activation of SIRT1 in β-cells should serve as a model for bridging islet development, insulin secretion, and glucose metabolism.
Supplementary Material
Additional File 1Supplementary figures and table.
Acknowledgements
We thank Dr. Douglas Melton for PDX1-Cre mice, and Dr. Kenneth S. Zaret for GFP-FOXA2 expression vector. We thank Drs. Oksana Gavrilova, Tyler J. Lahusen, Cuiling Li, and Cuiying Xiao for technical assistance. We also thank the members of the Deng lab for critical review of the manuscript. This research was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, USA.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Banks AS, Kon N, Knight C, Matsumoto M, Gutierrez-Juarez R, Rossetti L, Gu W, Accili D. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008;8:333-41
2. Ben-Shushan E, Marshak S, Shoshkes M, Cerasi E, Melloul D. A pancreatic beta -cell-specific enhancer in the human PDX-1 gene is regulated by hepatocyte nuclear factor 3beta (HNF-3beta ), HNF-1alpha, and SPs transcription factors. The Journal of biological chemistry. 2001;276:17533-40
3. Biason-Lauber A, Boni-Schnetzler M, Hubbard BP, Bouzakri K, Brunner A, Cavelti-Weder C, Keller C, Meyer-Boni M, Meier DT, Brorsson C, Timper K, Leibowitz G, Patrignani A, Bruggmann R, Boily G, Zulewski H, Geier A, Cermak JM, Elliott P, Ellis JL, Westphal C, Knobel U, Eloranta JJ, Kerr-Conte J, Pattou F, Konrad D, Matter CM, Fontana A, Rogler G, Schlapbach R, Regairaz C, Carballido JM, Glaser B, McBurney MW, Pociot F, Sinclair DA, Donath MY. Identification of a SIRT1 mutation in a family with type 1 diabetes. Cell metabolism. 2013;17:448-55
4. Bordone L, Motta MC, Picard F, Robinson A, Jhala US, Apfeld J, McDonagh T, Lemieux M, McBurney M, Szilvasi A, Easlon EJ, Lin SJ, Guarente L. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 2006;4:e31
5. Carrasco M, Delgado I, Soria B, Martin F, Rojas A. GATA4 and GATA6 control mouse pancreas organogenesis. The Journal of clinical investigation. 2012;122:3504-15
6. Chen D, Bruno J, Easlon E, Lin SJ, Cheng HL, Alt FW, Guarente L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008;22:1753-7
7. Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100:10794-9
8. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834-40
9. Cohen DE, Supinski AM, Bonkowski MS, Donmez G, Guarente LP. Neuronal SIRT1 regulates endocrine and behavioral responses to calorie restriction. Genes Dev. 2009;23:2812-7
10. Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390-2
11. Deng CX. SIRT1, is it a tumor promoter or tumor suppressor? Int J Biol Sci. 2009;5:147-52
12. Donmez G, Wang D, Cohen DE, Guarente L. SIRT1 suppresses beta-amyloid production by activating the alpha-secretase gene ADAM10. Cell. 2010;142:320-32
13. Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587-91
14. Gao N, LeLay J, Vatamaniuk MZ, Rieck S, Friedman JR, Kaestner KH. Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev. 2008;22:3435-48
15. Gasser SM, Cockell MM. The molecular biology of the SIR proteins. Gene. 2001;279:1-16
16. Gerrish K, Gannon M, Shih D, Henderson E, Stoffel M, Wright CV, Stein R. Pancreatic beta cell-specific transcription of the pdx-1 gene. The role of conserved upstream control regions and their hepatic nuclear factor 3beta sites. J Biol Chem. 2000;275:3485-92
17. Gerrish K, Van Velkinburgh JC, Stein R. Conserved transcriptional regulatory domains of the pdx-1 gene. Mol Endocrinol. 2004;18:533-48
18. Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447-57
19. Guarante LP. Regulation of Aging by SIR2. Ann N Y Acad Sci. 2005;1055:222
20. Guarente L. Sirtuins in aging and disease. Cold Spring Harb Symp Quant Biol. 2007;72:483-8
21. Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253-95
22. Halperin-Sheinfeld M, Gertler A, Okun E, Sredni B, Cohen HY. The Tellurium compound, AS101, increases SIRT1 level and activity and prevents type 2 diabetes. Aging (Albany NY). 2012;4:436-47
23. Herranz D, Serrano M. SIRT1: recent lessons from mouse models. Nat Rev Cancer. 2010;10:819-23
24. Ishihara H, Asano T, Tsukuda K, Katagiri H, Inukai K, Anai M, Kikuchi M, Yazaki Y, Miyazaki JI, Oka Y. Pancreatic beta cell line MIN6 exhibits characteristics of glucose metabolism and glucose-stimulated insulin secretion similar to those of normal islets. Diabetologia. 1993;36:1139-45
25. Jacquemin P, Lemaigre FP, Rousseau GG. The Onecut transcription factor HNF-6 (OC-1) is required for timely specification of the pancreas and acts upstream of Pdx-1 in the specification cascade. Developmental biology. 2003;258:105-16
26. Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371:606-9
27. Kaneto H, Matsuoka TA, Miyatsuka T, Kawamori D, Katakami N, Yamasaki Y, Matsuhisa M. PDX-1 functions as a master factor in the pancreas. Front Biosci. 2008;13:6406-20
28. Kitamura T, Nakae J, Kitamura Y, Kido Y, Biggs WH 3rd, Wright CV, White MF, Arden KC, Accili D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest. 2002;110:1839-47
29. Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996;93:5860-5
30. Lavu S, Boss O, Elliott PJ, Lambert PD. Sirtuins--novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discov. 2008;7:841-53
31. Lee CS, Sund NJ, Behr R, Herrera PL, Kaestner KH. Foxa2 is required for the differentiation of pancreatic alpha-cells. Developmental biology. 2005;278:484-95
32. Lee CS, Sund NJ, Vatamaniuk MZ, Matschinsky FM, Stoffers DA, Kaestner KH. Foxa2 controls Pdx1 gene expression in pancreatic beta-cells in vivo. Diabetes. 2002;51:2546-51
33. Lee JH, Song MY, Song EK, Kim EK, Moon WS, Han MK, Park JW, Kwon KB, Park BH. Overexpression of SIRT1 protects pancreatic beta-cells against cytokine toxicity by suppressing the nuclear factor-kappaB signaling pathway. Diabetes. 2009;58:344-51
34. Li C, Xu X, Nelson DK, Williams T, Kuehn MR, Deng CX. FGFR1 function at the earliest stages of mouse limb development plays an indispensable role in subsequent autopod morphogenesis. Development. 2005;132:4755-64
35. Li H, Rajendran GK, Liu N, Ware C, Rubin BP, Gu Y. SirT1 modulates the estrogen-insulin-like growth factor-1 signaling for postnatal development of mammary gland in mice. Breast Cancer Res. 2007;9:R1
36. Li X, Kazgan N. Mammalian sirtuins and energy metabolism. International journal of biological sciences. 2011;7:575-87
37. Marshak S, Benshushan E, Shoshkes M, Havin L, Cerasi E, Melloul D. Functional conservation of regulatory elements in the pdx-1 gene: PDX-1 and hepatocyte nuclear factor 3beta transcription factors mediate beta-cell-specific expression. Molecular and cellular biology. 2000;20:7583-90
38. McBurney MW, Yang X, Jardine K, Hixon M, Boekelheide K, Webb JR, Lansdorp PM, Lemieux M. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol. 2003;23:38-54
39. Morris BJ. Seven sirtuins for seven deadly diseases of aging. Free radical biology & medicine. 2013;56:133-71
40. Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Meneur C, Permutt MA, Imai S. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005;2:105-17
41. Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, Hogan BL, Wright CV. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122:983-95
42. Oliver-Krasinski JM, Kasner MT, Yang J, Crutchlow MF, Rustgi AK, Kaestner KH, Stoffers DA. The diabetes gene Pdx1 regulates the transcriptional network of pancreatic endocrine progenitor cells in mice. J Clin Invest. 2009;119:1888-98
43. Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009;9:327-38
44. Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S, Hottiger MO, Gupta MP. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol Cell Biol. 2009;29:4116-29
45. Samaras SE, Cissell MA, Gerrish K, Wright CV, Gannon M, Stein R. Conserved sequences in a tissue-specific regulatory region of the pdx-1 gene mediate transcription in Pancreatic beta cells: role for hepatocyte nuclear factor 3 beta and Pax6. Mol Cell Biol. 2002;22:4702-13
46. Samaras SE, Cissell MA, Gerrish K, Wright CV, Gannon M, Stein R. Conserved sequences in a tissue-specific regulatory region of the pdx-1 gene mediate transcription in Pancreatic beta cells: role for hepatocyte nuclear factor 3 beta and Pax6. Molecular and cellular biology. 2002;22:4702-13
47. Samaras SE, Zhao L, Means A, Henderson E, Matsuoka TA, Stein R. The islet beta cell-enriched RIPE3b1/Maf transcription factor regulates pdx-1 expression. The Journal of biological chemistry. 2003;278:12263-70
48. Saunders LR, Verdin E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene. 2007;26:5489-504
49. Sekiya T, Muthurajan UM, Luger K, Tulin AV, Zaret KS. Nucleosome-binding affinity as a primary determinant of the nuclear mobility of the pioneer transcription factor FoxA. Genes Dev. 2009;23:804-9
50. Shih DQ, Stoffel M. Molecular etiologies of MODY and other early-onset forms of diabetes. Curr Diab Rep. 2002;2:125-34
51. Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet. 1997;17:138-9
52. Stoffers DA, Heller RS, Miller CP, Habener JF. Developmental expression of the homeodomain protein IDX-1 in mice transgenic for an IDX-1 promoter/lacZ transcriptional reporter. Endocrinology. 1999;140:5374-81
53. Tuteja G, White P, Schug J, Kaestner KH. Extracting transcription factor targets from ChIP-Seq data. Nucleic acids research. 2009;37:e113
54. von Meyenn F, Porstmann T, Gasser E, Selevsek N, Schmidt A, Aebersold R, Stoffel M. Glucagon-induced acetylation of Foxa2 regulates hepatic lipid metabolism. Cell metabolism. 2013;17:436-47
55. Wang RH, Kim HS, Xiao C, Xu X, Gavrilova O, Deng CX. Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. The Journal of clinical investigation. 2011;121:4477-90
56. Wang RH, Li C, Deng CX. Liver Steatosis and Increased ChREBP Expression in Mice Carrying a Liver Specific SIRT1 Null Mutation under a Normal Feeding Condition. Int J Biol Sci. 2010;6:682-90
57. Wang RH, Li C, Xu X, Zheng Y, Xiao C, Zerfas P, Cooperman S, Eckhaus M, Rouault T, Mishra L, Deng CX. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005;2:399-409
58. Wang RH, Sengupta K, Li C, Kim HS, Cao L, Xiao C, Kim S, Xu X, Zheng Y, Chilton B, Jia R, Zheng ZM, Appella E, Wang XW, Ried T, Deng CX. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14:312-23
59. Wang RH, Yu H, Deng CX. A requirement for breast-cancer-associated gene 1 (BRCA1) in the spindle checkpoint. Proc Natl Acad Sci U S A. 2004;101:17108-13
60. Wang RH, Zheng Y, Kim HS, Xu X, Cao L, Luhasen T, Lee MH, Xiao C, Vassilopoulos A, Chen W, Gardner K, Man YG, Hung MC, Finkel T, Deng CX. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol Cell. 2008;32:11-20
61. Wu KL, Gannon M, Peshavaria M, Offield MF, Henderson E, Ray M, Marks A, Gamer LW, Wright CV, Stein R. Hepatocyte nuclear factor 3beta is involved in pancreatic beta-cell-specific transcription of the pdx-1 gene. Mol Cell Biol. 1997;17:6002-13
62. Xu X, Ehdaie B, Ohara N, Yoshino T, Deng CX. Synergistic action of Smad4 and Pten in suppressing pancreatic ductal adenocarcinoma formation in mice. Oncogene. 2010;29:674-86
63. Xu X, Li C, Garrett-Beal L, Larson D, Wynshaw-Boris A, Deng CX. Direct removal in the mouse of a floxed neo gene from a three-loxP conditional knockout allele by two novel approaches. Genesis. 2001;30:1-6
64. Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T, Hennighausen L, Wynshaw-Boris A, Deng CX. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation [see comments]. Nat Genet. 1999;22:37-43
65. Xuan S, Borok MJ, Decker KJ, Battle MA, Duncan SA, Hale MA, Macdonald RJ, Sussel L. Pancreas-specific deletion of mouse Gata4 and Gata6 causes pancreatic agenesis. The Journal of clinical investigation. 2012;122:3516-28
66. Yu J, Auwerx J. Protein deacetylation by SIRT1: an emerging key post-translational modification in metabolic regulation. Pharmacol Res. 2010;62:35-41
67. Zhang J, Chen L, Zheng J, Zeng T, Li H, Xiao H, Deng X, Hu X. The protective effect of resveratrol on islet insulin secretion and morphology in mice on a high-fat diet. Diabetes Res Clin Pract. 2012;97:474-82
Author contact
![]() Corresponding author: Tel: (301) 402-7225; Fax: (301) 480-1135 Email: chuxiadniddk.nih.gov.
Corresponding author: Tel: (301) 402-7225; Fax: (301) 480-1135 Email: chuxiadniddk.nih.gov.