Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(10):1099-1107. doi:10.7150/ijbs.7268 This issue Cite
Research Paper
Development of a Rapid Streptavidin Capture-Based Assay for the Tyrosine Phosphorylated CSF-1R in Peripheral Blood Mononuclear Cells
Shalini Chaturvedi1 ![]() , Elayne Dell1, Derick Siegel1, Gregory Brittingham2, Shobha Seetharam1
, Elayne Dell1, Derick Siegel1, Gregory Brittingham2, Shobha Seetharam1
1. Janssen Pharmaceutical Companies of Johnson & Johnson 1400 McKean Rd, Spring House, PA 19477, USA
2. Department of Biochemistry and Molecular Biology, Gettysburg College, 300 North Washington Street, Gettysburg, PA 17325, USA
Received 2013-7-26; Accepted 2013-11-7; Published 2013-11-22
Abstract
A novel assay was developed to measure ratio of p-FMS (phospho FMS) to FMS using the Meso Scale Discovery® (MSD) technology and compared to the routinely used, IP-Western based approach. The existing IP-Western assay used lysed PBMCs (Peripheral Blood Mononuclear Cells) that were immunoprecipitated (IP) overnight, and assayed qualitatively by Western analysis. This procedure takes three days for completion. The novel IP-MSD method described in this paper employed immunoprecipitation of the samples for one hour, followed by assessment of the samples by a ruthenium labeled secondary antibody on a 96-well Streptavidin-coated MSD plate. This IP-MSD method was semi-quantitative, could be run in less than a day, required one-eighth the volume of sample, and compared well to the IP-Western method. In order to measure p-FMS/FMS, samples from healthy volunteers (HV) were first stimulated with CSF-1(Macrophage colony-stimulating factor) to initiate the changes in the phosphotyrosyl signaling complexes in FMS. The objective of the present work was to develop a high throughput assay that measured p-FMS/FMS semi-quantitatively, with minimal sample requirement, and most importantly compared well to the current IP-Western assay.
Keywords: Colony stimulating factor - 1 receptor, IP-MSD assay, CSF-1
Introduction
There is a growing body of evidence showing a positive correlation between angiogenesis, tumor progression and number of tumor associated macrophages, which comprises 5-50% of cells in many tumors [1]. Macrophages participate in many cellular functions including immune defense, tissue development, production of cytokines and chemokines [2, 3], and phagocytosis of foreign material or apoptotic cells [4]. Colony Stimulating Factor -1 (CSF-1) is expressed in many tumors, is a growth factor for macrophages, and mediates osteoclast differentiation, which mediates bone erosions and fracture in metastatic bone disease [5, 6].
CSF-1 was initially discovered as a haematopoietic cytokine influencing proliferation, differentiation and survival of the monocyte-macrophage cell lineage, and later on recognized as an important factor in pathogenesis of a number of malignancies, including HL (Hodgkin's lymphoma) [7-9]. Cancer cells of different origin were shown to produce CSF-1 and/or express its receptor CSF-1R, also called FMS or c-FMS (McDonough feline sarcoma viral (v-fms) oncogene homolog) [10-12]. CSF-1 may contribute to cancer progression by driving cancer cell proliferation in an autocrine fashion and also by inducing macrophage infiltration, survival, differentiation and polarization towards the cancer-promoting M2 macrophage phenotype [13, 14].
Colony stimulating factor-1 receptor is encoded by the c-fms proto-oncogene. It has an extracellular ligand binding domain which binds CSF-1, a trans-membrane domain, and an intra-cellular tyrosine domain that contains a kinase insert domain [15]. Effects of CSF-1, which exists as a disulfide homodimer, are mediated by binding to FMS and stabilizing FMS dimerization, thereby activating the receptor. The dimerized receptor is phosphorylated (p-FMS), which initiates a cascade of steps resulting in gene transcription, protein translation, and the control of growth and differentiation [16, 17]. CSF-1 is expressed by several tumors and in metastatic bone disease, and has also been linked to systemic elevated levels of CSF-1 in plasma [18, 19]. CSF-1 inhibition, by targeting the receptor, attracts new interest in the context of research on anticancer therapies [20].
Classical HL is lymphoid tumor characterized by prevalence of malignant Hodgkin and Reed-Sternberg (H-RS) cells dominated by an abundant background of non-neoplastic cells. HL is regarded as an inflammatory disease, and H-RS cells derived from mature B cells have characteristically lost the B-cell phenotype and demonstrate several abnormalities in signaling pathways, and deregulation at transcription and epigenetic levels [21]. Overexpression of cytokines and cytokine receptors in H-RS cell lines and in primary H-RS cells, and abnormal levels of these molecules in vivo in HL patients, was correlated to the development of clinical characteristics of the disease [22-24]. Given that CSF-1 and FMS offer excellent targeted therapy for cancer, it was important to develop a good, reliable assay to measure inhibition of p-FMS. The current approach has been an IP-Western assay that measures ratios between p-FMS and total FMS expression from human blood. This procedure consisted of first stimulating whole blood ex-vivo with CSF-1, followed by isolation and lysis of the PBMCs, immunoprecipitation of total FMS (both the phosphorylated and unphosphorylated forms), electrophoresis by SDS-PAGE, and Western analysis using antibodies specific for total FMS and p-FMS, respectively. Even though the assay performed well, it was a lengthy procedure- lasted 3 days to complete one run- and was limited by the number of samples that could be analyzed by Western Blot. Therefore, it was important to develop another assay that utilized less sample volume and was faster, with higher throughput.
In this study, we developed a novel assay- the IP-MSD- to measure the ratio between p-FMS and total FMS. This entailed immunoprecipitation of the samples with two separate antibodies specific for p-FMS and total FMS, capture of the precipitate on an MSD plate, and analysis with a polyclonal ruthenium-labeled detection antibody. This assay required less sample volume, could be performed faster, and sample throughput could be increased and accommodated on a 96-well plate.
Materials and Methods
Matched sets of human serum and plasma from HL and healthy volunteers were purchased from Bioreclamation, Inc. All blood was collected in various Vacutainer® tubes purchased from Becton, Dickinson and Company (BD). Recombinant human CSF-1 (216-MC-005), human M-CSF R mAb (3292) and human M-CSF R antibody (AF329) were purchased from R&D Systems, Inc. BSA (Bovine Serum Albumin, Blocker A, R93AA-1), SULFO-TAG™ NHS Ester (R91AN-1), Streptavidin Gold Multi Array 96 well plates (L15SA-1), MSD Read Buffer (R92TC-1) and SECTOR Imager 6000 were purchased from Meso Scale Discovery. DMSO (Dimethyl sulfoxide ,D2650), ZnCl2 (39059), β Mercaptoethanol (63689) and Phosphatase Inhibitor Cocktail (P5726) were purchased from Sigma. The milk buffer was made using instant nonfat dry milk. JNJ-DRUG, a small molecule inhibitor of colony stimulating factor receptor (CSF-1R or FMS) was acquired from Janssen R&D, Inc. Tween-20 (X251-07) was purchased from J.T. Baker. NaCl (7532-04) was purchased from Macron. PBS (21-031-CM) was purchased from CellGro. Tris-HCl (15567-027), NuPAGE Novex 4-12% Bis-Tris gels (NP0336BOX) and nitrocellulose membrane (LC2009) were purchased from Invitrogen. Complete Protease Inhibitor Tab, EDTA-free (04693132001) was purchased from Roche. TrueBlot anti-Mouse Ig IP Beads (00881125) were purchanced from eBioscience. Laemmli Sample Buffer (1610737) was purchased from Bio-Rad. The rabbit anti-human c-Fms/CSF-1R mAb (Cat# sc-692, Lot # K0911) and goat α-rabbit IgG-HRP (sc-2004) were purchased from Santa Cruz Biotechnology, Inc. The rabbit anti-human phospho-M-CSF Receptor (Tyr723) polyclonal antibody (3155S) was purchased from Cell Signaling Technology. The 4G10 Platinum, Anti-Phosphotyrosine, Biotin Conjugate (16-452) was purchased from Millipore. NP-40 (28324), SuperSignal West Femto Maximum Sensitivity Substrate (34096), EZ-Link Sulfo-NHS-LC-Biotin reagent (21327) and BCA Protein Assay Kit (23225) were purchased from Thermo-Pierce. SoftMax Pro 4.6 Enterprise software was purchased from Molecular Devices.
PBMC Preparation and CSF-1 Stimulation
PBMCs lysates were prepared as outlined in figure 1. Whole blood was collected from healthy subjects (n=5) at Janssen Pharmaceutical Companies of Johnson & Johnson in Radnor, PA. Informed consent was obtained from the volunteers, and the study protocol for employee volunteer participation in sample donation for research was approved by Quorum Review Independent Review Board, QR File# 23337/1. For each of the eight conditions to be tested, eight ml of blood was collected into a CPT (Cell Preparation Tube) with Sodium Citrate. Two tubes received 2.0 μM JNJ-DRUG (1.6 μL of 10mM stock in DMSO), two tubes received 0.2 μM JNJ-DRUG (1.6 μL of 1 mM stock in DMSO), two tubes received 1.6 μL DMSO, and two tubes were untreated. Tubes were capped and immediately inverted five times, and then incubated at room temperature for 30 minutes. Following incubation, tubes were opened and 50 μL CSF-1 (8 μg/mL in PBS) was added to 1 tube for each test condition, and 50 μL vehicle (0.1% BSA in PBS) was added to the 4 remaining tubes for each test condition. Tubes were capped, inverted five times, and centrifuged for 30 minutes at room temperature in a swinging bucket rotor at 1800 RCF. Most of the clear plasma was removed from above the peripheral blood mononuclear cell (PBMC) layer, and PBMCs were transferred to a 15 mL conical tube containing 10 mL ice cold PBS. Cells were pelleted at 300 RCF for 15 minutes at 4°C, and supernatant was removed. The cell pellets were resuspended in 1 mL ice cold PBS, and the tubes were filled with ice cold PBS for a second wash. Cells were pelleted as above, supernatant was removed, and cells were resuspended in 360 μL ice cold complete lysis buffer, containing 10 mM Tris (pH 7.5), 50 mM NaCl, 0.1% BSA, 0.5% NP-40, 5 μM ZnCl2, Complete Protease Inhibitor Tab and Phosphatase Inhibitor Cocktail II. Cells were incubated on ice for 15 minutes to enhance lysis, and then the lysates were centrifuged at 13,000 RCF to pellet cell debris. Subsequently, 20 μL from each lysate was transferred to a fresh microtube for MSD analysis; the remaining 340 μL was transferred to a separate microtube for IP-Western analysis. Lysates were frozen at -70 °C until ready to be assayed.
Schematic Diagram of PBMC Lysate Preparation from Whole Blood for 1 Subject
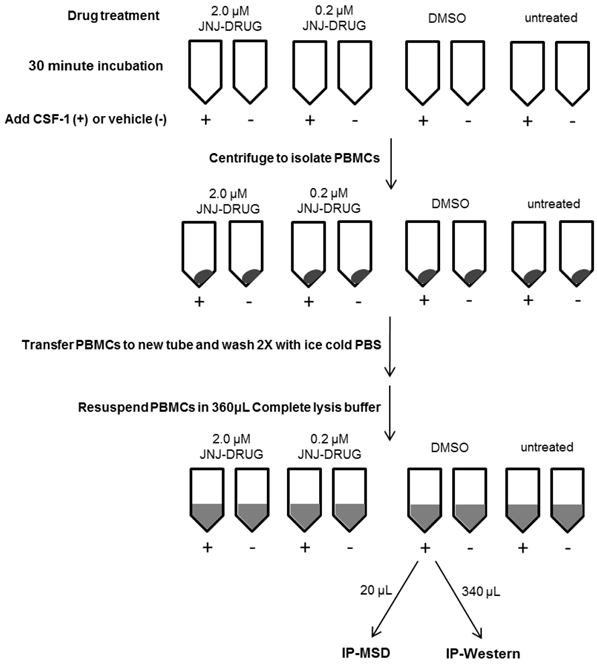
Immunoprecipitation of samples for IP-Western assay
TrueBlot anti-Mouse Ig IP Beads were removed from vial, transferred to a 15 mL tube, washed with 10 mL complete lysis buffer, and centrifuged at 2,000 RPM for 3 minutes at 4 °C. Supernatant was aspirated and beads were washed again as above. Supernatant was aspirated until beads were dry, and beads were resuspended in 2.5 mL complete lysis buffer. Lysates for IP-Western were thawed on ice, and an additional 300 μL complete lysis buffer was added to each tube. Lysates were precleared by adding 50 μL Ig IP beads, and rotating tubes for 30 minutes at 4 °C. Beads were then pelleted and supernatants were transferred to new microtubes. IP antibody- Human M-CSF R mAb was added at 5 μg per sample and samples were rotated for 90 minutes at 4 °C. Ig IP beads were added at 50 μL per tube and samples were rotated overnight at 4 °C. To serve as a negative bead control, 50 μL of beads were combined with 500 μL complete lysis buffer and rotated overnight along with the test samples. The following day, tubes were centrifuged at 10,000 RCF for 2 minutes at 4 °C, and supernatant was aspirated from the beads. Beads were resuspended in 1 mL complete lysis buffer and pelleted again at 10,000 RCF for 2 minutes. Supernatant was removed, wash was repeated, and beads were aspirated dry. Protein was eluted from the beads by adding 50 μL of 2X Laemmli Sample Buffer containing 5% beta-mercaptoethanol and incubating samples at 95 °C for 10 minutes. Bead control was treated likewise. Tubes were centrifuged at 10,000 RCF for 5 minutes to separate supernatant from beads.
Western Blots of p-FMS and FMS for IP-Western assay
For Western Blots to be analyzed for p-FMS expression, 20 μL of protein were separated on NuPAGE Novex 4-12% Bis-Tris gels, while 10 μL of protein were loaded on gels for Western Blot evaluation of FMS expression. To serve as a positive control, 3 μg of lysate consisting of FMS-transfected, CSF-1 stimulated HEK293 cells was run on p-FMS and FMS gels; 20 μL and 10 μL of negative bead control were loaded on p-FMS and FMS gels, respectively. All samples from a single subject were resolved on a single membrane. Resolved proteins were transferred to a nitrocellulose membrane and membranes were blocked for 1 hour at room temperature in milk buffer consisting of TBS + 0.1% Tween 20 + 5% nonfat dry milk. FMS blots were probed with c-Fms/CSF-1R antibody at 1:2000 in milk buffer; p-FMS blots were probed with phospho-M-CSF Receptor (Tyr723) antibody at 1:1000 in TBS + 0.1% Tween 20 + 5% BSA, both overnight at 4 °C. Membranes were washed and probed with goat α-rabbit IgG-HRP at 1:20,000 in milk buffer. Membranes were washed again and developed using SuperSignal Substrate. Chemiluminescence was detected using the ChemiDoc XRS+ System with supercooled CCD camera. Band intensities were measured at ~150-165kDa, the estimated molecular weight for CSF-1R after accounting for glycosylation, and quantified with the Image Lab image acquisition and analysis software (Bio-Rad).
Labeling of antibodies for MSD
The biotinylated p-FMS capture antibody, 4G10 Platinum, Anti-Phosphotyrosine Antibody, did not require additional conjugation. The labeling procedures for the other antibodies required in this protocol utilize reagents that attach their label at primary amines on the antibody. The FMS capture antibody, human M-CSF R mAb (3292), was biotinylated using EZ-Link Biotin reagent at 20:1 molar excess. Excess biotin was quenched with NH4Cl and removed by centrifugal filtration. The detection antibody for FMS and p-FMS, antibody AF329, was Ruthenium labeled using SULFO-TAG™ NHS-Ester at 12:1 molar excess. Free ruthenium was removed by centrifugal filtration. Labeled antibody concentrations were determined by BCA assay.
Immunoprecipitation of MSD Samples
For each sample and control to be assayed, immunoprecipitation was carried out in individual wells of a 96-well round bottom plate. For FMS immunoprecipitation, 50 ng biotinylated Human M-CSF R capture antibody was dispensed per well. For p-FMS immunoprecipitation, biotin-conjugated Anti-Phosphotyrosine capture antibody was diluted 50-fold, and 5 μL was dispensed per well. To each well containing FMS or p-FMS capture antibody, 70 μL of PBMC lysate was dispensed.
To serve as a positive control for FMS and p-FMS expression, cell lysates from FMS-transfected, CSF-1-stimulated HEK293 cells were included. HEK293 cells were transfected with the FMS gene under the CMV promoter, stimulated for 10 minutes with 50 ng/ml of CSF-1, and lysed. Identical cell lysates were created without CSF-1 stimulation to serve as a control for basal level FMS and p-FMS expression. Untransfected HEK293 cell lysates were also generated to serve as a negative control. Lysate controls were diluted in complete lysis buffer to 10 μg /70 μL, and then immunoprecipitated. To establish background for FMS and p-FMS, another negative control consisting of 70 μL of lysis buffer was dispensed into 1 well containing specified capture antibody. Plate was incubated at room temperature for 1 hour.
MSD Assay for FMS and p-FMS
Streptavidin plates were blocked with 100 μL complete lysis buffer at room temperature for 1 hour. The plates were washed and 25 μL of each immunoprecipitated sample and control were dispensed in duplicate. Plates were incubated for 30 minutes at room temperature, with agitation. Plates were washed, 25 μL of ruthenium labeled human M-CSF R antibody was dispensed at 1 μg/mL, and plates were incubated in the dark for 2 hours at 4 °C, with agitation. Plates were washed again, 150 μL of MSD 1 X Read Buffer was dispensed, and plates were read on the SECTOR Imager 6000. Data, captured in relative light units (RLUs), were copied and analyzed using Softmax Pro 4.6 Enterprise software.
Dilution Linearity
2 X 16 mL of whole blood was collected from donors (N=2), in which one set was stimulated with recombinant human CSF-1, and the other set was not stimulated. PBMCs were processed as described previously (PBMC Preparation and CSF-1 Stimulation), and the lysates were resuspended in 180 μL of complete lysis buffer, instead of 360 μL, to get a more concentrated sample for dilutional linearity. Samples were serially diluted (1:2) in complete lysis buffer (90 μL sample mixed with 90 μL complete lysis buffer), and assayed for FMS and p-FMS using the MSD protocol as described earlier.
Data Calculations
For Western Blots, background from the density measured at ~150-165 kDa in the bead control lane was subtracted from band densities measured in p-FMS and FMS blots. For the MSD assay, RLUs from duplicate sample and control wells were averaged; RLUs from the lysis buffer background controls were subtracted from the averaged readings of sample wells. In both assays, the p-FMS values were normalized against the total FMS values by determining the p-FMS/FMS ratio. The resulting p-FMS/FMS ratios for CSF-1 stimulated samples were divided by p-FMS/FMS ratio for unstimulated samples to calculate the fold difference in expression between basal level p-FMS, and p-FMS expression upon CSF-1 stimulation.
Results
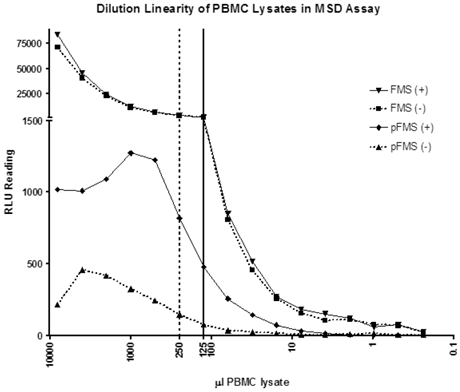
Dilutional linearity using the IP-MSD method to show the minimum volume of whole blood required for minimal matrix effect
A dilution linearity experiment was performed using the IP-MSD method to determine the minimum volume of whole blood required to minimize matrix effects and deliver consistent p-FMS/FMS ratios. As shown by the solid line in Figure 2, RLU counts dilute linearly from 125 μL onwards for FMS, indicating that PBMC lysate prepared from 125 μL of whole blood can accurately measure the FMS portion of the signal. Evident from the figure is that FMS signal is the same and dilutes similarly, whether the samples are stimulated (triangle pointing down) or unstimulated (squares). However, with p-FMS, there is a matrix effect until PBMCs from 1000 μL of whole blood are processed, as shown by the p-FMS stimulated signal (diamonds), and there is a clear difference between stimulated (diamond) and unstimulated (triangle). p-FMS signal evaluated using PBMCs from 250 μL of whole blood can be used to evaluate the ratios as shown by the dotted line in Figure 2. p-FMS dilutes linearly from 500 μL whole blood to 125 μL, after which the RLU counts approach background levels, and signal is too low to be quantified. Therefore, going forward, PBMCs made from 250 μL of whole blood would be sufficient to determine counts for p-FMS and 125 μL would be sufficient for FMS.
Dilutional linearity of PBMC lysates in IP-MSD assay. PBMC lysates were divided into two groups- CSF-1 stimulated (+) and non-stimulated (-). FMS and p-FMS were measured for both the samples using the MSD assay. The samples were sequentially diluted 50% in lysis buffer and each of the diluted samples analyzed in both the FMS and p-FMS assays. The dotted and solid vertical lines display the minimum PBMC lysate volumes (μL) needed for the p-FMS and FMS assays, respectively.
In order to compare the trends seen in the IP-Western assay and IP-MSD assay, a study was designed so that the exact same samples of PBMC lysates were run in both assays. PBMCs were processed from whole blood, as described in 'Materials and Methods' section, and resuspended in 360 μl complete lysis buffer. For the MSD assay, 5 μL was run on the FMS plate and 10 μL was run on the p-FMS plate; 340 μL of PBMC lysate was run by IP-Western.
p-FMS/FMS in IP-Western versus IP-MSD method upon stimulation with CSF-1
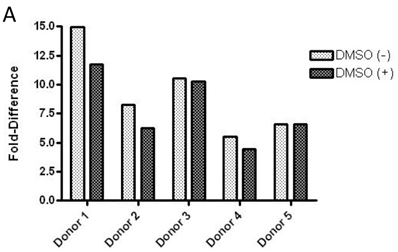
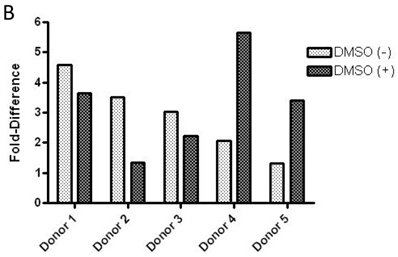
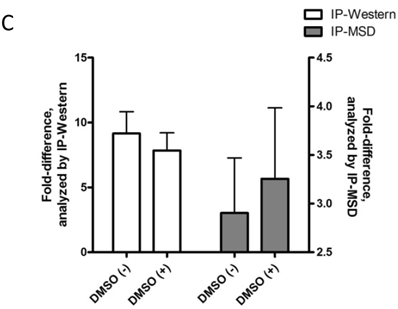
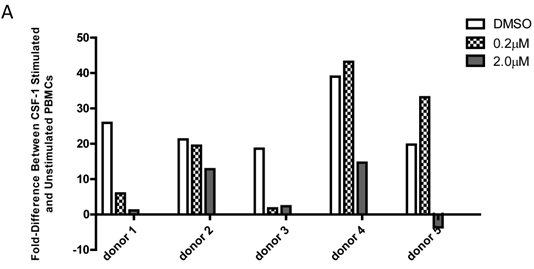
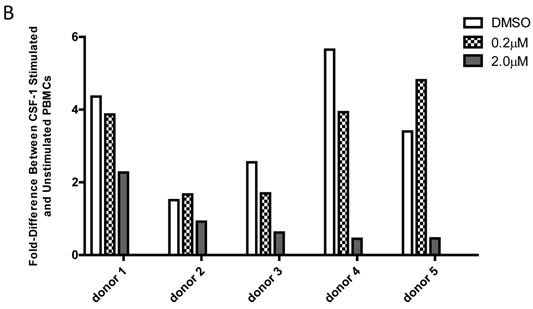
Baseline value of p-FMS cannot be measured unless recombinant human CSF-1 is added to fresh whole human blood, prior to processing PBMCs to stimulate the signal. Band intensity was assayed for both p-FMS and total FMS, and p-FMS intensities were normalized against total FMS intensity in order to compare the signal between CSF-1 stimulated and non-stimulated. An increase in the quantity of p-FMS/FMS was detected upon stimulation, as compared to unstimulated samples. This was referred to as the fold-difference by IP-Western assay, as shown in figure 3A for all donors tested (N=5), ranging from 14.9 - 5.49 in absence of DMSO (average = 9.15) and 11.75 - 4.40 in presence of DMSO (average = 7.84). It was important to measure fold difference in presence and absence of DMSO, since it was the medium used to dissolve the extremely lipophilic drug. Shown in figure 3B, similar trends were observed using the IP-MSD assay ranging from 4.59 - 1.3 1(average = 2.90) fold difference in absence of DMSO and ranging from 5.65 - 1.34 (average = 3.25) in the presence of DMSO. The average fold difference (N=5) as captured in figure 3A and 3B was plotted in figure 3C, where the average IP-Western fold difference was shown on the left y-axis and the average IP-MSD fold difference was shown on the right y-axis. The trends observed were comparable in both assays, but higher fold ratios were observed with the IP-MSD assay.
Comparison of p-FMS/FMS ratios between CSF-1 stimulated and unstimulated samples from whole blood. 3A. IP-Western; 3B. IP-MSD assay; 3C. Comparison of the average values of the five donors, for both conditions, in both assays. Fold difference reflects the effect of CSF-1. PBMCs from whole blood were collected, and samples were divided into two categories: containing DMSO (+) and without DMSO (-). Each sample was further sub-divided into two- stimulated and unstimulated- and for each sample p-FMS and FMS were both assayed. Ratio between p-FMS and FMS was determined for each sample and ratios of p-FMS/FMS between stimulated and unstimulated samples were assessed to indicate the fold difference.
JNJ-DRUG causes inhibition of p-FMS in PBMCs when stimulated with CSF-1, as seen by IP-Western and IP-MSD
JNJ-DRUG causes inhibition of p-FMS in PBMCs when stimulated with CSF-1, as seen by IP-Western and IP-MSD. In order to mimic drug-treated samples, freshly collected whole blood was incubated with varying amounts of JNJ-DRUG prior to inducing p-FMS with CSF-1, as described in the 'Materials and Methods' section. The JNJ-DRUG must be solubilized in DMSO. However, as shown in Figure 3C, DMSO at the concentration used (0.02%) does not interfere with either the IP-Western or the IP-MSD assay. Dose responses to drug treatment are shown in Figure 4.
Comparison of p-FMS/FMS ratios between CSF-1 stimulated and unstimulated PBMCs from whole blood. 4A. IP-Western; 4B. IP-MSD assay. JNJ-DRUG was dissolved in DMSO and a final concentration of 0.0 μM, 0.2 μM, and 2.0 μM was added to whole blood prior to stimulation for all donors (N=5). Comparison of the fold-difference of the five donors, for three conditions, in both assays.
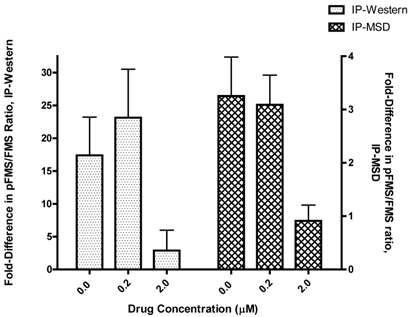
IP-Western results comparison with IP-MSD
In order for the IP-MSD assay to be used, it was important to demonstrate good correlation with the existing IP-Western assay. Since the two assays were based on very different methods, we were only looking to observe similar trends in fold difference upon stimulation with CSF-1, in the presence of increasing concentration of the drug JNJ-DRUG. Therefore, the calculated fold differences in p-FMS/FMS for all of the donors (N=5) for both assays was averaged for 0.0 μM, 0.2 μM and 2.0 μM of JNJ-DRUG (figure 5). Data from all five donors illustrated the same trend, and demonstrated good correlation between the two assays. Data indicated that 0.2 μM was an insufficient drug concentration to inhibit p-FMS/FMS ratio. However, 2.0 μM of JNJ-DRUG was sufficient to inhibit p-FMS/FMS, thereby confirming target engagement at that concentration.
Comparison of p-FMS/FMS between CSF-1 stimulated and unstimulated PBMCs. In order to compare the two assays, fold difference assessed in figure 4 was averaged between the donors (N=5) using the IP-Western and the IP-MSD assay and plotted as shown, with the left y-axis capturing the IP-Western data and the right y-axis capturing the IP-MSD data.
Discussion
In recent years, considerable progress has been made in designing novel assays to understand mechanism of action of drugs by obtaining biopsies, easily accessible bodily fluids e.g. serum/plasma, urine, saliva, etc. , and combining the data with clinical efficacy, so that these drug-target combinations provide a better understanding of the disease. JNJ-DRUG, a novel small molecule inhibitor of CSF-1R or FMS, has been identified as one such compound with high specificity to its target. In order to study and understand the mechanism of action of this compound, it was important to develop a very fast, sensitive, and robust assay that would demonstrate drug to target engagement, thereby inhibiting the downstream cascade of autophosphorylation and subsequent steps. The routinely used IP-Western was a qualitative assay, which required 16 mL of whole blood per time point (8 mL stimulated and 8 mL unstimulated), and lasted three days, beginning with the processing of PBMCs, followed by sample immunoprecipitation, and finally assaying on the Western blot. Described in this paper was a novel semi-quantitative assay, which was more sensitive, required only 750 μL of whole blood (250 μL*2 for stimulated and unstimulated p-FMS and 125 μL *2 for stimulated and unstimulated FMS), took only 4 hours to conduct, and exhibited the same trend as the IP-Western assay.
The IP-MSD assay takes advantage of the first step of immunoprecipitating the lysates with a capture antibody already delineated in the IP-Western method. Following immunoprecipiation step, this antigen-antibody conjugate is captured on a streptavidin MSD plate, which is then detected using a ruthenium labeled antibody in a sandwich format. Several rounds of experiments with various antibody combinations- both as capture and detection- were carried out, varying the concentrations and time of addition (stepwise versus one step) of the antibodies; the time of incubation (1 hour at room temperature to overnight at 4 oC); the temperature of incubation (4 oC or room temperature); cooling the wash buffer to 4 oC prior to washing the plates; and also including or excluding protease and phosphatase inhibitors in the wash buffer. The results of these experiments led to a procedure involving two separate antibodies for p-FMS and total FMS for immunoprecipitation, capture of the antibodies on a streptavidin MSD plate, and finally detection using the same ruthenium labeled antibody for both the assays. The amount of p-FMS captured by RLU was normalized to the RLU counts from total FMS.
With the development of this new IP-MSD assay, it is important to understand that the results from IP-Western and IP-MSD could not be identical matches since the two assays are based on distinct principles, and different antibodies were used for analysis of the samples. It is more important to match trends between the two assays to ensure that the two assays match qualitatively, rather than quantitatively. It has been shown consistently that after stimulation with CSF-1, FMS internalizes and degrades [25]. In the IP-Western assay, the samples were immunoprecipitated and run on an SDS gel, during which all of the degraded or smaller molecular weight FMS proteins are separated, and only the full-length FMS is used for analysis. Therefore, the total FMS measured on the IP-Western is quantitatively less (but more homogenous relative to molecular weight) than the total FMS read by the MSD method. In the MSD method, all of the FMS protein is captured on the streptavidin-MSD plate, whether it is degraded or not, since there is no step to separate the full-length FMS molecules from the smaller, degraded molecules. Hence, when capturing the ratios of p-FMS/FMS, IP-Western always generated a larger ratio than the IP-MSD method. Since IP-Western is a qualitative method and IP-MSD is a semi-quantitative, we are expected to look at similar trends between the assays. Going forward, since the values of total FMS do not change much in presence or absence of stimulation, we propose to measure only p-FMS using the IP-MSD assay and compare the ratios between stimulated and unstimulated.
The authors acknowledge that there are some limitations inherent to this study. These limitations are primarily due to the relatively low number of donors tested (n=5). One difficulty of comparing the IP-MSD assay to the IP-Western assay is that a large volume of very fresh blood (approximately 64 mL) is required from the same individual at the same time point to make the stimulated and unstimulated PBMCs. Even then, the IP-Western assay can be performed only once with the samples, making it difficult to perform multiple runs and calculate error bars. However, several experimental comparisons have shown similar trends between the two assays, which the authors believe show the validity of the IP-MSD assay as a potential replacement for the IP-Western assay. Further platform comparison testing of additional healthy donors as well as diseased patients will be needed to further validate the utility of the IP-MSD assay.
In this paper, we have described a novel semi-quantitative assay to measure p-FMS/FMS in PBMCs derived from whole blood and stimulated with CSF-1. This innovative IP-MSD method is extremely fast, sensitive, and utilizes only 750 μL whole blood, as compared to the 16 mL whole blood used in the IP-Western assay. Using this novel IP-MSD assay, we can easily confirm mechanism of action of JNJ-DRUG with minimal sample, and for all time points in the study. Importantly, for our long term goals, this method can be employed for other drugs as well, and a similar assay can be worked out with the appropriate combination of antibodies.
Acknowledgements
This work has been supported by the Janssen Pharmaceutical Companies of Johnson & Johnson. We would like to thank Carol F. Franks, Stanley Belkowski, and Carl L. Manthey for their advice during assay development.
Conflict of Interest
Authors have declared no conflict of interest.
References
1. Khatami M. Standardizing cancer biomarkers criteria: data elements as a foundation for a database. Inflammatory mediator/M-CSF as model marker. Cell biochemistry and biophysics. 2007;47:187-98
2. Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG. et al. Macrophage polarization in tumour progression. Seminars in cancer biology. 2008;18:349-55 doi:10.1016/j.semcancer.2008.03.004
3. Biswas SK, Sica A, Lewis CE. Plasticity of macrophage function during tumor progression: regulation by distinct molecular mechanisms. J Immunol. 2008;180:2011-7
4. Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. The Journal of pathology. 2002;196:254-65 doi:10.1002/path.1027
5. Pollard JW, Stanley ER. Pleiotropic Roles for CSF-1 in Development Defined by the Mouse Mutation Osteopetrotic. In: (ed.) Paul MW. Advances in Developmental Biochemistry. Academic Press. 1996:153-93
6. Boyce BF, Rosenberg E, de Papp AE, Duong le T. The osteoclast, bone remodelling and treatment of metabolic bone disease. European journal of clinical investigation. 2012;42:1332-41 doi:10.1111/j.1365-2362.2012.02717.x
7. Paietta E, Racevskis J, Stanley ER, Andreeff M, Papenhausen P, Wiernik PH. Expression of the macrophage growth factor, CSF-1 and its receptor c-fms by a Hodgkin's disease-derived cell line and its variants. Cancer research. 1990;50:2049-55
8. Janowska-Wieczorek A, Belch AR, Jacobs A, Bowen D, Padua RA, Paietta E. et al. Increased circulating colony-stimulating factor-1 in patients with preleukemia, leukemia, and lymphoid malignancies. Blood. 1991;77:1796-803
9. Lamprecht B, Walter K, Kreher S, Kumar R, Hummel M, Lenze D. et al. Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nature medicine. 2010;16:571-9 doi:10.1038/nm.2129
10. Filderman AE, Bruckner A, Kacinski BM, Deng N, Remold HG. Macrophage colony-stimulating factor (CSF-1) enhances invasiveness in CSF-1 receptor-positive carcinoma cell lines. Cancer research. 1992;52:3661-6
11. Kacinski BM. CSF-1 and its receptor in ovarian, endometrial and breast cancer. Annals of medicine. 1995;27:79-85
12. Pei XH, Nakanishi Y, Takayama K, Bai F, Hara N. Granulocyte, granulocyte-macrophage, and macrophage colony-stimulating factors can stimulate the invasive capacity of human lung cancer cells. British journal of cancer. 1999;79:40-6 doi:10.1038/sj.bjc.6690009
13. Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer metastasis reviews. 2006;25:315-22 doi:10.1007/s10555-006-9001-7
14. Kawamura K, Komohara Y, Takaishi K, Katabuchi H, Takeya M. Detection of M2 macrophages and colony-stimulating factor 1 expression in serous and mucinous ovarian epithelial tumors. Pathology international. 2009;59:300-5 doi:10.1111/j.1440-1827.2009.02369.x
15. Hercus TR, Broughton SE, Ekert PG, Ramshaw HS, Perugini M, Grimbaldeston M. et al. The GM-CSF receptor family: mechanism of activation and implications for disease. Growth factors. 2012;30:63-75 doi:10.3109/08977194.2011.649919
16. Pixley FJ, Stanley ER. CSF-1 regulation of the wandering macrophage: complexity in action. Trends in cell biology. 2004;14:628-38 doi:10.1016/j.tcb.2004.09.016
17. Yeung YG, Stanley ER. Proteomic approaches to the analysis of early events in colony-stimulating factor-1 signal transduction. Molecular & cellular proteomics: MCP. 2003;2:1143-55 doi:10.1074/mcp.R300009-MCP200
18. Kascinski B. Expression of CSF-1 and its receptor CSF-1R in non-hematopoietic neoplasms. Cancer treatment and research. 2002;107:285-92
19. Ide H, Hatake K, Terado Y, Tsukino H, Okegawa T, Nutahara K. et al. Serum level of macrophage colony-stimulating factor is increased in prostate cancer patients with bone metastasis. Human cell. 2008;21:1-6 doi:10.1111/j.1749-0774.2007.00042.x
20. Wilson KJ, Illig CR, Chen J, Wall MJ, Ballentine SK, DesJarlais RL. et al. Reducing ion channel activity in a series of 4-heterocyclic arylamide FMS inhibitors. Bioorganic & medicinal chemistry letters. 2010;20:3925-9 doi:10.1016/j.bmcl.2010.05.013
21. Kuppers R. Molecular biology of Hodgkin lymphoma. Hematology / the Education Program of the American Society of Hematology American Society of Hematology Education Program. 2009:491-6 doi:10.1182/asheducation-2009.1.491
22. Gorschluter M, Bohlen H, Hasenclever D, Diehl V, Tesch H. Serum cytokine levels correlate with clinical parameters in Hodgkin's disease. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 1995;6:477-82
23. Vener C, Guffanti A, Pomati M, Colombi M, Alietti A, La Targia ML. et al. Soluble cytokine levels correlate with the activity and clinical stage of Hodgkin's disease at diagnosis. Leukemia & lymphoma. 2000;37:333-9 doi:10.3109/10428190009089433
24. Karube K, Ohshima K, Suzumiya J, Kawano R, Kikuchi M, Harada M. Gene expression profile of cytokines and chemokines in microdissected primary Hodgkin and Reed-Sternberg (HRS) cells: high expression of interleukin-11 receptor alpha. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2006;17:110-6 doi:10.1093/annonc/mdj064
25. Li W, Stanley ER. Role of dimerization and modification of the CSF-1 receptor in its activation and internalization during the CSF-1 response. The EMBO journal. 1991;10:277-88
Author contact
![]() Corresponding author: Tel.: 215-628-5206; Fax: 215-540-4763; E-mail: schatur1jnj.com
Corresponding author: Tel.: 215-628-5206; Fax: 215-540-4763; E-mail: schatur1jnj.com