Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
INTRODUCTION
TYPES OF AUTOPHAGY
REGULATION OF MACROAUTOPHAGY
MEASURING THE AUTOPHAGIC FLUX
AUTOPHAGY REGULATING SIGNALING...
THE ROLE OF AUTOPHAGY IN NORMAL...
CONCLUSION AND PERSPECTIVES
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(10):1121-1133. doi:10.7150/ijbs.6666 This issue Cite
Review
The Regulation of the Autophagic Network and Its Implications for Human Disease
Jing Yang1,2, Serena Carra1, Wei-Guo Zhu2, Harm H. Kampinga1 ![]()
1. Department of Cell Biology; University Medical Center Groningen, University of Groningen; Groningen, The Netherlands.
2. Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education); Department of Biochemistry and Molecular Biology; Peking University Health Science Center, Beijing 100191, China;
3. University of Modena and Reggio Emilia, Department of Biomedical, Metabolic and Neuronal Sciences, Giuseppe Campi 287, 41125 Modena, Italy
Received 2013-5-10; Accepted 2013-6-28; Published 2013-12-1
Abstract
Autophagy has attracted a lot of attention in recent years. More and more proteins and signaling pathways have been discovered that somehow feed into the autophagy regulatory pathways. Regulation of autophagy is complex and condition-specific, and in several diseases, autophagic fluxes are changed. Here, we review the most well-established concepts in this field as well as the reported signaling pathways or components which steer the autophagy machinery. Furthermore, we will highlight how autophagic fluxes are changed in various diseases either as cause for or as response to deal with an altered cellular homeostasis and how modulation of autophagy might be used as potential therapy for such diseases.
Keywords: Autophagy, ATGs, mTOR, UPR, FOXO1, HSF1, Heat Shock Proteins (HSP), Human diseases.
INTRODUCTION
Autophagy, as a dynamic and fundamental process, has been reported to play roles in development, differentiation, cell repair, immunologic defense as well as protein quality control. Ensuring the diverse and proper functioning of autophagy in cells has been highlighted as an important aspect for the maintenance of cellular homeostasis. Inversely, failures in autophagic regulation have been associated with many pathological processes, such as muscle atrophy, neurodegeneration, tumor development and aging.
It has been generally accepted that the dynamic autophagic process is mainly executed by a series of proteins, encoded by the so-called AuTophagy related Genes (ATGs). Although the number of ATG(s) is still increasing, the function of most ATG(s) has been well characterized to orchestrate the dynamic process of the formation of pre-autophagosome, the maturation of autophagosome and the fusion with lysosome. As the autophagic process can be affected either by producing autophagosomes or by degrading autophagosomes, the concept of the autophagic flux has been introduced which is generally used as an indicator of the activity of entire autophagic process.
The core machinery of autophagy, consisting of the ATG(s), is regulated by many signaling pathways such as the mTOR pathway, the insulin pathway and the ER stress response. How the signals are transferred to lead to activation of autophagy is still far from being understood and they may occur via both direct interactions with pre-existing ATGs and transcriptionally up-regulation of the ATGs (1-4).
Based on what has been explored about the autophagic network, we review here: 1) the core machinery of ATG(s), 2) the signaling pathways affecting the autophagic flux and 3) the implications of autophagy in human diseases.
TYPES OF AUTOPHAGY
Since the 1960s, autophagy has been gradually recognized as an important catabolic process. The main aim of autophagy is to recycle and reallocate cellular components to satisfy the change of energy requirement or to decrease the accumulation of toxic products. As such, autophagy is responsible for removing long-lived proteins, damaged organelles and protein aggregates. So far, three types of autophagy have been reported, respectively named macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) (see figure 1). The common feature they all share is that the cargo is eventually delivered into lysosomes for degradation. However, the way of cargo delivery is different.
Overview of the different forms of autophagy in mammals. Macroautophagy, microautophagy and chaperone-mediated autophagy are three types of autophagy in mammals. Here, the main steps in these processes as well as the most important characteristic structures and the related mediators are presented.

Macroautophagy is a multi-step and time-consuming journey for cargo wrapping and delivery. A complete process includes membrane initiation, nucleation, elongation (expansion), autophagosome maturation and the fusion with lysosome (see figure 1A). Notably, the double-membrane structure in autophagy is similar to those of other cellular plasma membranes, such as the endoplasmic reticulum, the trans-Golgi network, and mitochondria. The origin of autophagosomal membranes however is still obscure. Because of its complexity, autophagy has to be controlled precisely. On one hand, the main regulation of autophagy requires a specific group of molecules named Autophagy-related genes (ATGs). Until now, more than 30 ATGs have been discovered in yeast (5) and they are highly conserved among eukaryotes (6). On the other hand, there are many signaling pathways involved in macroautophagic induction such as the mTOR pathway, endoplasmic reticulum (ER) stress signals, the insulin pathway, and calcium signaling.
In microautophagy, the cargo is directly engulfed by the lysosome itself. Here, the targeted region of lysosomal membrane undergoes invagination, protrusion and/or septation to enclose the cargo for degradation (7) (see figure 1B). Under both basal and stress conditions, microautophagy has been reported to be involved in energy balance maintenance (8, 9).
Chaperone-mediated autophagy (CMA) uses KFERQ-like sequences in proteins to deliver them into the lysosome (10). The recognition of cargo and its delivery to the lysosome-associated membrane protein type-2A (LAMP-2A) is mediated by the HSPA8 (Hsc70)-containing chaperone complex (11-13) (see figure 1C).
Macroautophagy, the bulky and low-selective form of autophagy is the most investigated type, especially in relation to cellular health and pathology. Therefore, in this review we will mainly focus on the regulation of macroautophagy (hereafter generally referred to as autophagy) as well as its involvement in human diseases.
REGULATION OF MACROAUTOPHAGY
Considering the multiple roles of autophagy in both physiological and pathological processes, better understanding how autophagy is regulated in cells may enlarge our knowledge of the maintenance of homeostasis, as well as facilitate the development of efficient therapies for patients.
Autophagosomal initiation, nucleation and maturation
The precise collaboration of ATGs is essential for performing autophagy successfully. Based on their functions, especially derived from detailed studies in yeast, ATGs have been divided into 6 groups: 1) the ATG1/ULK1 kinase complex, 2) ATG9, 3) the class III phosphatidylinositol (PI)3-kinase complex (ATG6-ATG14-Vps15-Vps34), 4) the PI(3)P-binding ATG2-ATG18 complex, 5) the ATG12 conjugation system (ATG12-ATG5-ATG16), and 6) the ATG8 conjugation system (ATG8-phosphatidylethanolamine (PE)). (See figure 2 and table 1).
The nomenclature and functions of ATGs in yeast and mammals.
| Yeast | Mammals | Functions |
|---|---|---|
| Atg1 | ULK1, ULK2 | Serine/threonine protein kinase required for vesicle formation |
| Atg2 | ATG2A, ATG2B | Forms a complex with Atg18 |
| Atg3 | ATG3 | An E2-like enzyme, which catalyzes the lipidation of Atg8-like proteins (GABARAP, GABARAPL1, GABARAPL2 or MAP1LC3A); which also autocatalyzes itself to form Atg12-Atg3 complex for maintaining mitochondrial homeostasis. |
| Atg4 | ATG4A, B, C, D | Cysteine protease: Atg8 processing and activation. |
| Atg5 | ATG5 | Forms the Atg12-Atg5 complex, which plays E3-like activity for Atg8 lipidation |
| Atg6 | Beclin-1 | Forms the Vps34 PI3K complex |
| Atg7 | ATG7 | An E1 enzyme that mediates Atg8 lipidation and Atg12-Atg5/Atg3 interaction |
| Atg8 | LC3/MAP1LC3A, GATE-16/GABARAP | Modifier, involves in formation of autophagosomal vacuoles through conjugating to PE. |
| Atg9 | ATG9A, ATG9B | Cycles between a juxta-nuclear trans-Golgi network compartment and late endosomes, and interacts with the Atg2-Atg18 complex. |
| Atg10 | ATG10 | An E2-like enzyme that catalyzes the conjugation of Atg12 to Atg5. |
| Atg12 | ATG12 | Conjugated to Atg3 and Atg5 |
| Atg13 | ATG13 | Downstream target of mTOR signaling pathway: the Atg1-Atg13-Atg17-Atg101 complex |
| Atg14 | ATG14 | Component of Vps34 PI3K complex |
| Atg16 | ATG16L | Forms the Atg16L-Atg12-Atg5 complex |
| Atg17 | RB1CC1/FIP200 | Component of Atg1/ULK1 complex: Atg1-Atg13-Atg17-Atg29-Atg31 in yeast, and ULK1/2-Atg13-FIP200-Atg101 in mammals |
| Atg18 | WIPI-1,2,3,4 | Forms the complex with Atg2. |
| Atg29 | ? | Component of Atg1 complex |
| Atg101 | ATG101 | Component if the ULK1/2 complex |
The ATGs uncovered so far as well as ATG functions are listed. Abbreviations: GABARAP is GABA(A) receptor‐associated protein; MAP1LC3A/MAP1LC3B is microtubule‐associated proteins 1A/1B light chain 3. ATG16L: autophagy related Atg 16‐like; RB1CC1 is RB1‐inducible coiled‐coil 1. FIP200 is 200 kDa FAK family kinase‐interacting protein. WIPI‐1,2,3,4 is WD repeat domain, phosphoinositide interacting 1.
Overview of the main steps in autophagy and the main genes involved.
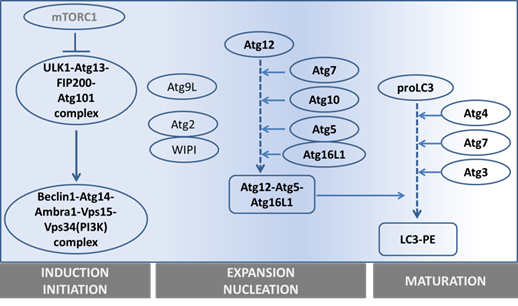
1) Autophagy is initiated by forming a lipid bilayer, which is either newly synthesized or budded from existing membranes in the cytoplasm (14). In Saccharomyces cerevisiae, the initiation site is defined as the pre-autophagosomal structure (PAS) (15). The serine/threonine-protein kinase ATG1 containing complex, supported by ATG13-FIP200-ATG101 (16-18), works as the most upstream component of the core autophagy machinery, especially after starvation (19). Unc-51 like kinase 1 and 2 (ULK1 and ULK2) are the mammalian homologues of ATG1. In yeast, ATG1 forms a complex with ATG13, ATG17, ATG29 and ATG31. However, the mammalian ULK complex has different components, including ULK1/2, ATG13, FIP200 and ATG101.
2) To facilitate membrane transport for the expansion step, ATG9, the autophagy-specific phosphatidylinositol 3-kinase (AS-PI3K) complex, ATG2-ATG18 and ATG12-ATG5-ATG16 are required. ATG9, the sole integral membrane protein, facilitates autophagic membrane transport (20).
3) The components of the AS-PI3K complex are different in yeast and mammals. In yeast, there are two PI3K complexes, ATG30-ATG14-Vps15-Vps34 and ATG30-Vps38-Vps15-Vps34, which respectively function in autophagy and vacuole protein sorting (21, 22). Among these components, Vps34 is the one known to have PI3K activity. Similarly, the mammalian homologue of Vps34, also known as PI3K class III (PI3KC3), is involved in both autophagy and endosomal trafficking. The mammalian AS-PI3K complexes are more elaborate than in yeast. They consist of Vps15, Vps34, ATG14, Beclin-1, UVRAG, Rubicon and probably more components (23, 24). Among them, Beclin-1 (the mammalian homologue of ATG6) is the crucial one differentiating the autophagic PI3KC3 complex from the endosomal one, and it is essential for autophagosome formation (25). Beclin-1 can either up-regulate or down-regulate autophagic activity through binding with different partners. Ambra1, Bif-1, and VMP-1 are Beclin-1 partners that activate autophagy, whereas, its partners Bcl-2, ICP34.5 and M11 inhibit autophagy (26-28).
4) The ATG2-ATG18 complex is required for nucleation. Especially its PtdIns(3)P-binding ability is critical for the complex to localize to autophagic membranes, endosomes or vacuolar membrane (29).
5) The ATG12-ATG5-ATG16 complex mediates ATG8 lipidation (see next paragraph). The activating process of ATG12 shares similarities with the ubiquitin-ligation process (consisting of an E1-ubiquitin activating enzyme and E2-ubiquitin-conjugating enzymes): ATG7, as E1-like protein, firstly catalyses ATG12 to become an active form, and next ATG10 (an E2-like protein) facilitates the active ATG12 to bind with ATG5. ATG12-ATG5 further assembles into a 350kD complex by interacting with the small coiled-coil protein ATG16 which is essential for autophagy (30).
6) The ATG8 conjugation to phosphatidylethanolamine (PE) is crucial for autophagosomal expansion and maturation. Therefore, ATG8 or its mammalian homologue, LC3, is generally used as a marker to trace the autophagic process (see next section) (31, 32). To fulfill ATG8-PE conjugation (ATG8 lipidation), two possible routes can be used. One is via assisted assembly by the ATG12-ATG5-ATG16 complex mentioned above (33); another is via processing LC3 itself. Newly synthesized LC3 is firstly cleaved by ATG4 to become a cytosolic form (LC3 I), then a fraction of LC3I is further activated by ATG7 (an E1-like protein) (34) and ATG3 (an E2-like protein) (35) to become LC3II (the membrane-binding form). Even though the core machinery of autophagy has been rolled out as being complex already, the number of components discovered to be involved in this network still increases (36).
MEASURING THE AUTOPHAGIC FLUX
The activity of autophagy is controlled by autophagosomal formation (input) and autophagosomal degradation (output). Therefore, the speed of the autophagosomal turnover is defined as autophagic flux, which is the real indicator of autophagic activity. How to measure the autophagic flux has been excellently described elsewhere (37).
Here, we only shortly summarize the markers and inhibitors that are most commonly used.
1) As mentioned above, LC3 lipidation is essential for autophagy to proceed. This lipidation of LC3 can be demonstrated by western blot analysis (WB) (38). Upon an increase in autophagosomes, the band of LC3 will shift from LC3I (18kDa) to LC3II (16kDa) on SDS-PAGE. Upon time after autophagic activation, levels of LC3II will decline again because it is co-degraded with the autophagic cargo in lysosomes.
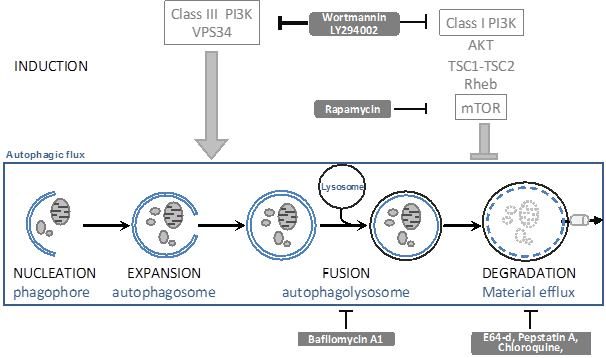
Whilst LC3 data are indicative of a changed autophagic flux, they cannot distinguish whether more autophagy is induced or whether autophagosomal-lysosomal fusion is impaired, unless accurate kinetics are studied. To distinguish between these possibilities, one can block either the synthesis or degradation of LC3II using autophagic inhibitors (see figure 3). Wortmannin and LY294002 can inhibit the autophagosome formation: if LC3 is still lipidated by the experimental condition in the presence of these inhibitors, this implies that autophagosomal-lysosomal fusion was impaired. Bafilomycin A1 and the inhibitors of lysosomal proteases (such as E64, pepstatin A, chloroquine) stop LC3II degradation, This in itself will further increase LC3II levels; if under the experimental condition LC3II levels even further increase, this then implies that the condition has led to an increased autophagosome formation.
The autophagic flux and generally used inhibitors. Inside the square, the autophagic flux is depicted. This is a dynamic process that can be modulated by the indicated drugs (grey text boxes) by affecting autophagosome induction and expansion (ON-rate) or by affecting autophagosome fusion or degradation (OFF-rate).
2) p62/SQSTM1 is thought to be responsible for cargo selecting and transport of proteins or protein aggregates (not organelles), and is also co-degraded with its cargo in lysosomes (39). Thus, p62 degradation can also be used to measure the autophagic flux, which should then be delayed by bafilomycin A1 and the inhibitors of lysosome proteases.
Besides measuring LC3II and p62 biochemically, one can also use microscopic analyses as they form characteristic puncta. The number of these puncta can be counted as (semi) quantitative analyses of autophagy.
3) Still the best way to analyze the induction of autophagy and to demonstrate that the observed puncta/vesicles are indeed autophagosomes is by using Electron Microscopy(EM) in which it can be recognized as a vesicle surrounded by a double membrane that contains cytosol, protein aggregates, and/or organelles that look morphologically intact.
AUTOPHAGY REGULATING SIGNALING PATHWAYS
Autophagy is regulated by many signaling pathways. The most established ones of these are discussed below.
The mTOR pathway
The mTOR (mammalian target of rapamycin) serine/threonine kinase is a well-characterized and critical node of cell metabolism (40) and (besides controlling autophagy) mainly regulates ribosomal biogenesis and protein translation (41). The mTOR pathway is the most extensively studied network for autophagy regulation because of its ability to timely sense the nutrient state, growth factors availability and stress (42). It is still unclear how mTOR itself is regulated, although the phosphatidylinositol 3-kinase (PI3K)-AKT/protein kinase B (PKB) (43), TSC1/TSC2 (44, 45), Rheb (Ras homolog enriched in brain) (43), PRAS40 (46) and DEPTOR (47) have been found as its regulators under different conditions.
So far, two distinct mTOR complexes have been found in mammals (see figure 4).
Regulation of the two mTOR complexes. mTORC1 is able to sense the amino acid concentration and energy state directly, but needs the PI3K-AKT axis to sense growth factor status. The downstream effects of mTORC1 are mainly mediated by 4E-BP1 and S6K1, which can be inhibited by the rapamycin-FKBP12 complex. mTORC2, as a brake-like inhibitor, decreases the extent of AKT-induced mTORC1 activation.
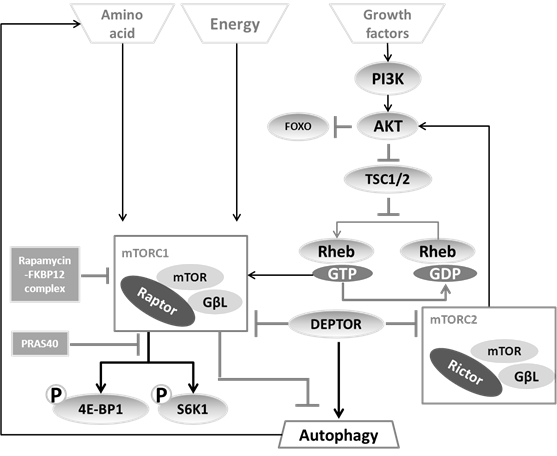
(i) The first complex, mTORC1, includes mTOR, the mammalian LST8/G-protein β-subunit like protein (mLST8/GβL), and the regulatory-associated protein of mTOR (Raptor). Based on the observation that the FKBP12-rapamycin complex can destabilize the mTOR-GβL-Raptor complex (41), rapamycin has been generally used as a mTORC1 inhibitor leading to autophagy induction (48, 49). As substrates of mTORC1, S6K1 (50) and 4E-BP1 (51) can be modulated to change the recruitment of ribosomal subunits as well as the efficiency of cellular translation, thereby balance the protein synthesis to fit with cellular requirement for energy. For this reason, the phosphorylated state of S6K1 and 4E-BP has been considered as a convenient indicator of mTORC1 activation (52).
(ii) The second complex, mTORC2 consists of mLST8/GβL and Rictor (rapamycin-insensitive companion of mTOR). This branch regulates the Akt-FOXO axis and the PKCα pathway, but not S6K1 or 4EBP1 (53). mTORC2 has been considered as rapamycin-insensitive complex (54). Nevertheless upon prolonged treatment, rapamycin also impedes on mTORC2 (55).
Calcium signaling pathway
Calcium signaling is another generally studied, but still obscure pathway by which autophagy can be regulated. In cells, a stable calcium concentration is maintained by Ca2+ channels through two independent but closely linked processes (56): 1) a slow, sustained entry of extracellular calcium, and 2) a rapid, transient release of calcium from stores in the ER. To replenish or maintain Ca2+ stores, Voltage-Operated Channels (VOCs), Receptor-Operated channels (ROCs) and Store-Operated Channels (SOCs) are required (56, 57). Importantly, Ca2+ release is only mediated by inositol 1,4,5-trisphosphate (InsP3/IP3) receptors (IP3Rs) in the ER that thus play a critical role in transient Ca2+ signaling (56). Not surprisingly, IP3R and TRPML3 (a member of the TRPML subfamily of the transient receptor potential (TRP) channel superfamily) have been shown to respectively inhibit and promote autophagy (58, 59). In line with this, it has been also shown that the expanding ER is a potent stimulator of autophagy (60).
Importantly, the concentration of calcium directly links with the activity of calcium-sensitive kinases and proteases. On one hand, Calmodulin-Dependent Kinase Kinase-β (CaMKKβ) (61), elongation factor-2 kinase (eEF-2 kinase; Ca2+/calmodulin-dependent kinase III) (62) and AMP-activated protein kinase (AMPK) (63) will be activated by abnormal cytoplasmic concentrations of calcium under stressed conditions. Each of these upstream sensors will immediately transmit the signals to downstream substrates for adjusting cellular metabolism in new microenvironment and autophagy will be activated to release more amino acids under these conditions. On the other hand, higher Ca2+ concentration increases activity of calpain which can cleave ATG5, thereby inhibits autophagosome formation (64).
The Unfolded Protein Response
Except being an important regulator of cytosolic calcium concentration, the ER is the main cellular protein factory that synthesizes membrane bound and secretory proteins (65). Protein homeostasis in the ER is controlled by the Unfolded Protein Response (UPR) that consists of three parallel branches: activating transcription factor 6 (ATF6), double-stranded RNA-activated protein kinase (PKR)-like ER kinase (PERK), and inositol requiring enzyme 1 (IRE1). These branches serve to not only increase the folding capability of the ER (through induction of chaperones), but also enhance the degradation capability. This involves both ER-associated proteasomal degradation (ERAD) as well as autophagy (see figure 5) (66).
The different branches of the Unfolded Protein Response (UPR) and autophagy. The UPR is mediated by the ATF6, PERK, and IRE1 pathways. 1) After cleavage, ATF6 is able to up-regulate the expression of chaperones. 2) PERK is responsible for eIF2α activation. Activated, phosphorylated eIF2α-p then inhibits translation initiation, meanwhile stimulates ATF4 to transcriptionally activate CHOP and Atg12. In addition, ATF4 also activates GADD34 to decrease the activity of eIF2α-p, which forms a feedback loop. 3) IRE1 cleaves XBP1 to increase the expression of chaperones as well as speed up ERAD.
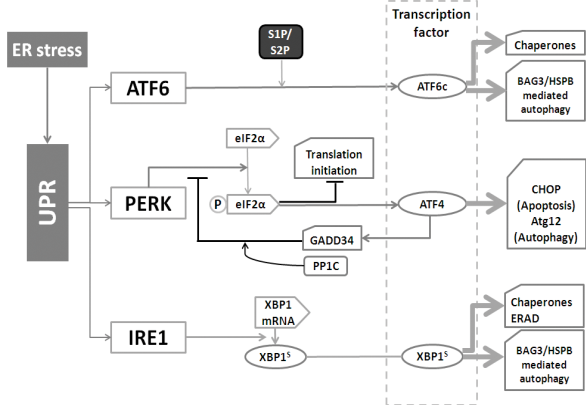
(i) IRE1 (an endoribonuclease, presents in the ER membrane) is the most conserved branch of the UPR (67). Once activated, IRE1 cleaves the mRNA of XBP1 (X-box binding protein 1). This step produces XBP1s (the spliced mRNA) which transcriptionally up-regulates UPR related genes. Generally, IRE1 is responsible as the driving force of the entire UPR gene program(68). XBP1 also up-regulates the BAG3/HSPB8 complex (69), which recently has been found to be involved in the induction of autophagy (70).
(ii) ER located ATF6 can be cleaved by site-1 and site-2 protease (S1P and S2P) to yield a cytosolic fragment (ATF6c) that can translocates into the nucleus for up-regulating UPR genes, such as BiP (the ER-resident chaperone of the heat shock protein HSP70 family), protein disulfide isomerase (PDI) and GRP94 (glucose-regulated protein 94, the ER-resident chaperone of the Hsp90 family) (71). Whilst the ATF6 pathway primarily serves to induce the expression of ER chaperones, ATF6 can also up-regulate the autophagy-inducing BAG3/HSPB8 complex under certain conditions (Minoia et al., unpublished), indicating that also this UPR-branch can increase the autophagic flux.
(iii) The 3rd UPR branch, PERK, has as main function to decrease the burden of newly synthesized ER proteins. PERK phosphorylates and inactivates translation initiation factor 2α (eIF2α), which causes a general block in mRNA translation. Meanwhile, ATF4 is specially induced by eIF2α through recognizing its 5׳-untranslated region, allowing the selective up-regulation of transcription factor C/EBP homologous protein (CHOP) and the activation of growth arrest and DNA damage-inducible 34 (GADD34). GADD34 then plays as a negative feedback loop for the response essential for cell survival (72, 73). Interestingly, however, ATF4 is also involved in autophagy activation as it leads to the induction of ATG12 (74).
Insulin (IS) pathway
As one of the most important signaling pathways in cell metabolism, the Insulin/IGF (insulin-like growth factors) axis is responsible for promotion of cell proliferation and inhibition of cell death, which are mediated mainly by the PI3K-AKT/PKB pathway and the Ras-mitogen-activated protein kinase (MAPK) pathway (75-77). The Insulin/IGF axis has many substrates, two of which being the forkhead box O class transcription factors (FOXOs) and the heat shock transcription factor 1 (HSF1) (78). Whilst the FOXO pathway has in various ways been linked up with protein degradation systems, especially autophagy (1, 79), the HSF1 branch of this network has been mainly thought to be involved in enhancing the protein folding capability of cells (80). Yet, also HSF1 activity can lead to enhancement in autophagic activity (see below).
Most of the pioneering studies on FOXO and HSF1 were done in C. elegans. Both DAF16 (FOXO homologue in C. elegans) and HSF1 were found to be crucial for the downstream effects of the insulin pathway on life span and the ability to handle diseases associated (misfolded) proteins (81-84). Below, we will shortly summarize those data.
(a) FOXO
While there is only one DAF-16 in C. elegans and one dFOXO in flies, the mammalian FOXO family includes 4 members, FOXO1/FKHR, FOXO3/FKHRL1, FOXO4/AFX, and FOXO6 (85). FOXO6, expressed in a specific temporal and spatial pattern, is believed to be mainly involving in brain development (86). Currently, FOXO1, FOXO3 and FOXO4 are known to share the most elementary and conserved sequence motifs as well as modification features (87): 1) the DNA-binding domain (or forkhead (FKH) domain), which recognizes (G/C)(T/C)AAA(C/T)AA or TT(G/A)TTT(G/A)(G/C) in the promoter of target genes in mice; 2) the three AKT1-phosphorylation sites (Thr24, Ser256, and Ser319 in human FOXO1) that mediate nuclear-cytoplasm shuttling (88, 89). Generally, the members of the family can compensate for each other (90). However, in knock-out mice different FOXOs do show some specific functions and expression patterns: FOXO1 in vascular development, FOXO3 in ovarian follicular development, and FOXO4 without any distinguishable phenotype(91).
Nuclear (active)-Cytoplasm (inactive) shuttling is a critical and generally accepted model for regulating the transcriptional activity of FOXOs. Except for the PI3K-AKT/PKB pathway that mediates the nuclear export of FOXO, many other upstream modulators do in parallel affect the location of FOXOs, including various kinases and phosphatases (SGK, CK1, AMPK, JNK, MST1, IKKβ, DYRK1, CDK2, and PP2A), acetylases and deacetylases (CBP/P300, PCAF, SIRT1/2) and ubiquitinases and deubiquitinases (USP7/HAUSP, and Skp2)(87, 92). Irrespective of how FOXOs are activated, the consequence will be a change in the expression profile of its large set of target genes that regulate many cellular processes including autophagy (93) (see table 2). In line, alterations in FOXOs activity have also been related to many pathological processes, such as diabetes, cancer, infertility, cardiovascular disorder, immune system dysfunction, neurodegeneration and ageing (94-97).
The downstream targets of FOXOs.
| Target genes (ref) | Cellular processes | Consequences |
|---|---|---|
| p21Cip1, p27Kip1 (98) | Cell cycle | Oncogenesis |
| FasL, Bim, Bnip3 (98) | Apoptosis | |
| LC3, Gabarapl1, Vps34, ULK2, and ATG12 (99) | Autophagy | Longevity Suppression of neurodegeneration |
| Catalase, Sod2, Gadd45, DDB1 (98), caveolin-1 (100) | Stress resistance | |
| Sprouty2, PBX1 (90), Cited2 (101) | Angiogenesis | Differentiation |
| BTG1, KLF4 (102), | Differentiation | |
| NPY (neuropeptide Y ), Agrp (Agouti-related protein) (103), Pomc (104) | Food intake | Metabolism |
| Atrogin-1, MuRF1 (105) | Ubiquitin-proteasome | Muscular Degeneration |
Regarding the role of FOXOs in autophagy two models of action have been described. In line with its function as transcription factor, FOXO3 can induce autophagy through transcriptionally up-regulating LC3, Gabarapl1, Vps34, ULK2 and ATG12(99). FOXO3 induced autophagy entirely seems to depend on FOXO1 since lack FOXO1 prevents FOXO3's function in autophagy (106). Surprisingly, however, FOXO1 (also) can boost the autophagic flux in a transcription independent manner by direct binding to ATG7 in the cytosol (1). This novel and unprecedented action of FOXO1 might explain to some extent why DAF-16 mediates such a large life span expansion and leads to pronounced protection against neurodegenerative protein aggregation diseases in C. elegans (107, 108) (also see below).
(b) HSF1
As another downstream target of the Insulin/IGF axis, HSF1 is essential for development and stress resistance (109). HSF1 is known as the major factor involved in the up-regulation of heat shock proteins serving to restore proteostasis in the cytosol (81, 110). Under stress conditions, HSF1 is thought to release from the Hsp90 docking complex that normally keeps it inactive under non-stress conditions, and may undergo some structural changes by not yet elucidated mechanisms. Then, HSF1 translocates into the nucleus where it gets hyperphosphorylated and binds to HSE in promoter of target genes (mainly HSPs). Next, the acetylation of HSF1 seems required for its release from DNA and the turn-off of gene expression (111).
In mammalian cells, HSPs are divided into HSPC (HSP90), HSPA (HSP70), DNAJ (HSP40), HSPB (small HSP) and chaperonins (CCT, GroES/EL) (112). Whilst HSF1 regulated HSPs generally seem to serve in either protein folding or proteasomal degradation, recent evidence has suggested that some of its target HSP genes are also linked to autophagy. E.g. Carra et al demonstrated that HSPB8 in complex with BAG3 and Hsp70 can increase the autophagic flux and thereby enhance the clearance of protein aggregates which arise during neurodegenerative diseases (70, 113) (see figure 6). Except as UPR target genes (see figure 5), HSPB8 and BAG3 can also be induced by activation of HSF1 (114). Another small HSP, HSPB7, also serves in autophagic clearance of model aggregation putatively through improving loading of cargo (the aggregates) into autophagosome (115). How HSPB7 is regulated, however, is still unknown.
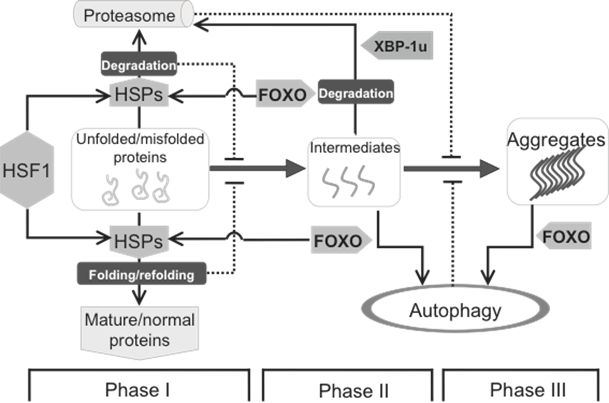
Protein homeostasis is mainly maintained by HSF1 and FOXO, two downstream targets of the insulin pathway. The formation of biochemistry-detectable aggregates is a dynamic process, which is initiated by accumulation of unfolded/misfolded proteins (Phase I), facilitated by the formation of intermediates (Phase II) leading to the appearance of detectable aggregates (Phase III). This process can be intervened by different cellular strategies, including HSPs mediated folding/refolding, as well as proteasome- and autophagy-mediated degradation. HSPs, up-regulated by HSF1 and FOXO, are mainly responsible for Phase I, in which the misfolded proteins can be either refolded to become functional normal proteins or degraded if refolding is unsuccessful. Once the intermediates are formed, autophagy starts playing a more important role. At the beginning of Phase II, the intermediates can still be handled by either HSPs or proteasome, and this response supposedly requires FOXO. At both Phase II and III, FOXO is also involved the induction of autophagy to clear large(r) protein aggregates.
THE ROLE OF AUTOPHAGY IN NORMAL PHYSIOLOGY AND DISEASE
Physiological roles of autophagy
Autophagy has mostly been studied as a mean of cells to cope with acute or chronic stresses, where metabolism needs to be adjusted rapidly to fit with the changed microenvironments (116-118). However, autophagy is also indispensable under normal-growth conditions (64, 119) and the physiological roles of autophagy include:
(a) Nutrients recycling
Through degradation of cellular materials, autophagy facilitates an efficient and dynamic energy conversion of amino acids, glucose, lipids and nuclear acid, especially under conditions of starvation (118, 120, 121).
(b) Cell self-Repair
Autophagy is required for removal of damaged mitochondria (mitophagy), peroxisomes (pexophagy) and protein aggregates (aggrephagy) as arise during normal cell growth (39, 122).
(c) Protein quality control
Protein quality control is mastered accurately by balancing protein synthesis, protein folding and protein degradation. Unlike steric considerations for proteasomal degradation, autophagy shows more general or non-selective property. Autophagic and proteasomal degradation are closely linked with each other, which is exemplified by a boost of autophagy after proteasome inhibition (123, 124).
(d) Cell maturation and differentiation
As evidenced from the phenotype in ATG knock-out mice, autophagy also affects differentiation (125). Similarly, in Drosophila, autophagy is essential for remodeling of the larval mid-gut as well as the development of the larval salivary gland (126).
(e) Infection and immunity
Autophagy is involved in both bacteria-related and virus-related infectious processes (127-129). In addition, Beclin1/Atg6 mediated autophagy impedes on lymphocyte development (130).
(f) Cell death
Controlling cell fate properly is important for development, differentiation and tumor suppression. Autophagy initially provides the adaptive response to micro-environmental change or stress. However, if the stress persist or the amount of inflicted damage goes beyond of cellular tolerance, exaggerated autophagy will lead to cell death and thus remove the damaged cells to maintain tissue and organismal integrity. As such, autophagy parallels apoptosis which also primarily serves as a cell remodeling process under stresses, but if over-activated helps cell elimination (131).
Autophagy and diseases
Deregulation of autophagy has been linked with loss of cell function leading to organismal pathologies (132, 133). In many cases, it is not clear whether deregulation of autophagy itself is the trigger for such pathologies or just a mere consequence and reflection of alterations in the amount of cargo for the autophagic degradation. Below, a few links between altered autophagy and diseases will be summarized.
(a) Tumor development
Tumorigenesis is a multi-step process, involving oncogenes and tumor suppressor genes. Several of these genes, e.g. p53, FOXOs, TOR, SirT1/2, Bcl-2, PTEN, Ras, NF-κB and series of micro RNAs, also have effects on autophagy (134). On one hand, autophagy could directly play a role in tumor suppression. Firstly, mutations in or deletion of Beclin-1, which is the essential ATG for autophagy initiation are found in 40-75% of cases with human breast, ovarian or prostate cancer (25). Even more so, deficiency of Beclin-1 enhances tumorigenesis of lymphomas, lung carcinomas, hepatocellular carcinomas and mammary precancerous lesions in mice (135). Secondly, FOXO1, except as transcription factor to induce the expression of tumor suppression genes, alternatively can bind to ATG7 to activate the autophagic cell death, hereby antagonizing tumor development (1). Why and how this FOXO1 related function is silenced during tumorigenesis is yet unclear. However, it was recently found by Zhao et al (136) that the unspliced form of XBP-1(XBP-1u) can promote ubiquitin-independent proteasomal degradation of FOXO1 and may thus be considered as an oncogene stimulating tumor initiation.
On the other hand, autophagy can be up-regulated to support tumor development, e.g. under hypoxic conditions, tumor cells require autophagy to cope with the low nutrient availability (137). In addition, autophagy may provide tumor cells with resistance to chemotherapy (138).
So, it seems that impaired autophagy can both enhance and lower the risk of tumorigenesis (139). Which of these effects prevail likely is dependent on the stage of tumor, tumor physiology and the tissue from which the tumor originated.
(b) Liver diseases
The liver, the most important tissue for organismal metabolism, is responsible for: 1) amino acid metabolism (synthesis and degradation), 2) glucose metabolism (gluconeogenesis, glycogenolysis and glycogenesis), 3) lipid metabolism (cholesterol synthesis, lipogenesis and lipoproteins synthesis), 4) blood cell regeneration, blood ingredient replenishment and coagulation reaction, 5) endocrine system homeostasis (secretion of IGF-1, breakdown of insulin and other hormones), 6) detoxification (endogenous toxic metabolic products and exogenous medicine) and 7) completion of immunological effects. Not surprisingly, autophagy is a critical process in hepatocytes, and abnormal autophagy has been observed in many pathological liver-related disorders, such as insulin resistance, nonalcoholic steatohepatitis (NASH), cirrhosis, viral hepatitis, hepatic steatosis, mutant α1-Antitrypsin Z protein related diseases and liver cancer (140). What kind of role autophagy is playing in these diverse liver pathologies is still largely unknown. Recently, we revealed that in polycystic liver disease (PCLD), autophagy is activated by the deficiency of hepatocystin, of which mutants have been shown to cause PCLD (141). Therefore, in PCLD, autophagy inhibition might be a strategy to delay pathogenesis.
(c) Brain and neurodegenerative diseases
Autophagy has been shown as an essential process for the maintenance of neuronal function (124). This theory is supported by the observation that in neurodegeneration such as Alzheimer's, Parkinson's, Huntington's disease and amyotrophic lateral sclerosis (ALS) protein aggregates have accumulated indicative of disturbed proteostasis, including impaired autophagy. Indeed, several lines of evidence support that boosting autophagy in such diseases may have beneficial effects (142-144). However, over-stimulating autophagy has the risk to become cytotoxic and pro-degenerative. Therefore, besides increasing the autophagic flux, improving cargo (protein aggregate) delivery into autophagosomes seems a promising strategy to counteract neurodegeneration. This might be accomplished by e.g. stimulating proteins like Alfy (145), or boosting certain HSP like HSPB7 (115, 146).
CONCLUSION AND PERSPECTIVES
Autophagy is not merely a strategy of cellular catabolism to recycle proteins and other nutrients, but also responsible for many other cellular functions, such as cellular quality control which requires proper clearance of misfolded proteins, damaged organelles, and sometimes ensuring clearance of damaged cells. Although the regulation of the autophagic network has been comprehensively investigated in the recent decades, it is still controversial whether a dysfunctional autophagy is actually the cause for a disease or just an indicator of upstream protective response that has failed to protect cells and tissues from an injury. Nevertheless, boosting the autophagic flux has shown the significant benefits in various disease models (116, 119, 132, 143), implying that its activity at least does influence progression of certain diseases. How to modulate the autophagic network precisely to do so without impeding on normal cellular physiology, i.e. avoiding toxicity, will however remain a major challenge. Precise knowledge on the many different signaling pathways that are involved in the regulation of the autophagic network will therefore be crucial and require co-induction of upstream protective pathways such that damage clearance is also accompanied by preventing further damage. As such, the coordinated inhibition of the insulin pathways probably sets the best example. Here, longevity and stress resistance seems is not associated with induction of autophagy, but also with an increase in molecular chaperones, and improved proteolytic activity (figure 6) (147, 148).
Acknowledgements
This work was supported by the sandwich PhD program of the University Medical Center in Groningen (awarded to Dr. J. Yang), a 111 project from the Ministry of Education of China awarded to Dr. W.G. Zhu, the Rita Levi Montalcini Prize (2011) awarded to Dr. S. Carra, and a grant from Senter Novem (IOP-IGE07004) awarded to Dr. H. H. Kampinga.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S. et al. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol. 2010;12(7):665-75
2. Nakatogawa H, Ohbayashi S, Sakoh-Nakatogawa M, Kakuta S, Suzuki SW, Kirisako H. et al. The Autophagy-related Protein Kinase Atg1 Interacts with the Ubiquitin-like Protein Atg8 via the Atg8 Family Interacting Motif to Facilitate Autophagosome Formation. J Biol Chem. 2012;287(34):28503-7
3. Backues SK, Lynch-Day MA, Klionsky DJ. The Ume6-Sin3-Rpd3 complex regulates ATG8 transcription to control autophagosome size. Autophagy. 2012;8(12):1835-6
4. Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J. et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465(7300):942-6
5. Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10(7):458-67
6. Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9(10):1102-9
7. Kunz JB, Schwarz H, Mayer A. Determination of four sequential stages during microautophagy in vitro. J Biol Chem. 2004;279(11):9987-96
8. Dubouloz F, Deloche O, Wanke V, Cameroni E, De Virgilio C. The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol Cell. 2005;19(1):15-26
9. Mortimore GE, Lardeux BR, Adams CE. Regulation of microautophagy and basal protein turnover in rat liver. Effects of short-term starvation. J Biol Chem. 1988;263(5):2506-12
10. Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. 1990;15(8):305-9
11. Schatz G, Dobberstein B. Common principles of protein translocation across membranes. Science. 1996;271(5255):1519-26
12. Agarraberes FA, Dice JF. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci. 2001;114(Pt 13):2491-9
13. Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273(5274):501-3
14. Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol. 2010;12(9):831-5
15. Suzuki K, Ohsumi Y. Current knowledge of the pre-autophagosomal structure (PAS). FEBS Letters. 2010;584(7):1280-6
16. Noboru M. The role of the Atg1/ULK1 complex in autophagy regulation. Current Opinion in Cell Biology. 2010;22(2):132-9
17. Kim J, Kundu M, Viollet B, Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132-41
18. Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy. 2009;5(7):973-9
19. Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284(18):12297-305
20. He C, Song H, Yorimitsu T, Monastyrska I, Yen W-L, Legakis JE. et al. Recruitment of Atg9 to the preautophagosomal structure by Atg11 is essential for selective autophagy in budding yeast. The Journal of Cell Biology. 2006;175(6):925-35
21. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11(5):329-41
22. Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152(3):519-30
23. Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT. et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11(4):468-76
24. Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 Forms Two Distinct Phosphatidylinositol 3-Kinase Complexes with Mammalian Atg14 and UVRAG. Molecular Biology of the Cell. 2008;19(12):5360-72
25. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100(25):15077-82
26. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N. et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927-39
27. Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W. et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1(1):23-35
28. Sinha S, Colbert CL, Becker N, Wei Y, Levine B. Molecular basis of the regulation of Beclin 1-dependent autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy. 2008;4(8):989-97
29. Obara K, Sekito T, Niimi K, Ohsumi Y. The Atg18-Atg2 Complex Is Recruited to Autophagic Membranes via Phosphatidylinositol 3-Phosphate and Exerts an Essential Function. J Biol Chem. 2008;283(35):23972-80
30. Kuma A, Mizushima N, Ishihara N, Ohsumi Y. Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J Biol Chem. 2002;277(21):18619-25
31. Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. Embo J. 2010;29(11):1792-802
32. Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3(6):542-5
33. Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T. et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282(52):37298-302
34. Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I. et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169(3):425-34
35. Hanada T, Satomi Y, Takao T, Ohsumi Y. The amino-terminal region of Atg3 is essential for association with phosphatidylethanolamine in Atg8 lipidation. FEBS Lett. 2009;583(7):1078-83
36. Chen Y, Klionsky DJ. The regulation of autophagy - unanswered questions. J Cell Sci. 2011;124(Pt 2):161-70
37. Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007;3(3):181-206
38. Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1(2):84-91
39. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H. et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131-45
40. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124(3):471-84
41. Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17(6):596-603
42. Jung CH, Ro S-H, Cao J, Otto NM, Kim D-H. mTOR regulation of autophagy. FEBS Letters. 2010;584(7):1287-95
43. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926-45
44. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4(9):648-57
45. Huang J, Dibble CC, Matsuzaki M, Manning BD. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol Cell Biol. 2008;28(12):4104-15
46. Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E. et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25(6):903-15
47. Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM. et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137(5):873-86
48. Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105(45):17414-9
49. Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000;150(6):1507-13
50. Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123(4):569-80
51. Gingras AC, Kennedy SG, O'Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 1998;12(4):502-13
52. Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J. et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20(7):1992-2003
53. Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J. et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11(6):859-71
54. Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A. et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6(11):1122-8
55. Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF. et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22(2):159-68
56. Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1(1):11-21
57. Venkatachalam K, van Rossum DB, Patterson RL, Ma HT, Gill DL. The cellular and molecular basis of store-operated calcium entry. Nat Cell Biol. 2002;4(11):E263-72
58. Kim HJ, Soyombo AA, Tjon-Kon-Sang S, So I, Muallem S. The Ca(2+) channel TRPML3 regulates membrane trafficking and autophagy. Traffic. 2009;10(8):1157-67
59. Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D. et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007;14(5):1029-39
60. Li J, Ni M, Lee B, Barron E, Hinton DR, Lee AS. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ. 2008;15(9):1460-71
61. Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T. et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25(2):193-205
62. Py BF, Boyce M, Yuan J. A critical role of eEF-2K in mediating autophagy in response to multiple cellular stresses. Autophagy. 2009;5(3):393-6
63. Kroemer G, Mariño G, Levine B. Autophagy and the Integrated Stress Response. Molecular Cell. 2010;40(2):280-93
64. Xia H-G, Zhang L, Chen G, Zhang T, Liu J, Jin M. et al. Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy. 2010;6(1):61-6
65. Sitia R, Braakman I. Quality control in the endoplasmic reticulum protein factory. Nature. 2003;426(6968):891-4
66. Walter P, Ron D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science. 2011;334(6059):1081-6
67. Mori K. Signalling Pathways in the Unfolded Protein Response: Development from Yeast to Mammals. Journal of Biochemistry. 2009;146(6):743-50
68. Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B. et al. IRE1 Signaling Affects Cell Fate During the Unfolded Protein Response. Science. 2007;318(5852):944-9
69. Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C. et al. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell. 2007;27(1):53-66
70. Carra S, Brunsting JF, Lambert H, Landry J, Kampinga HH. HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2{alpha} phosphorylation. J Biol Chem. 2009;284(9):5523-32
71. Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10(11):3787-99
72. Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H. et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12(7):982-95
73. Kojima E, Takeuchi A, Haneda M, Yagi A, Hasegawa T, Yamaki K. et al. The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress: elucidation by GADD34-deficient mice. FASEB J. 2003;17(11):1573-5
74. Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H. et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14(2):230-9
75. Peruzzi F, Prisco M, Dews M, Salomoni P, Grassilli E, Romano G. et al. Multiple signaling pathways of the insulin-like growth factor 1 receptor in protection from apoptosis. Mol Cell Biol. 1999;19(10):7203-15
76. Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7(2):85-96
77. Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799-806
78. Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300(5622):1142-5
79. Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P. et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6(6):458-71
80. Morley JF, Morimoto RI. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell. 2004;15(2):657-64
81. Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319(5865):916-9
82. Henis-Korenblit S, Zhang P, Hansen M, McCormick M, Lee SJ, Cary M. et al. Insulin/IGF-1 signaling mutants reprogram ER stress response regulators to promote longevity. Proc Natl Acad Sci U S A. 2010;107(21):9730-5
83. Lee SJ, Murphy CT, Kenyon C. Glucose shortens the life span of C. elegans by downregulating DAF-16/FOXO activity and aquaporin gene expression. Cell Metab. 2009;10(5):379-91
84. Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120(4):449-60
85. Tran H, Brunet A, Griffith EC, Greenberg ME. The many forks in FOXO's road. Sci STKE. 2003;2003(172):RE5
86. Jacobs FM, van der Heide LP, Wijchers PJ, Burbach JP, Hoekman MF, Smidt MP. FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J Biol Chem. 2003;278(38):35959-67
87. Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27(16):2276-88
88. Xuan Z, Zhang MQ. From worm to human: bioinformatics approaches to identify FOXO target genes. Mech Ageing Dev. 2005;126(1):209-15
89. Van Der Heide LP, Hoekman MF, Smidt MP. The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem J. 2004;380(Pt 2):297-309
90. Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z. et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128(2):309-23
91. Hosaka T, Biggs WH 3rd, Tieu D, Boyer AD, Varki NM, Cavenee WK. et al. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci U S A. 2004;101(9):2975-80
92. Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci. 2007;120(Pt 15):2479-87
93. Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol. 2008;20(2):126-36
94. van der Horst A, Burgering BM. Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol. 2007;8(6):440-50
95. Castrillon DH, Miao L, Kollipara R, Horner JW, DePinho RA. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science. 2003;301(5630):215-8
96. Peng SL. Foxo in the immune system. Oncogene. 2008;27(16):2337-44
97. Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24(50):7410-25
98. Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117(4):421-6
99. Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S. et al. FoxO3 Coordinately Activates Protein Degradation by the Autophagic/Lysosomal and Proteasomal Pathways in Atrophying Muscle Cells. Cell Metabolism. 2007;6(6):472-83
100. van den Heuvel AP, Schulze A, Burgering BM. Direct control of caveolin-1 expression by FOXO transcription factors. Biochem J. 2005;385(Pt 3):795-802
101. Bakker WJ, Harris IS, Mak TW. FOXO3a is activated in response to hypoxic stress and inhibits HIF1-induced apoptosis via regulation of CITED2. Mol Cell. 2007;28(6):941-53
102. Bakker WJ, Blazquez-Domingo M, Kolbus A, Besooyen J, Steinlein P, Beug H. et al. FoxO3a regulates erythroid differentiation and induces BTG1, an activator of protein arginine methyl transferase 1. J Cell Biol. 2004;164(2):175-84
103. Kim M-S, Pak YK, Jang P-G, Namkoong C, Choi Y-S, Won J-C. et al. Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat Neurosci. 2006;9(7):901-6
104. Belgardt BF, Husch A, Rother E, Ernst MB, Wunderlich FT, Hampel B. et al. PDK1 deficiency in POMC-expressing cells reveals FOXO1-dependent and -independent pathways in control of energy homeostasis and stress response. Cell Metab. 2008;7(4):291-301
105. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A. et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117(3):399-412
106. Zhou J, Liao W, Yang J, Ma K, Li X, Wang Y, Wang D. et al. FOXO3 induces FOXO1-dependent autophagy by activating the AKT1 signaling pathway. Autophagy. 2012;8(12):1712-23
107. Cohen E, Dillin A. The insulin paradox: aging, proteotoxicity and neurodegeneration. Nat Rev Neurosci. 2008;9(10):759-67
108. Hsu A-L, Murphy CT, Kenyon C. Regulation of Aging and Age-Related Disease by DAF-16 and Heat-Shock Factor. Science. 2003;300(5622):1142-5
109. Ahn SG, Thiele DJ. Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev. 2003;17(4):516-28
110. Ben-Zvi A, Miller EA, Morimoto RI. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci U S A. 2009;106(35):14914-9
111. Westerheide SD, Anckar J, Stevens SM Jr, Sistonen L, Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323(5917):1063-6
112. Kampinga H, Hageman J, Vos M, Kubota H, Tanguay R, Bruford E. et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress and Chaperones. 2009;14(1):105-11
113. Carra S, Crippa V, Rusmini P, Boncoraglio A, Minoia M, Giorgetti E. et al. Alteration of protein folding and degradation in motor neuron diseases: Implications and protective functions of small heat shock proteins. Prog Neurobiol. 2012;97(2):83-100
114. Du ZX, Zhang HY, Meng X, Gao YY, Zou RL, Liu BQ. et al. Proteasome inhibitor MG132 induces BAG3 expression through activation of heat shock factor 1. J Cell Physiol. 2009;218(3):631-7
115. Vos MJ, Zijlstra MP, Kanon B, van Waarde-Verhagen MA, Brunt ER, Oosterveld-Hut HM. et al. HSPB7 is the most potent polyQ aggregation suppressor within the HSPB family of molecular chaperones. Hum Mol Genet. 2010;19(23):4677-93
116. Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S. et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21(13):1621-35
117. Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S. et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26(24):9220-31
118. Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330(6009):1344-8
119. Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R. et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885-9
120. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M. et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131-5
121. Heydrick SJ, Lardeux BR, Mortimore GE. Uptake and degradation of cytoplasmic RNA by hepatic lysosomes. Quantitative relationship to RNA turnover. J Biol Chem. 1991;266(14):8790-6
122. Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7(3):279-96
123. Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33(4):517-27
124. Mizushima N, Hara T. Intracellular quality control by autophagy: how does autophagy prevent neurodegeneration? Autophagy. 2006;2(4):302-4
125. Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12(9):823-30
126. Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131(6):1137-48
127. Orvedahl A, Levine B. Autophagy and viral neurovirulence. Cell Microbiol. 2008;10(9):1747-56
128. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC. et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222-30
129. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T. et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456(7219):264-8
130. Arsov I, Li X, Matthews G, Coradin J, Hartmann B, Simon AK. et al. BAC-mediated transgenic expression of fluorescent autophagic protein Beclin 1 reveals a role for Beclin 1 in lymphocyte development. Cell Death Differ. 2008;15(9):1385-95
131. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(9):741-52
132. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27-42
133. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069-75
134. Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432(7015):307-15
135. Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A. et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809-20
136. Zhao Y, Li X, Cai MY, Ma K, Yang J, Zhou J. et al. XBP-1u suppresses autophagy by promoting the degradation of FoxO1 in cancer cells. Cell Res. 2013;23(4):491-507
137. Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7(12):961-7
138. Kang R, Tang D, Schapiro NE, Livesey KM, Farkas A, Loughran P. et al. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010;17(4):666-76
139. Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306(5698):990-5
140. Rautou PE, Mansouri A, Lebrec D, Durand F, Valla D, Moreau R. Autophagy in liver diseases. J Hepatol. 2010;53(6):1123-34
141. Yang J, Zhao Y, Ma K, Jiang FJ, Liao W, Zhang P. et al. Deficiency of hepatocystin induces autophagy through an mTOR-dependent pathway. Autophagy. 2011;7(7):748-59
142. Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy. 2008;4(2):176-84
143. Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH. et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci. 2008;28(27):6926-37
144. Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11(9):1107-17
145. Filimonenko M, Isakson P, Finley KD, Anderson M, Jeong H, Melia TJ. et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell. 2010;38(2):265-79
146. Vos MJ, Zijlstra MP, Carra S, Sibon OC, Kampinga HH. Small heat shock proteins, protein degradation and protein aggregation diseases. Autophagy. 2011;7(1):101-3
147. Vilchez D, Morantte I, Liu Z, Douglas PM, Merkwirth C, Rodrigues AP. et al. RPN-6 determines C. elegans longevity under proteotoxic stress conditions. Nature. 2012;489(7415):263-8
148. Walker GA, Lithgow GJ. Lifespan extension in C. elegans by a molecular chaperone dependent upon insulin-like signals. Aging Cell. 2003;2(2):131-9
Author contact
![]() Corresponding author: Harm H. Kampinga. Phone: +31-50-3632903; Email: h.h.kampinganl
Corresponding author: Harm H. Kampinga. Phone: +31-50-3632903; Email: h.h.kampinganl