Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(4):457-465. doi:10.7150/ijbs.7562 This issue Cite
Research Paper
Genistein Attenuates Brain Damage induced by Transient Cerebral Ischemia Through Up-regulation of ERK Activity in Ovariectomized Mice
Shiquan Wang1, 4, Haidong Wei2, 4, Min Cai3, Yan Lu1, Wugang Hou1, Qianzi Yang1, Hailong Dong1 ![]() , Lize Xiong1
, Lize Xiong1 ![]()
1. Department of Anesthesiology, Xijing Hospital, Forth Military Medical University, Xi'an, China;
2. Department of Anesthesiology, the Second Affiliated Hospital of Xi'an Jiaotong University College of Medicine, Xi'an, China;
3. Department of Psychosomatic Medicine, Xijing Hospital, Fourth Military Medical University, Xi'an, China;
4. Dr Shiquan Wang and Dr Haidong Wei equally contributed to this work.
Received 2013-9-2; Accepted 2014-3-23; Published 2014-4-8
Abstract
Stroke has severe consequences in postmenopausal women. As replacement therapy of estrogen have various adverse effects and the undermined outcomes. Genistein, a natural phytoestrogen, has been suggested to be a potential neuroprotective agent for such stroke patients. However, the role of genistein and its underlying mechanism in ovariectomized mice has not yet been evaluated. In the present study, ovariectomized mice were treated with genistein (10 mg/kg) or vehicle daily for two weeks before developing transient cerebral ischemia (middle cerebral artery occlusion). The neurological manifestation was evaluated, and infarct volumes were demonstrated by 2,3,5-triphenyltetrazolium chloride staining at 24 h after reperfusion. In addition, phosphorylation of extracellular signal-regulated kinase (ERK) was detected by Western blotting and immunofluorescence staining, and cellular apoptosis was evaluated in the ischemic penumbra. We found that treatment with genistein reduced infarct volumes, improved neurological outcomes and attenuated cellular apoptosis at 24 h after reperfusion. ERK1/2 showed increased phosphorylation by genistein treatment after reperfusion, and an ERK1/2 inhibitor U0126 abolished this protective effect of genistein in terms of infarct volumes, neurological scores and cellular apoptosis. Our findings indicate that treatment with genistein can reduce the severity of subsequent stroke episodes, and that this beneficial function is associated with ERK activation.
Keywords: cerebral ischemia, extracellular signal-regulated protein kinase, genistein, phytoestrogen, postmenopausal stroke.
Introduction
The incidence of stroke in postmenopausal women is similar to that in men. However, worse outcomes (almost 60% stroke-related deaths) have been reported in the former, and as postmenopausal women are more likely to suffer from stroke, it is a matter of serious concern for this population (1). The effectiveness of estrogen replacement therapy in reducing cerebral ischemic injury in experimental animals has been well established (2-4). This led to the thought that estrogen may also be effective for stroke prevention in postmenopausal women. However, estrogen therapy was also accompanied by adverse effects. Moreover, long-term estrogen replacement therapy may lead to more undesirable consequences (5). Thus, in recent years, extensive research has been conducted to find alternatives to conventional hormone replacement therapy. Novel selective estrogen receptor (ER) modulators may serve as a potential treatment with no or less adverse effects (6). Among these, the natural phytoestrogen isoflavone has shown a promising beneficial effect in postmenopausal women (7).
Genistein, a plant-derived isoflavone, is a polyphenolic non-steroidal compound. Since genistein has estrogen-like biological activity (8, 9), its biological effects have been explored in cancer, inflammation, apoptosis, etc. (10-12). Accumulating evidence showed that treatment with genistein could reduce the injury induced by global cerebral ischemia, focal cerebral ischemia or neuronal oxygen-glucose deprivation. The protective effect of genistein supplementation is governed by various processes including mitochondria-dependent apoptosis pathway, inflammation reaction, estrogen receptor-β signaling and oxidative stress (12-14). However, it has not been verified whether genistein supplementation is still beneficial for experimental stroke in ovariectomized mice. More specific molecular mechanisms should be investigated for exploration of the possible clinical targets of stroke.
P44/42 extracellular signal-regulated protein kinase (ERK1/2), a member of the mitogen-activated protein kinase (MAPK) family, plays a key role in cerebral ischemia (15). 17-β-estradiol (E2) supplementation attenuates the delayed neuronal cell death induced by glutamate toxicity, while ERK inhibition blocks the neuroprotective effect of E2 (16). Similarly, the activation of ERK pathway seems to be implicated in the effect of genistein in vitro (17, 18). All these studies suggested that ERK activation may be a primary mechanism of E2-induced ischemic tolerance. Accordingly, we tried to determine whether treatment with genistein prior to middle cerebral artery occlusion (MCAO) is protective against cerebral ischemia in ovariectomized mice, and to investigate the possible role of ERK in this beneficial process.
Materials and Methods
All procedures involving the use of animals were approved by the Ethics Committee for Animal Experimentation of Fourth Military Medical University (Xi'an, China), and proceeded in accordance with the Guidelines for Animal Experimentation of the same university. Female Kunming mice aged 10-12 weeks and weighing 20-25 g were purchased from the Experimental Animal Center of Fourth Military Medical University. The mice were acclimatized for one week in a 12 h light/dark cycle before surgery. A total of 145 mice were used, 8 of which died after ovariectomy (OVX) and 17 were excluded because of inadequate blood flow to the different regions of their brains. Of the remaining 137 mice, 17 were excluded since they showed no cerebral damage despite cerebral ischemia, and 10 mice died within 24 h after reperfusion.
Experimental procedure
The first part of the experiment dealt with the determination of the effect of genistein treatment on the cerebral ischemic injury of the ovariectomized mice. The mice were randomly divided into three groups (n = 17 for each group; 7 were used for neurological score and infarct volume, 3 for terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate nick-end labeling (TUNEL) staining, 4 for Western blot analysis and 3 for immunofluorescence): control, which included mice subjected to MCAO; gen, which included mice subjected to MCAO who were treated with genistein and vehicle, which included mice subjected to MCAO who were treated with vehicle. In the gen group, genistein was administrated intraperitoneally at a dose of 10 mg/kg once daily for two weeks. Genistein dosage was decided based on the dosage used in a previous study (19). In the vehicle group, only vehicle was administered. All the mice were then subjected to a unilateral MCAO. The neurological score, infarct volume, TUNEL staining and protein Bcl-2/Bax ratio were analyzed 24 h post reperfusion and the effect of genistein therapy on ERK regulation at 24 h post ischemia was investigated. ERK activity was examined by Western blotting and immunofluorescence staining for phosphorylated ERK.
The second part of the experiment dealt with the evaluation of the effect of the ERK inhibitor U0126 in order to further testify the role of ERK signaling pathway in neuroprotection induced by genistein. The mice were subjected to MCAO and then randomly assigned to 3 groups (n = 14 for each group; 7 were used for neurological score and infarct volume evaluation, 4 for Western blot analysis and 3 for TUNEL staining): gen, gen + vehicle (genistein plus vehicle of U0126) and gen + U0126 (genistein plus U0126). U0126 (0.5 mg/kg) was injected intraperitoneally 120 min before MCAO. The neurological score and brain infarct volume were evaluated 24 h following reperfusion. Protein expression of Bcl-2 and Bax was detected by Western blotting, followed by TUNEL staining.
Ovariectomy
Ovariectomy (OVX) was executed by making a dorsolateral incision bilaterally on the frontal wall of the abdomen, and the two ovaries were removed as previously described (20). Mice belonging to the control group were subjected to the same operation; however, their ovaries were kept intact. Five days after OVX, vaginal smears of the mice were obtained to confirm cessation of the estrous cycle (21).
Focal cerebral ischemia and reperfusion
The mice were fasted overnight with free access to tap water before surgery. Cerebral ischemia was induced by MCAO as previously described (22). The mice were then intraperitoneally anesthetized with pentobarbital sodium (60 mg/kg). A heat-blunted 6-0 nylon suture was inserted into the right common carotid artery and advanced through the internal carotid artery to obstruct the MCA. The right external carotid artery and the common carotid artery were simultaneously ligated. The suture remained in position for 1.5 h during this arterial occlusion, and was then removed to allow subsequent reperfusion. The body temperature of the mice was maintained at 37 ± 0.5°C with a heating pad controlled by a rectal probe. A laser Doppler flow probe was attached to the right side of the dorsal surface of the skull 2 mm caudal and 4 mm lateral to the bregma. An 80% drop and 70% recovery in the regional cerebral blood flow was considered as a successful ischemic injury.
Assessment of the neurological deficit score
Based on the score system of Garcia et al (23), the neurological behavior of the mice was assessed 24 h post reperfusion by an observer who was blind to the animal grouping. This system consisted of the following six tests: spontaneous activity, symmetry in the movement of the four limbs, forepaw outstretching, climbing, body proprioception and response to vibrissal touch. The final score given to each mouse at the completion of the evaluation was the summation of all six individual test scores. The minimum neurological score was 3 and the maximum was 18.
Infarct volume evaluation
The infarct volume was evaluated as previously described (24). After neurological behavior assessment, the mice were decapitated under deep anesthesia with an overdose of pentobarbital sodium. Six slices of 1-mm thick coronal sections obtained from the entire brain were stained in 2% 2,3,5-triphenyltetrazolium chloride at 37°C for 20 min. The slices were subsequently photographed with a digital camera (Nikon, IXUS 220HS, Japan), and the infarct volume was analyzed with Adobe Photoshop CS5. A relative infarct volume was obtained with correction for edema as: infarct volume = 100% × (contralateral hemisphere volume - non-infarct ipsilateral hemisphere volume) / contralateral hemisphere volume.
TUNEL staining
Cellular apoptosis was evaluated 24 h post reperfusion by an in situ terminal TUNEL kit (TUNEL, Roche Diagnostics, Germany). For this purpose, mice brains were fixed and embedded in paraffin. The tissue was then cut into 4-μm thick coronal sections approximately 1.33 mm from the rostrum to the bregma. The slides were stained according to the manufacturer's instructions. The positive cells were acquired using a 40× objective lens from areas in the ischemic penumbra, and the number of positive cells was expressed as number per 100 μm2. The definition of ischemic penumbra area was as previously described (25).
Western blotting
The brain tissue of the ischemic penumbra was dissected. The sample was then homogenized in a RIPA lysis buffer (Beyotime, Nantong, China) containing whole proteinase inhibitor cocktail on ice. A BCA protein assay kit (Beyotime, Nantong, China) was used to determine the protein concentration. An equivalent amount of protein (30 μg per lane) was loaded and separated by a 12% SDS-PAGE gel. After electrophoresis, the protein was transferred to a polyvinylidene difluoride membrane. The membrane was blocked with 2% bovine serum albumin (BSA) in TBST. After overnight incubation at 4°C with the primary antibodies p-ERK1/2, ERK1/2, Bcl-2, Bax and β-Tubulin (1:1000, Abcam, Cambridge, United Kingdom), the membrane was incubated with horseradish peroxidase-conjugated secondary goat anti-rabbit antibody (1:5000, Pierce, Rockford, IL) for 1 h at room temperature. Program Image J was used to analyze the quantification of the optical density of each band.
Immunofluorescence staining
The sample was fixed with 4% paraformaldehyde at 24 h post reperfusion. After dehydration with 30% sucrose, the brain was frozen and cut into 12-μm sections approximately 1.33 mm from the rostrum to the bregma. The slices were then washed with TBST, incubated with 0.3% TritonX-100 for 5 min at room temperature, and blocked with 5% fetal bovine serum and 3% BSA for 30 min. The primary antibody was rabbit against p-ERK antibody (1:200, Abcam, Cambridge, United Kingdom). After overnight incubation at 4°C, Alexa Fluor 488-conjugated donkey anti-rabbit antibody and NeuroTrace red (1:2000, Molecular Probes; a dye for staining of neurons) were used for fluorescence detection. The samples were visualized under a microscope (OLYMPUS, BX51) through the program DP2-BSW.
Statistical analysis
Data was presented as mean ± standard error of mean, and statistical analysis was performed using the Statistical Package for the Social Sciences (SPSS) Version 16.0 (SPSS Inc, Chicago, IL, USA) for windows, except for neurological scores which were expressed as median with interquartile range and analyzed by Kruskal-Wallis test followed by the Mann-Whitney U test. The Bonferroni test was used as a correction method. Other values were analyzed by one-way ANOVA analysis followed by post hoc Tukey test. P < 0.05 was considered statistically significant.
Results
Physical variables of the animals in the experimental groups
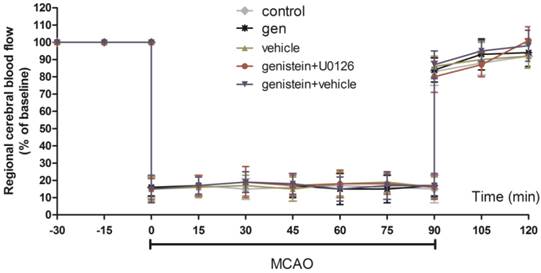
As shown in Fig. 1, there was no difference in the regional cerebral blood flow among the three groups prior to ischemia, during ischemia or after reperfusion. Table 1 shows the gasometric parameters and blood glucose levels in each group. No other parameter except for PaO2 showed any difference in the corresponding values in the control group.
Gasometric parameters and blood glucose levels in arterial blood samples of mice from all experimental groups.
| Control | Genistein | Genistein + U0126 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Basal | Ischemia | Reperfusion | Basal | Ischemia | Reperfusion | Basal | Ischemia | Reperfusion | |
| pH | 7.46 ± 0.02 | 7.38 ± 0.02 | 7.40 ± 0.02 | 7.45 ± 0.02 | 7.36 ± 0.01 | 7.42 ± 0.02 | 7.45 ± 0.02 | 7.37 ± 0.02 | 7.39 ± 0.02 |
| PaCO2(mmHg) | 35 ± 2.0 | 39 ± 1.8 | 30 ± 1.9 | 37 ± 1.9 | 41 ± 1.7 | 31 ± 2.0 | 36 ± 2.0 | 42 ± 1.9 | 29 ± 1.8 |
| PaO2(mmHg) | 282 ± 6.9 | 213 ± 8.3 | 291 ± 7.6 | 279 ± 5.4 | 236 ± 5.9* | 285 ± 6.5 | 290 ± 7.4 | 229 ± 4.9* | 287 ± 6.6 |
| Glucose (mg/dl) | 79 ± 3.6 | 68±4.1 | 83 ± 3.2 | 81 ± 2.9 | 72 ± 3.1 | 85 ± 4.2 | 78 ± 3.3 | 70 ± 2.8 | 82 ± 4.0 |
Data are expressed as mean ± SEM. *Ρ < 0.05 compared with the corresponding values in the control group.
Regional cerebral blood flow in ischemic hemispheres of mice during MCAO surgery Data are expressed as mean ± SEM.
Genistein treatment induced cerebral ischemic tolerance
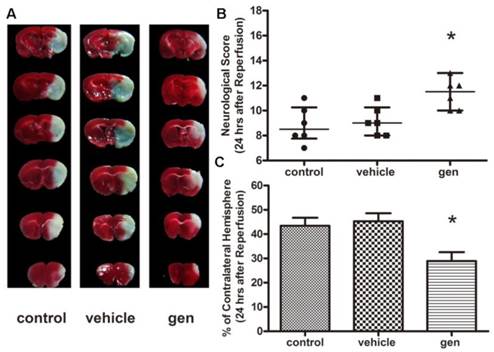
As illustrated in Fig. 2B, as compared to the control group, mice treated with genistein showed better neurological behavior outcomes (P < 0.05), while vehicle had no effect on the neurological score. The infarct volume assay was performed 24 h post reperfusion (Fig. 2C). The infarct volumes in mice subjected to MCAO in the control and vehicle groups were 43.4% ± 3.34% and 45.3% ± 3.35%, respectively. Genistein treatment significantly reduced the infarct volume (28.9% ± 3.63%) as compared to the control group (P < 0.05).
Effect of genistein pretreatment on cerebral ischemia in ovariectomized mice (n = 7). A, representative TTC staining; B, neurological scores; C, infarct volumes 24 h after reperfusion. Genistein pretreatment significantly improved the neurological scores and infarct volumes as compared with the control group. *Ρ < 0.05 versus control group.
Genistein treatment attenuated neuronal apoptosis in cerebral ischemic penumbra
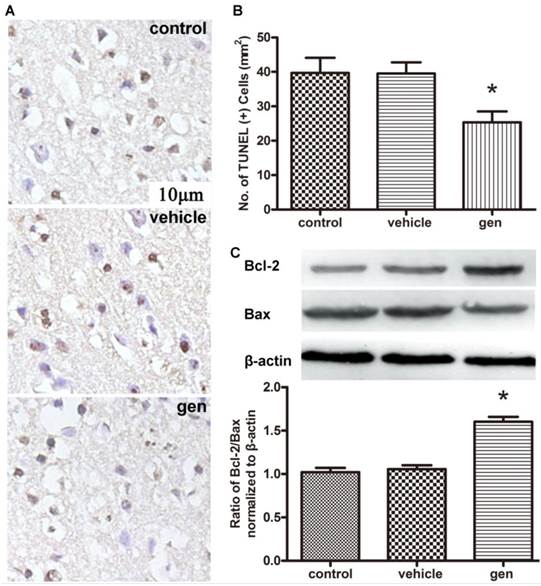
Cellular apoptosis was analyzed to confirm the neuroprotective effect of genistein. The typical photomicrographs of TUNEL staining in ischemic penumbra area and TUNEL-positive cell quantification analysis are shown in Fig. 3A, 3B. Genistein induced a 36.7% reduction in TUNEL-positive cells as compared to the control group (P < 0.05), while vehicle had no effect on cellular apoptosis (42 ± 8 vs. 40 ± 6 per 100 μm2). Detection of apoptosis-associated protein by Western blotting is shown in Fig. 3C. Compared to the control group, genistein increased the ratio of protein Bcl-2/Bax (P < 0.05), while the vehicle showed no such effect.
Effect of genistein pretreatment on cellular apoptosis after reperfusion. A, TUNEL staining of the penumbra slices; B, counting of TUNEL-positive cells at 24 h after reperfusion (n = 3); C, expression of Bcl-2 and Bax in the ischemic penumbra (n = 4). Genistein significantly decreased cellular apoptosis compared with the control group. *Ρ < 0.05 versus control group.
Administration of genistein prompted ERK activation in ischemic penumbra
As shown in Fig. 4B, phosphorylated ERK1/2 content, which represents the active form of the protein, was increased in the group receiving genistein treatment as compared to the control group 24 h following reperfusion (P < 0.01). Administration of vehicle itself had no effect on the level of phosphorylated ERK1/2 in the ischemic penumbra area. Western blotting analysis result was confirmed by the immunofluorescence result. Moreover, the phosphorylated ERK was primarily co-localized with the neuron (Fig. 4A).
Effect of genistein pretreatment on ERK phosphorylation after reperfusion. A, immunofluorescence of p-ERK (n = 3); B, Western blotting of p-ERK (n = 4); Scale bar = 10 μm Genistein treatment increased ERK phosphorylation as compared with the control group. *P < 0.05 compared with the control group.

ERK1/2 inhibition abolished the protection induced by genistein therapy
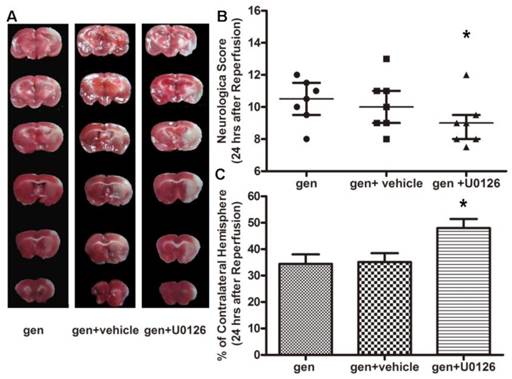
As seen in Fig. 5B, U0126, an inhibitor to the upstream kinase MEK1 of ERK1/2, reversed the neurological improvement by genistein (gen + U0126 vs. gen, P < 0.05). While the neurological score in the vehicle-treated group was similar to that in the genistein-treated group, the infarct volume in genistein + U0126 group was more than that in the genistein group (P < 0.01). Vehicle + U0126 had no effect on the infarct volume (Fig. 5C). ERK1/2 inhibitor U0126 attenuated the reduction of TUNEL-positive cells induced by genistein. The protein ratio of Bcl-2/Bax was also reversed, as seen in Fig. 6 (P < 0.01, gen + vehicle vs. gen + U0126).
Effect of U0126 on genistein-induced neuroprotection (n = 7). A, representative TTC staining; B, neurological scores; C, infarct volumes 24 h after reperfusion The improvement in neurological score and infarct volume induced by genistein was reversed by U0126. *Ρ < 0.05 versus genistein-treated group.
Effect of U0126 on cellular apoptosis when treated with genistein. A, TUNEL staining of the penumbra slices; B, counting of TUNEL-positive cells at 24 h after reperfusion (n = 3); C, expression of Bcl-2 and Bax in the ischemic penumbra (n = 4) U0126 reversed the anti-apoptosis effect of genistein pretreatment. *Ρ < 0.05 versus genistein-treated group.

Discussion
The present study evaluated the neuroprotective effect of genistein against cerebral ischemia in ovariectomized mice. This beneficial effect is thought to be brought about by the activation of ERK signal, as the administration of ERK1/2 inhibitor U0126 dampened the protection induced by genistein. Thus, it can be speculated that genistein treatment induces cerebral ischemic tolerance through activation of ERK.
Accumulating evidences support that E2 pretreatment can induce neuroprotection in brain injuries (2). However, clinical trials have generated controversial reports regarding the effect of estrogen replacement therapy on stroke in postmenopausal women (26, 27). Also, since estrogen replacement therapy is also associated with some undesirable adverse effects, it may not be a suitable alternative in this population (28, 29).
Some plant-derived pharmacological agents such as soy isoflavones (a selective estrogen receptor modulator) have been studied for their treatment potential in ischemic stroke. The ability of isoflavone to cross the blood-brain barrier and its neuroprotective properties without any toxic effects have been well demonstrated in animal models (30). Although genistein has shown a potency similar to isoflavone in brain ischemia (12, 14), its role in protection from stroke in postmenopausal women is unknown. Thus, we tried to determine the effect of genistein treatment on ischemic stroke in ovariectomized mice. Our results revealed that genistein pretreatment could induce ischemic tolerance in ovariectomized mice subjected to MCAO. Genistein treatment at 10 mg/kg dose significantly improved neurological outcome, reduced infarct volume and decreased cellular apoptosis. Hence, we speculate that genistein may be as effective a neuroprotective agent as E2 against cerebral ischemia in ovariectomized mice (31). However, postmenopausal animals show fewer ER, and hence, E2 loses its protective effect in such animals (32). Therefore, it is likely that genistein treatment may not be effective in postmenopausal animals.
For the current study, we selected a dose of 10 mg/kg genistein based on previous results, which showed that intraperitoneal administration of 10 mg/kg genistein improved neurological sores and reduced infarct volumes (19). This dose could also prevent the apoptosis in CA1 area after transient global cerebral ischemia (33). Genistein is quickly and almost completely absorbed orally, and its plasma peak could be reached as quickly as 0.5-6 h in rodents and 5-8 h in humans (34, 35). This high permeability of genistein may be attributed to its low molecular weight and favorable lipophilic property, making passive diffusion its major transport mechanism. The total bioavailability of genistein is considerably high; however, its absolute bioavailability is even lower than 20% in humans, as when ingested, genistein is readily converted to genistein aglycone, its major active form (36). Further, genistein absorption could be altered by breast cancer resistance protein, which complicates the intake of genistein (37). Moreover, the double plasma peaks and a longer terminal half-time of genistein in oral administration as compared to intravenous administration indicate that substantial enterohepatic recycling occurs after oral administration (38). After absorption from the gastrointestinal tract, genistein is metabolized in the enterocytes and delivered to the liver for further hepatic metabolism. Glucuronidation and sulfation are the major pathways for genistein metabolism (36), following which it is primarily redistributed to the reproductive organs. Genistein concentration in the brain tissue is very low (39). The elimination half-time of orally ingested genistein is 6-9 h in rodents and 7-11 h in humans. This half-time is very inconsistent after intravenous administration. However, this refers to the excretion time of total genistein including the metabolites, while the absolute excretion time of genistein aglycone may be much shorter (36). The nonlinear pharmacokinetics of genistein complicates its dose-response effect and plasma or target tissue concentration after administration. Although the pharmacokinetics of genistein has been reported in previous studies, the accurate process that genistein undergoes in the body is still unclear.
MAPKs are important regulators for neuronal survival or death in response to cerebral ischemic injury (40). ERK1/2, a member of the MAPK family which is a critical kinase in the survival signaling pathway, serves as an early indicator of cellular stress response (41). Recent studies suggested that ERK1/2 signaling pathway is important in the biological effects of estrogen, including neuroprotection (42, 43). As a phytoestrogen, isoflavone is structurally similar to estrogen and can mimic its biological effect (44). In the present study, the activation of ERK1/2 was measured at 24 h after reperfusion according to our previous study (45). Genistein was found to increase the phosphorylation of ERK1/2. In order to further explore the role of ERK1/2 in the neuroprotection of genistein, we applied ERK1/2 inhibitor U0126, a commonly used tool drug for the detection of ERK signal. The time point of administration and dose of U0126 was based on the same in our previous study (45). In the current study, we found that the ischemic tolerance induced by genistein was blocked by U0126. Thus, our findings demonstrate that ERK1/2 signaling pathway is essential to the ischemic tolerance induced by genistein pretreatment in ovariectomized mice. However, some researchers reported that U0126 could induce neuroprotection against cerebral ischemia, which contradicts our findings. Some other results regarding the role of ERK in cerebral ischemia are comparable to our findings. We speculate that these controversial findings could be attributed to the differences in the models of cerebral ischemia, the areas of the brain and cell types (40).
In summary, the current study indicates that genistein pretreatment induces neuroprotection against focal cerebral ischemia reperfusion injury in ovariectomized mice. The anti-apoptotic function of genistein through ERK1/2 activation may be involved in its neuroprotective action.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant 81371446 and Grant 43411C1071). The authors greatly appreciate the help rendered by Miss Yaoying Yu, and thank Dr Bairen Wang for carefully reading the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB. et al. Heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127(1):e6-e245
2. McCullough LD, Hurn PD. Estrogen and ischemic neuroprotection: an integrated view. Trends Endocrinol Metab. 2003;14(5):228-35
3. Liu SB, Zhao MG. Neuroprotective effect of estrogen: role of nonsynaptic NR2B-containing NMDA receptors. Brain Res Bull. 2013;93:27-31
4. Merchenthaler I, Dellovade TL, Shughrue PJ. Neuroprotection by estrogen in animal models of global and focal ischemia. Ann N Y Acad Sci. 2003;1007:89-100
5. Viscoli CM, Brass LM, Kernan WN, Sarrel PM, Suissa S, Horwitz RI. A clinical trial of estrogen-replacement therapy after ischemic stroke. N Engl J Med. 2001;345(17):1243-9
6. Musa MA, Khan MO, Cooperwood JS. Medicinal chemistry and emerging strategies applied to the development of selective estrogen receptor modulators (SERMs). Curr Med Chem. 2007;14(11):1249-61
7. Glazier MG, Bowman MA. A review of the evidence for the use of phytoestrogens as a replacement for traditional estrogen replacement therapy. Arch Intern Med. 2001;161(9):1161-72
8. Li W, Liu YH. Effects of phytoestrogen genistein on genioglossus function and oestrogen receptors expression in ovariectomized rats. Arch Oral Biol. 2009;54(11):1029-34
9. Somjen D, Amir-Zaltsman Y, Gayer B, Kulik T, Knoll E, Stern N. et al. 6-Carboxymethyl genistein: a novel selective oestrogen receptor modulator (SERM) with unique, differential effects on the vasculature, bone and uterus. J Endocrinol. 2002;173(3):415-27
10. Lee J, Ju J, Park S, Hong SJ, Yoon S. Inhibition of IGF-1 signaling by genistein: modulation of E-cadherin expression and downregulation of beta-catenin signaling in hormone refractory PC-3 prostate cancer cells. Nutr Cancer. 2012;64(1):153-62
11. Valles SL, Dolz-Gaiton P, Gambini J, Borras C, Lloret A, Pallardo FV. et al. Estradiol or genistein prevent Alzheimer's disease-associated inflammation correlating with an increase PPAR gamma expression in cultured astrocytes. Brain Res. 2010;1312:138-44
12. Qian Y, Guan T, Huang M, Cao L, Li Y, Cheng H. et al. Neuroprotection by the soy isoflavone, genistein, via inhibition of mitochondria-dependent apoptosis pathways and reactive oxygen induced-NF-kappaB activation in a cerebral ischemia mouse model. Neurochem Int. 2012;60(8):759-67
13. Donzelli A, Braida D, Finardi A, Capurro V, Valsecchi AE, Colleoni M. et al. Neuroprotective effects of genistein in Mongolian gerbils: estrogen receptor-beta involvement. J Pharmacol Sci. 2010;114(2):158-67
14. Liang HW, Qiu SF, Shen J, Sun LN, Wang JY, Bruce IC. et al. Genistein attenuates oxidative stress and neuronal damage following transient global cerebral ischemia in rat hippocampus. Neurosci Lett. 2008;438(1):116-20
15. Friguls B, Petegnief V, Justicia C, Pallas M, Planas AM. Activation of ERK and Akt signaling in focal cerebral ischemia: modulation by TGF-alpha and involvement of NMDA receptor. Neurobiol Dis. 2002;11(3):443-56
16. Jover-Mengual T, Zukin RS, Etgen AM. MAPK signaling is critical to estradiol protection of CA1 neurons in global ischemia. Endocrinology. 2007;148(3):1131-43
17. Borras C, Gambini J, Gomez-Cabrera MC, Sastre J, Pallardo FV, Mann GE. et al. Genistein, a soy isoflavone, up-regulates expression of antioxidant genes: involvement of estrogen receptors, ERK1/2, and NFkappaB. FASEB J. 2006;20(12):2136-8
18. Xu SZ, Zhong W, Ghavideldarestani M, Saurabh R, Lindow SW, Atkin SL. Multiple mechanisms of soy isoflavones against oxidative stress-induced endothelium injury. Free Radic Biol Med. 2009;47(2):167-75
19. Castello-Ruiz M, Torregrosa G, Burguete MC, Salom JB, Gil JV, Miranda FJ. et al. Soy-derived phytoestrogens as preventive and acute neuroprotectors in experimental ischemic stroke: influence of rat strain. Phytomedicine. 2011;18(6):513-5
20. Lasota A, Danowska-Klonowska D. Experimental osteoporosis--different methods of ovariectomy in female white rats. Rocz Akad Med Bialymst. 2004;49(Suppl 1):129-31
21. Inderdeo DS, Edwards DR, Han VK, Khokha R. Temporal and spatial expression of tissue inhibitors of metalloproteinases during the natural ovulatory cycle of the mouse. Biol Reprod. 1996;55(3):498-508
22. Yang Q, Yan W, Li X, Hou L, Dong H, Wang Q. et al. Activation of canonical notch signaling pathway is involved in the ischemic tolerance induced by sevoflurane preconditioning in mice. Anesthesiology. 2012;117(5):996-1005
23. Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26(4):627-34 discussion 635
24. Xiong L, Zheng Y, Wu M, Hou L, Zhu Z, Zhang X. et al. Preconditioning with isoflurane produces dose-dependent neuroprotection via activation of adenosine triphosphate-regulated potassium channels after focal cerebral ischemia in rats. Anesth Analg. 2003;96(1):233-7
25. Ashwal S, Tone B, Tian HR, Cole DJ, Pearce WJ. Core and penumbral nitric oxide synthase activity during cerebral ischemia and reperfusion. Stroke. 1998;29(5):1037-46 discussion 1047
26. Rapp SR, Espeland MA, Shumaker SA, Henderson VW, Brunner RL, Manson JE. et al. Effect of estrogen plus progestin on global cognitive function in postmenopausal women: the Women's Health Initiative Memory Study: a randomized controlled trial. JAMA. 2003;289(20):2663-72
27. Wassertheil-Smoller S, Hendrix SL, Limacher M, Heiss G, Kooperberg C, Baird A. et al. Effect of estrogen plus progestin on stroke in postmenopausal women: the Women's Health Initiative: a randomized trial. JAMA. 2003;289(20):2673-84
28. Bushnell C. Stroke Hormones and Outcomes in Women (SHOW) study: is the 'healthy-user effect' valid for women after stroke? Womens Health (Lond Engl). 2009;5(5):485-96
29. Renoux C, Suissa S. Hormone therapy administration in postmenopausal women and risk of stroke. Womens Health (Lond Engl). 2011;7(3):355-61
30. Siow RC, Mann GE. Dietary isoflavones and vascular protection: activation of cellular antioxidant defenses by SERMs or hormesis? Mol Aspects Med. 2010;31(6):468-77
31. Strom JO, Theodorsson E, Holm L, Theodorsson A. Different methods for administering 17beta-estradiol to ovariectomized rats result in opposite effects on ischemic brain damage. BMC Neurosci. 2010;11:39
32. Cai M, Ma YL, Qin P, Li Y, Zhang LX, Nie H. et al. The loss of estrogen efficacy against cerebral ischemia in aged postmenopausal female mice. Neurosci Lett. 2013
33. Liang HW, Qiu SF, Shen J, Sun LN, Wang JY, Bruce IC. et al. Genistein attenuates oxidative stress and neuronal damage following transient global cerebral ischemia in rat hippocampus. Neurosci Lett. 2008;438(1):116-20
34. Kwon SH, Kang MJ, Huh JS, Ha KW, Lee JR, Lee SK. et al. Comparison of oral bioavailability of genistein and genistin in rats. Int J Pharm. 2007;337(1-2):148-54
35. Takimoto CH, Glover K, Huang X, Hayes SA, Gallot L, Quinn M. et al. Phase I pharmacokinetic and pharmacodynamic analysis of unconjugated soy isoflavones administered to individuals with cancer. Cancer Epidemiol Biomarkers Prev. 2003;12(11 Pt 1):1213-21
36. Yang Z, Kulkarni K, Zhu W, Hu M. Bioavailability and pharmacokinetics of genistein: mechanistic studies on its ADME. Anticancer Agents Med Chem. 2012;12(10):1264-80
37. Yang Z, Zhu W, Gao S, Yin T, Jiang W, Hu M. Breast cancer resistance protein (ABCG2) determines distribution of genistein phase II metabolites: reevaluation of the roles of ABCG2 in the disposition of genistein. Drug Metab Dispos. 2012;40(10):1883-93
38. Zhou S, Hu Y, Zhang B, Teng Z, Gan H, Yang Z. et al. Dose-dependent absorption, metabolism, and excretion of genistein in rats. J Agric Food Chem. 2008;56(18):8354-9
39. Schlosser PM, Borghoff SJ, Coldham NG, David JA, Ghosh SK. Physiologically-based pharmacokinetic modeling of genistein in rats, Part I: Model development. Risk Anal. 2006;26(2):483-500
40. Sawe N, Steinberg G, Zhao H. Dual roles of the MAPK/ERK1/2 cell signaling pathway after stroke. J Neurosci Res. 2008;86(8):1659-69
41. Shamloo M, Rytter A, Wieloch T. Activation of the extracellular signal-regulated protein kinase cascade in the hippocampal CA1 region in a rat model of global cerebral ischemic preconditioning. Neuroscience. 1999;93(1):81-8
42. Busch S, Ryden L, Stal O, Jirstrom K, Landberg G. Low ERK phosphorylation in cancer-associated fibroblasts is associated with tamoxifen resistance in pre-menopausal breast cancer. PLoS One. 2012;7(9):e45669
43. Wang S, Ren P, Li X, Guan Y, Zhang YA. 17beta-estradiol protects dopaminergic neurons in organotypic slice of mesencephalon by MAPK-mediated activation of anti-apoptosis gene BCL2. J Mol Neurosci. 2011;45(2):236-45
44. Cederroth CR, Nef S. Soy, phytoestrogens and metabolism: A review. Mol Cell Endocrinol. 2009;304(1-2):30-42
45. Du J, Wang Q, Hu B, Peng Z, Zhao Y, Ma L. et al. Involvement of ERK 1/2 activation in electroacupuncture pretreatment via cannabinoid CB1 receptor in rats. Brain Res. 2010;1360:1-7
Author contact
![]() Corresponding author: Dr Hailong Dong and Dr Lize Xiong, Department of Anesthesiology, Xijing Hospital, The Fourth Military Medical University, Xi'an, 710032 Shaanxi Province, China. Tel: +86-29-8477 5337, Fax: +86-29-8477 1262, E-mail: hldong6com (H. D.), lxiongedu.cn (L. X.).
Corresponding author: Dr Hailong Dong and Dr Lize Xiong, Department of Anesthesiology, Xijing Hospital, The Fourth Military Medical University, Xi'an, 710032 Shaanxi Province, China. Tel: +86-29-8477 5337, Fax: +86-29-8477 1262, E-mail: hldong6com (H. D.), lxiongedu.cn (L. X.).